

Abstract
Amyotrophic lateral sclerosis (ALS) is characterized by progressive degeneration of motoneurons. One potential mechanism is excitotoxicity. We studied the behaviors of spinal neurons using an in vitro preparation of the sacral cord from the G93A SOD1 mouse model of ALS. Measurements were conducted at presymptomatic [approximately postnatal day 50 (∼P50)], early (∼P90), and late (>P120) stages of the disease. Short-latency reflexes (SRs) in ventral roots, presumably monosynaptic, were evoked by electrical stimulation of a dorsal root. The fraction of motoneurons capable of responding to this activation was evaluated by measuring the compound action potential [total motor activity (TMA)] evoked by antidromic stimulation of the distal ventral root. In mutant SOD1 (mSOD1) mice, both the SR and the TMA decreased with age compared with nontransgenic littermates, ruling out the SR as a source of increasing excitotoxicity. Spinal interneuron activity was assessed using the synchronized ventral root bursts generated by both bath application of blockers of inhibitory neurotransmitters (glycine, GABAA) and agonists of glutamate receptors (especially NMDA receptors). After symptom onset, a higher percentage of preparations from mSOD1 mice exhibited bursting, and these bursts exhibited more sub-bursts and a more disorganized pattern. In mSOD1 mice with clear muscle tremor, the ventral roots exhibited spontaneous synchronized bursts, which were highly sensitive to the blockade of NMDA receptors. These data suggest that although short-latency sensory input does not increase as symptoms develop, interneuron activity does increase and may contribute to excitotoxicity.
Introduction
Excitotoxicity is one of several mechanisms being investigated for motoneuron degeneration in amyotrophic lateral sclerosis (ALS) (Cleveland and Rothstein, 2001; Rao and Weiss, 2004; Pasinelli and Brown, 2006). It is generally thought that excitotoxicity arises from excessive Ca2+ entry into motoneurons (Arundine and Tymianski, 2003; Wang and Zhang, 2005; Van Den Bosch et al., 2006), and is exacerbated by the poor motoneuronal Ca2+ buffering (Vanselow and Keller, 2000; von Lewinski and Keller, 2005). Ca2+ entry occurs primarily through two mechanisms: the opening of Ca2+-permeable ligand-gated channels and the activation of voltage-sensitive channels during action potential (AP) generation. Glutamatergic synaptic inputs allow Ca2+ entry through NMDA receptors (NMDARs) and AMPA receptors (AMPARs), although Ca2+ entry through AMPARs depends on the presence of the AMPA 2 subunit (Van Damme et al., 2003). Ca2+ entry also occurs during APs when voltage-sensitive Na+ channels depolarize the cell, leading to the activation of high-threshold voltage-sensitive Ca2+ channels (Powers and Binder, 2001). Therefore, changes in synaptic and intrinsic motoneuron excitability may play a key role in the ALS excitotoxicity hypothesis.
Recent studies suggest that both mechanisms of Ca2+ entry are altered at a very early age in the mSOD1 mouse model (G93A) of ALS. In motoneurons cultured from embryonic mSOD1 mice, an important component of the total Na+ current, the persistent Na+ current (Na PIC), is markedly upregulated (Kuo et al., 2004, 2005). This phenomenon is not restricted to cultured cells, as the Na PIC is also clearly upregulated in neonatal brainstem motoneurons (van Zundert et al., 2008). In addition, in this mSOD1 mouse model of ALS, the Na PIC in interneurons and synaptic input to motoneurons tend to be altered in neonatal preparations (van Zundert et al., 2008). Yet, there are also other changes that may counter the effects of interneuron and motoneuron hyperexcitability, including the abnormal growth of dendrites and the reduction in input conductance seen in neonatal motoneurons in G93A and G85R mice (Durand et al., 2006; Amendola and Durand, 2008; van Zundert et al., 2008).
Studies of spinal neuronal excitability in ALS thus far have been restricted to cell culture and neonates. We have recently developed an adult in vitro preparation of the sacral spinal cord that allowed us to measure the function of spinal motor system in adult mouse (Jiang and Heckman, 2006). In this study, we focused on the changes in the short-latency sensory input and the interneuronal input to motoneurons in the adult G93A mSOD1 mouse. We tested the hypothesis that synaptic inputs, which are involved in modulation of motoneuron activity, are altered in mSOD1 adult mice that range in age from presymptomatic (∼50 d old) to early (∼90 d) and late (>120 d) stages of the disease. Motor output via ventral roots was assessed in response to the short-latency, presumably monosynaptic input, and to the synchronized bursting generated by interneuronal circuits, as spinal interneurons are implicated in this disease (Berger et al., 1992; Nagao et al., 1998; Schütz, 2005; Stephens et al., 2006). The results indicate that the monosynaptic reflex decreases as the disease progresses, but that interneuronal excitability becomes higher than normal. These results suggest that increased excitability of spinal interneurons plays a role in the pathology of ALS, but that low-threshold short-latency sensory input does not.
Materials and Methods
Transgenic mice overexpressing the human mutant SOD1 gene (G93A, mSOD1) and their nontransgenic (NT) littermates were maintained at the Center for Comparative Medicine of Northwestern University Medical Center. All experimental procedures were reviewed and approved by the University Animal Research Committee and were in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals. In this study, we chose to record ventral root activity in sacral segments 1–3 (S1–S3) because their reliable activity can be maintained for several hours (Jiang and Heckman, 2006).
Sacral cord preparation.
For the dorsal root stimulation-evoked reflex experiments short-latency reflexes (SRs), animals of both NT and mSOD1 mice were divided into three age groups [postnatal day 50 (P50)–P60, P90–P100, and P120–P130]. Experiments measuring ventral root synchronized and periodic bursting, driven by spinal interneurons (Cowley and Schmidt, 1995; Hinckley et al., 2005), were performed on mSOD1 mice older than P90. The end stage criterion for mSOD1 mice was the inability to right themselves from a supine position, similar to the standards used by Gurney's laboratory (Chiu et al., 1995). Mice meeting this criterion were not included. We selected motoneurons in the S1–S3 segments based on our previous work and more importantly because ALS-resistant motoneurons are not located in this area (Hamson et al., 2002). It is likely, as in the rat sacral cord (Bennett et al., 1999), that studied motoneurons innervate the skeletal muscle of the tail. Our behavioral studies show that flaccidity develops in the tail during the progression of the disease (J. E. Schuster and C. J. Heckman, unpublished results). The surgical procedure is described in detail in Jiang and Heckman (2006). Briefly, animals were deeply anesthetized with intraperitoneal injections of urethane (0.18 g/100 g for all NT and P50–P60 mSOD1 mice). Significantly lower doses of urethane (∼0.06–0.12 g/100 g) were required for mSOD1 mice P90 and older. Supplemental anesthesia was determined by the animal's response to foot pinching with forceps.
During the surgery, animals were supplied with 95% O2/5% CO2 through a face mask. The spinal column was opened between the low thoracic and caudal segments, and the spinal cord was immediately superfused with modified artificial CSF (mACSF) composed of the following (in mm): 20 NaCl, 140 sucrose, 3 KCl, 1 NaH2PO4, 1.5 MgSO4, 1 CaCl2, 10 glucose, 26 NaHCO3, and 1 kynurenic acid. The osmolarity was adjusted to 340 mOsm by adding sucrose, and the pH was adjusted to 7.4 by adding NaHCO3 to the mACSF when bubbled with 95% O2/5% CO2. The fluid rate was adjusted to 5–7 ml/min, and the dura below lumbar segment was removed with eye-surgical scissors. Immediately after decapitation, the cord was transected at the L1–L2 region, and the dorsal/ventral roots at the caudal cord were cut at their spinal outlets. The transected spinal cord with the attached roots was quickly transferred to a 100 mm Petri dish filled with the mACSF and bubbled with 95% O2/5% CO2. After separating the ventral roots (S1–S3) and the dorsal root that enters at the S3 segment, the cord was cut at the sacral edge of the lumber enlargement and transferred to a recording chamber. The chamber in this experiment used two arrays of bipolar electrodes for the ventral roots (S1–S3). The proximal (recording) and distal (stimulating) electrodes were separated by 5 mm. In addition, one bipolar electrode was used to stimulate each dorsal root, one per side, which preferentially activated the S3 ventral roots. All stimulating and recording electrodes were made of stainless-steel wire coated with Teflon and mounted on a plastic plate (∼6 × 7 mm). The dorsal and ventral roots were placed on the electrodes and covered with a mineral/petroleum jelly (2:1) mixture. The chamber was perfused with ACSF composed of the following (in mm): 126 NaCl, 3 KCl, 1 NaH2PO4, 1.5 MgSO4, 2.5 CaCl2, 26 NaHCO3, and 10 glucose with osmolarity adjusted to 300 mOsm and pH to 7.4 by carefully adding NaHCO3. The ACSF was bubbled with 95% O2/5% CO2 and circulated at 2.5–3 ml/min.
Ventral root recording and protocols.
One root was stimulated at a time through an isolation unit (PSIU6E, Grass Instruments) that was connected to a stimulator (S88, Grass Instruments). The six recording electrodes on the proximal ventral roots were individually connected to six amplifiers (DAM 50, WPI) in differential mode with 1000× gain, high-pass filtering at 300 Hz, and low-pass filtering at 20 kHz. The outputs of these amplifiers were transferred to an analog-to-digital interface (DT1322A, Molecular Devices) and the analog signals were digitized at 20–50 kHz and acquired into a Dell computer controlled by pCLAMP software (version 9.1, Molecular Devices). Because the recording electrodes were of bipolar configuration, the motor output evoked from dorsal root stimulation arrived at the pole proximal to the cord first, while antidromically evoked compound APs from the distal ventral root stimulation arrived at the distal pole first. Therefore, the traces appear with opposite polarity.
The dorsal roots were stimulated with a 0.2 ms pulse at various intensities to evoke SRs in ventral roots. The amplitude of this evoked SR was measured from the peak-to-peak height of the initial reflex response (see Fig. 1). After the threshold of the SR was determined, the dorsal root was stimulated four times at intervals of 1 min, and this protocol was repeated for intensities 1.5, 2, 5, and 10 times threshold. The maximum SR responses (averaged from 5 and 10 times threshold) were compared between mSOD1 and NT mice across the three age groups. To confirm the monosynaptic nature of the evoked SR, the standard deviation (SD) of the latency was calculated (Doyle and Andresen, 2001) from >20 trials at each stimulating intensity. Beside this SR, evoked reflexes with longer latency were rare and therefore were not included in this paper. The total motoneuron activity (TMA) was assessed by stimulation of the distal portion of the ventral roots and recording proximally. For the TMA, the stimulating pulse was set to 0.05 ms to reduce the stimulus artifact, and the interstimulus interval was set to ∼10 s because the evoked TMA recovered much faster than the SR. The same protocol as in the SR stimulation was used to generate the intensity–response relationship for the TMA.

The evoked root-reflex and its difference between NT and mSOD1 mice. A , The root reflexes evoked at different stimulus intensities marked as 1–10 times threshold that just evokes a response. B , Inhibition by DNQX, the antagonist for AMPA receptor, on the root reflex. C , Summary plots showing the relationship between the stimulation intensity and the amplitude of the root reflexes for all three age groups. D , Plot of the maximum root reflexes between NT and mSOD1 mice for all three age groups. Animal number is indicated in each bar, and significant difference is indicated by asterisk (*p < 0.05, ***p < 0.001).
Burst activity was induced by bath application of 100 μm picrotoxin, a GABAA receptor antagonist, and 5 μm strychnine, a glycine receptor antagonist. On a scale of seconds, as shown in Figure 4, these bursts appear to occur simultaneously in all channels, and therefore are referred to as synchronized bursts. The cycle time of the synchronized bursting for each mouse was calculated from an average of 10–20 bursts. Sub-bursts were characterized by a synchronized increase in bursting amplitude before the ongoing burst completely terminated (see Fig. 4). When the time scale was expanded to the millisecond level (see Fig. 4 Bb), the initiation of the bursts for each segment became clear and the burst order from the S1 to S3 segments was ranked (1, 2, 3 as the first, second, and third burst). When burst initiation between segments was within 0.2 ms, the bursts were treated as starting at the same time and the same rank was assigned. For each segment (S1–S3), a percentage was calculated based on how many times that segment was the first to burst during synchronized activity. The calculations were made from at least 10 synchronized bursts. Differences in these parameters were compared between NT and mSOD1 mice. Due to difficulty in determining the end of each burst, the burst duration was not calculated in this study.
To investigate the origination and cellular mechanism of the synchronized bursts induced by picrotoxin and strychnine, Mg2+, AMPA and kainate acid/DNQX (the agonists/antagonist of AMPAR), and NMDA/APV (the agonist/antagonist of NMDA receptor) were used in various combinations. The characteristics of the resulting bursts were then compared between NT and mSOD1 mice.
All chemicals in this study were purchased from Sigma-Aldrich. The statistical analyses were performed with ANOVA, χ2 tests, Student's t tests, and univariate ANOVA using SPSS software.
Results
Short-latency reflexes are reduced in mSOD1 animals as symptoms appear
The changes in short-latency sensory input to spinal motoneurons in the mSOD1 mouse were evaluated by stimulating the dorsal root and recording the SR from the ventral root (Fig. 1). Because the S3 ventral root had the best response to the dorsal root stimulation in this experimental setting, only the evoked response recorded from the S3 ventral root was analyzed. The threshold for this SR was found to be 5–15 μA. The stimulus–response relationship showed that the maximum SR was mostly reached at an intensity of 2 times threshold with a slight, further increase up to 5–10 times threshold intensity, as illustrated in Figure 1, A and C. The latency for the SR was stable and short (∼2 ms) in both NT and mSOD1 mice, and therefore it is likely mediated through monosynaptic transmission, as reported by two other laboratories (Li et al., 2004; Moghaddasi et al., 2007). This was further supported by jitter analysis (Doyle and Andresen, 2001), which showed little variability in the SR latency (stimulation, mean latency ± SD: 1.5 times threshold, 2.43 ms ± 10 μs; 5 times threshold, 2.34 ms ± 20 μs; n = 4 mice). In four mice (2 NT and 2 mSOD1), the receptor that mediated the SR was tested. As shown in Figure 1 B, 3–5 min bath application of 10 μm DNQX completely blocked the SR, suggesting that this monosynaptic SR was mainly mediated through AMPAR. The averaged peak-to-peak amplitude of the SR at 5–10 times threshold was found to be significantly smaller only in the two older age groups of mSOD1 mice than NT mice (Fig. 1 D) (P90: p < 0.05; P120: p < 0.001).
Motoneuron death is the major pathological consequence in ALS. For this reason, the amplitude of the SR could be reduced due to progressive motoneuron death instead of loss of short-latency synaptic input. To evaluate this possibility, TMA was assessed by the antidromic response at the proximal ventral root electrode to electrical stimulation of the distal ventral root. It was found that the proximal ventral root threshold was reached in response to shorter stimulation times during antidromic activation, 0.05 ms versus 0.2 ms for the SR, while the stimulus intensity required remained the same, 5–10 μA. As seen in Figure 2, A and B, the stimulus–response relationship of the evoked TMA was similar to the SR in Figure 1. Comparison of the maximum TMA reveals that it was significantly smaller in all age groups of mSOD1 mice than that in NT mice (P50: p < 0.05; P90: p < 0.001; P120: p < 0.01), with the decrease progressing with age.

The antidromically activated ventral root TMA and its difference between NT and mSOD1 mice. A , An example of the antidromically evoked TMA at different intensities marked as 1–10 times threshold. Note that the compound action potentials are mirrored compared with the trace in Figure 1; see Materials and Methods for explanation. B , Summary plots showing the relationship between the stimulation intensity and the amplitude of the TMA in all three age groups. C , Plot of the evoked maximum TMA between NT and mSOD1 mice for three age groups. Animal number is indicated in each bar, and significant difference is indicated by asterisk (*p < 0.05, **p < 0.01, ***p < 0.001).
The above data showed progressive decreases in the SR and the TMA in mSOD1 mice with increased age compared with their NT littermates. Thus the decrease in the SR could be due to loss of motoneurons as reflected in the decreased TMA. If the loss of the SR was greater than the TMA, then the number of sensory synapses or their efficacy may decrease with age. Although it appears to be a steeper decline in the SR compared with TMA (compare Figs. 1 D, 2 C), this was not statistically significant (p = 0.062 by univariate ANOVA, 9–15 experiments per group). This result rules out the possibility that the SR is a source of increasing excitotoxic drive to motor neurons. One factor that might have influenced these results is that the mSOD1 are more prone to injury during the dissection process. The mSOD1 preparations, however, showed just as good survival times in vitro as did the NT preparations, both in terms of duration of reflex outputs and consistency of effects of dorsal root stimulation.
Bursting generated by interneuronal inputs is altered after symptom onset
A large proportion of the total synaptic input to motoneurons is mediated by spinal interneurons. Thus, a functional change in interneurons in ALS may also affect motoneuron excitability. To assess the excitability of interneuronal circuits, we analyzed the synchronized bursts recorded in ventral roots by blocking inhibitory synaptic transmission (Cowley and Schmidt, 1995; Hinckley et al., 2005). As shown in Figure 3 A, synchronized bursts were produced in some preparations by bath application of GABAA and glycine receptor antagonists, picrotoxin (100 μm) and strychnine (5 μm). The synchronous coordination between roots suggests involvement of excitatory spinal interneuronal network in the burst genesis, which was supported by the blocking effects of DNQX (Fig. 3 A) (n = 4) or APV (Fig. 3 C) (n = 3). For the cords without the inducible bursts, the unsynchronized bursts was produced by NMDAR activation under blocking AMPAR (Fig. 3 B) (n = 2), making the burst origin from strong gap junctions between motoneurons and recurrent excitation pathway unlikely in this experimental setting (see Discussion).
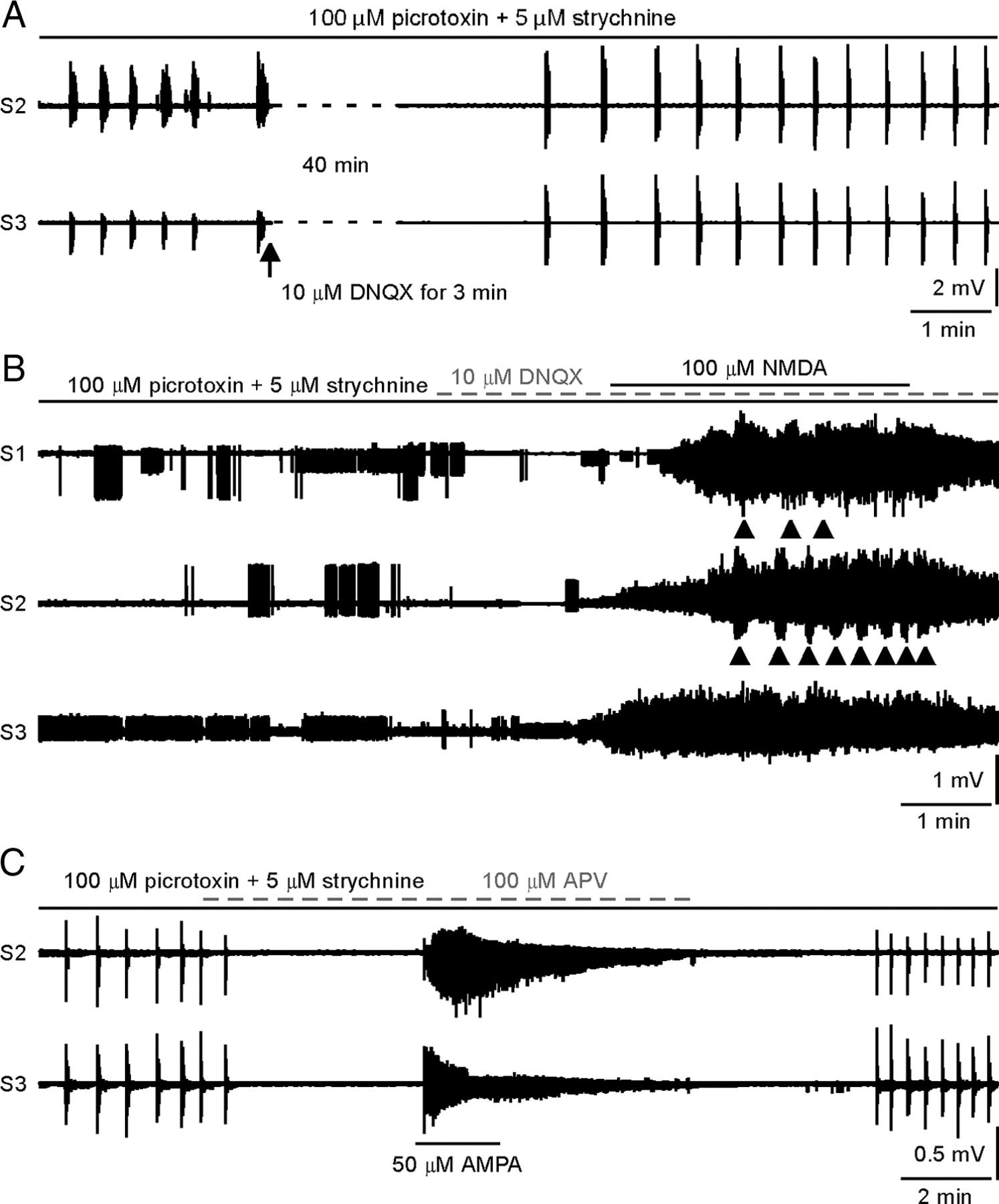
Bursting activity and its mechanism. A , Synchronized bursts were produced by bath application of picrotoxin and strychnine. Addition of DNQX blocked the bursts, suggesting the bursts originated from premotor interneurons. B , In this example, synchronized bursts did not occur after picrotoxin and strychnine application. Subsequent NMDA bath application produced spontaneous firing and unsynchronized bursts indicated by the arrows. C , Synchronized bursts were produced by bath application of picrotoxin and strychnine. Bursts were blocked with the NMDA receptor antagonist APV, subsequent AMPA receptor activation only produced tonic firing suggesting that synchronized bursting does not originate from AMPA receptors. S1–S3 ventral roots are from the same side of the spinal cord.
From the analysis of a total of 45 adult mice (in both the P90 and P120 groups), several features of these synchronized bursts were different between mSOD1 and their NT littermates. First, although not all preparations exhibited synchronized bursts following administration of picrotoxin and strychnine, the proportion of bursting preparations was higher in mSOD1 than NT (mSOD1: 21/26; NT: 10/19; χ2 test: χ2 = 4.06, p < 0.05). The lower occurrence of bursting in NT mice could be due to selective interneuron damage during the dissection. This is, however, highly unlikely since mSOD1 cords would be more susceptible to damage due to the disease. Second, as previously reported (Cowley and Schmidt, 1995), some bursts contained multiple sub-bursts, which is probably due to repetitive burst initiation from spinal interneurons. As illustrated in Figure 4 and summarized in Table 1, mSOD1 mice exhibited significantly more sub-bursts than NT mice (p < 0.05). Third, in NT mice, most bursts started at S3 and then quickly spread to S2 and S1. In contrast, a significantly higher portion of the synchronized bursts in mSOD1 mice started at S2 (Fig. 4 Bb, Table 2). In other words, the segmental order of excitability in mSOD1 interneurons is disrupted. However, the burst in mSOD1 mice does not occur at higher frequency than that in the NT mice (the averaged burst cycle, mean ± SD: for NT, 1.19 ± 0.22 min, n = 9; for mSOD1, 0.89 ± 0.65 min, n = 14; t test, p = 0.2).

Characteristics of the synchronized bursts. A , The examples show the difference in sub-bursting architecture between NT and mSOD1 mice. These bursts are produced by bath addition of 100 μm picrotoxin and 5 μm strychnine in the recording solution. B , The examples show initiation of the burst in S1–S3 ventral roots. Panels a show the regular displays of whole burstings including sub-bursting. When the time scale is expanded in b , timing differences in burst initiation in ventral roots S1–S3 become clear. The dashed lines indicate the starting time point. The sub-bursts are marked by the black dots. S1–S3 ventral roots are from the same side of the spinal cord.
Occurrence of sub-bursts
Distribution of burst initiation in S1–S3 ventral roots
NMDARs play an important role in bursting
It is known that the NMDAR has an important role in the generation of rhythmic bursting in the spinal cord (e.g., Schmidt et al., 1998; Bonnot et al., 2002). We tested whether this receptor contributed to the increased bursting activity generated by the interneurons in mSOD1 mice. Our data showed that administration of high doses of NMDA (100 μm) following block of AMPARs by DNQX can reinitiate synchronized bursting, but at an increased frequency compared with the pre-DNQX state (Fig. 5 A). The reverse experiment, blocking NMDAR and then adding AMPA, generated tonic firing but no bursting (Fig. 3 C) (n = 3). Because AMPARs are quickly desensitized following activation by AMPA (Mayer and Vyklicky, 1989), kainate was used to activate AMPARs in four additional experiments at doses of 25, 50, and 100 μm. In these experiments an initial burst was followed by tonic firing, but repetitive bursting was not seen (data not shown). In the preparations that did not exhibit bursts in the presence of picrotoxin and strychnine, administration of NMDA after DNQX produced tonic activity with unsynchronized bursts (Fig. 3 B) (n = 2). These results suggest that the NMDARs play a key role in the synchronized bursting following administration of picrotoxin and strychnine. The synchronized nature of the bursts suggests significant activation of interneurons by NMDA, though a role for NMDA acting directly on motoneurons to amplify these effects cannot be completely excluded.
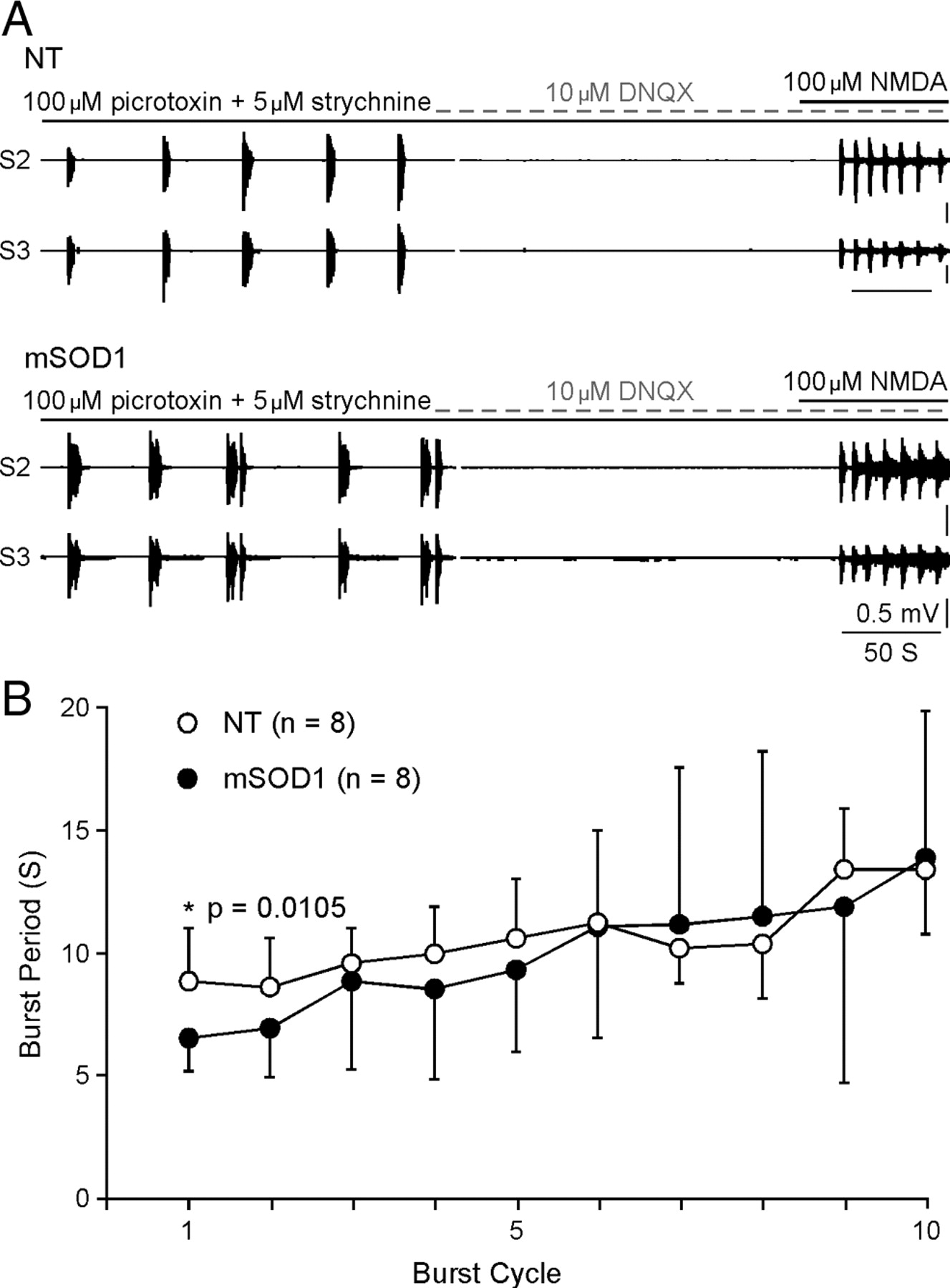
NMDA-induced burst. A , The examples show that NMDA produced synchronized bursts in both NT (top) and mSOD1 (bottom) mice. Before NMDA application, the application of picrotoxin/strychnine and DNQX demonstrates that the synchronized burst can be generated and is coordinated by premotor interneurons. B , The plot summarizes the time course of NMDA-induced bursts. Although the prolonged application of NMDA slows the bursts in both NT and mSOD1 mice, the first cycle of the burst is significantly shorter in mSOD1 mice than in NT mice. *p < 0.05. S2 and S3 ventral roots are from the same side of the spinal cord.
We then quantified the bursts cycle mediated by administration of NMDA following DNQX, picrotoxin, and strychnine in 16 mice (NT: n = 8, ages between P92 and P155; mSOD1: n = 8, ages between P88 and P133). On average, the burst cycle was 9.64 s/cycle in mSOD1 mice and 10.48 s/cycle in NT mice. It is noticeable that initial burst cycle was shorter than the following ones in all tested mice (Fig. 5 B). The reduction in the burst frequency with prolonged exposure to NMDA is likely due to receptor desensitization (Lester and Jahr, 1992; Sornarajah et al., 2008). Thus, the first bursting cycle may reflect better the intact functional state of the receptor. Statistics on the first burst cycle showed that it was significantly shorter in mSOD1 than in NT mice (NT: mean ± SD, 8.85 ± 2.15 s; mSOD1: 6.50 ± 1.9 s; p = 0.011), suggesting a significant change in the excitability of NMDAR in the interneurons of mSOD1 mice.
Spontaneous synchronized bursts emerge in animals well past symptom onset
In mSOD1 mice with clear muscle tremor (in the P120 group), spontaneously occurring, synchronized bursts were detected in the absence of any drugs (n = 7) (Fig. 6). In general, this type of bursting was much shorter in duration than the bursts induced by picrotoxin and strychnine. It generally occurred in various combinations of two to three ventral roots. In the four cords tested, reducing Mg2+ concentration to 1 mm (Fig. 6 A1, left) or to 0 mm (Fig. 6 A2, control) dramatically increased bursting activity, while increasing Mg concentration to 2 mm decreased it (Fig. 6 A1, right). Similarly to the induced burst in Figure 3 A, addition of 10 μm DNQX blocked the spontaneous bursts completely (Fig. 6 A2), suggesting that these bursts are driven by spontaneous activity in interneurons instead of motoneurons. In another two cords, the burst was dose-dependently inhibited by bath application of APV (Fig. 6 B). These results suggest that, similar to the induced bursts, these spontaneous bursts originate in spinal interneurons and that the NMDAR is at least one contributing factor in affecting its occurrence and frequency.

Spontaneous bursts in mSOD1 mice with clear tremor. A , The top ( 1 ) shows the occurrence of the synchronized burst varies with the concentration of magnesium. The bottom ( 2 ) on the left was recorded under 0 mm Mg2+. The burst was blocked by DNQX, indicating the burst was generated through the interneurons. The expansion on the right gives a better view of the synchronization between S2 and S3 ventral roots. B , A recording shows the spontaneous and synchronized burst was dose-dependently blocked by APV, the selective NMDAR antagonist. S2 and S3 ventral roots are from the same side of the spinal cord.
Discussion
Although ALS is characterized by loss of motoneurons in the spinal cord and corticospinal neurons in the motor cortex, loss or changes in other types of neurons may occur (Geracitano et al., 2003; Spalloni et al., 2006; Stephens et al., 2006; Bories et al., 2007; van Zundert et al., 2008). Increased activity in neurons that synapse onto motoneurons could lead to increased depolarization and subsequent excitotoxicity. Our results in the mutant SOD1 mouse model of ALS indicate that short-latency, presumably monosynaptic sensory input to motoneurons, is not a source of excess excitatory input. Instead, as symptoms develop, the reflex from group I muscle afferents declines in parallel with the number of motoneurons responding to the antidromic stimulation.
In contrast, the changes in synchronous bursting patterns suggest that overall activity of spinal interneurons does change as the disease progresses. The synchronous bursting in response to block of inhibitory receptors was more common in mutant animals, suggesting an overall increase in excitability of these cells or a change in their synaptic connectivity. Pattern changes in the bursting included increased sub-bursts per burst period and disrupted ordering of bursting. It was also striking that spontaneous bursts, occurring without administration of inhibitory blockers, emerged in mSOD1 mice with clear symptomatic tremors. Blocking either AMPARs or NMDARs eliminated bursts, but only administration of NMDA following AMPAR blockade reinduced bursting patterns. Moreover, the spontaneous bursting patterns that developed as the disease became more severe were sensitive to changes in extracellular Mg2+ concentration and APV, suggesting increased activity within circuits of interneurons that either have NMDARs or project to motoneurons via NMDARs. This change may be a major source of the tremor that characterizes the later stages of the disease. Further investigations are required to determine whether these spontaneous bursts are due to upregulation in NMDARs, changes in the intrinsic properties of the interneurons, or other modifications within these circuits. Behavioral testing has shown that the inhibition of NMDARs prolonged survival in mSOD1 mice (Wang and Zhang, 2005). The present study proposes a possible cellular mechanism for that observation.
Synchronous bursts are likely due to interneuronal circuits
The locomotor pattern is generated by an interneuronal network in which the excitatory interneurons initiate the rhythmic activity and the inhibitory interneurons are responsible for alternating the motor commands (Kiehn, 2006). Thus, synchronized bursts can be produced by selectively blocking the GABAA and/or glycine receptors in these inhibitory interneurons (e.g., Cowley and Schmidt, 1995; Bonnot and Morin, 1998; Hinckley et al., 2005), which provides a way to study the excitability state in interneurons of mSOD1 mice in our recording setting. In these experiments, we used high concentrations of both picrotoxin and strychnine to induce reliable bursting in the adult spinal cords (González-Forero and Alvarez, 2005). Premotor excitatory interneurons are clearly required to generate these synchronous bursts, as activity was coordinated across several spinal segments and the bursts were eliminated following block of ionotropic glutamate receptors. Previous studies in neonatal preparations have demonstrated that synchronized rhythmic activity on ventral roots may be partly due to gap junctions (Tresch and Kiehn, 2000; Kiehn and Tresch, 2002) or recurrent excitatory pathway (Machacek and Hochman, 2006). Although gap junctions are found in adult spinal interneurons and motoneurons (Rash et al., 2000), their function in neuronal communication in adult animal is not well established (Chang et al., 2000). Nonetheless a role for gap junctions cannot be excluded. If recurrent excitation played a role in the synchronous burst in this study, we would expect spontaneous firing after blocking synaptic transmission with DNQX, which was rarely seen in our induced and spontaneous burst. Neonatal motoneurons also exhibit TTX-resistant oscillation in response to administration of NMDA (Hochman et al., 1994). In our experiments however, block of synaptic input resulted in block of bursting, so it is unlikely to be due to intrinsic bursting in the motoneurons.
Possible mechanisms of changes in interneuronal circuits
The basic mechanism of the synchronized bursts appears to be similar in both NT and mSOD1 mice, in which both exhibited multiple sub-bursts per overall burst. Yet the increased tendency for sub-bursts in mSOD1 animals and the other changes in burst patterns noted above suggest substantial alterations occurred within the interneuronal circuits producing bursts. In this study, we provided clear evidence showing strong involvement of NMDAR in the altered bursts in mSOD1. Thus, one important mechanism to consider is that the NMDARs themselves may be altered. Detailed studies focused on the interneuronal NMDARs are needed to determine whether receptor density, subunit composition, or its phosphorylation state are responsible for the changes shown here. The role of NMDAR does not rule out other cellular mechanisms that may contribute to the bursts recorded in mSOD1 mice. There is a loss of spinal interneurons, including their synapses and fibers to motoneurons (Schütz, 2005; Stephens et al., 2006), but it is difficult to understand how this loss would cause a greater tendency for synchronized bursts or the emergence of spontaneous bursts later in the disease. Inhibition of AMPAR has prolonged survival of mSOD1 (G93A) mice (Van Damme et al., 2003; Andries et al., 2007). Although we showed that AMPAR itself is not involved in burst generation, it may accelerate the burst by depolarizing the cell and facilitating the opening of NMDARs.
Behavior and synaptic abnormalities in early life were reported for both mutant G85R and G93A mice (Amendola et al., 2004; Bories et al., 2007; van Zundert et al., 2008). Bories et al. (2007) showed that EPSPs evoked by stimulation between 1.3 and 2.0 times threshold were reduced in these very young mutant G85R mice. In contrast, our results with G93A mice show that the reflexes produced by this input were similar in NT and mutant SOD1 mice until symptoms developed (around postnatal day 90). This age likely corresponds to the time of degeneration onset of G93A motoneuron (Schütz, 2005). While the animal strains might have differences in their synaptic responses at different ages, it is more likely that the deficits in behavior and sensory inputs recover as the animal develops and then undergo a decline as symptoms progress. A similar early deficit and then recovery was noted in locomotor patterns by Amendola et al. (2004), although there may be differences in this regard in sacral versus lumbar motoneurons. Thus, early deficits in motoneurons and spinal interneurons occur, likely followed by some type of adaptation mechanism. The disease progression may be caused by the compensatory mechanisms or by failure of the compensatory mechanisms. Fundamentally important questions are as follows: what are the mechanisms of compensation, and how do these mechanisms affect disease progression?
As noted above, persistent Na PIC is upregulated in mSOD1 animals at a very early age (Kuo et al., 2004, 2005; van Zundert et al., 2008). The bursting pattern from the interneurons may also involve changes in interneuronal Na PIC. Recent studies have emphasized that Na PICs play a major role in bursting patterns for a wide variety of spinal interneurons (Tazerart et al., 2007; Wilson et al., 2007; Zhong et al., 2007; Ziskind-Conhaim et al., 2008). If, as in motoneurons, the Na PIC is upregulated in spinal interneurons, then this change would contribute to the increased tendency for bursting, manifested in the present study as a greater percentage of preparations showing synchronous bursts after blocking inhibition and the emergence of spontaneous bursts as symptoms become intense. On the other hand, increased Na PIC in motoneurons might increase the SR, which did not occur. Changes in Na PIC over time in both motoneurons and interneurons require further study.
Short-latency reflexes
The short-latency, presumably monosynaptic reflex decreased as the disease progressed, but most of this decrease could be accounted for by loss of motoneurons (as assessed from the antidromic stimulation of the ventral root). Thus this sensory input is not a source of increased excitotoxic drive as symptoms develop. Qualitative examination of human patients with ALS has recently shown that the equivalent reflex in human subjects, the H-reflex, is markedly decreased (T. Siddique, unpublished data). The degree to which the decrease in H-reflex also represents loss of motoneurons versus a loss in group I sensory input remains to be elucidated.
Summary
Excitotoxicity may be an important contributing mechanism causing degeneration of motoneurons in ALS. Our results indicate that NMDAR in spinal interneurons is a potential source of overexcitation of motoneurons as symptoms develop and could be a potential target for new therapeutic strategies.
Footnotes
-
This work was supported by The ALS Association Grant 1626, National Institutes of Health Grants (NS051462 and NS050641), Les Turner ALS Foundation, and Les Turner ALS Foundation/Herbert C. Wenske Foundation Professor.
- Correspondence should be addressed to Mingchen Jiang at the above address. m-jiang{at}northwestern.edu




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}