

Abstract
Aberrantly interconnected granule cells are characteristic of temporal lobe epilepsy. By reducing network stability, these abnormal neurons may contribute directly to disease development. Only subsets of granule cells, however, exhibit abnormalities. Why this is the case is not known. Ongoing neurogenesis in the adult hippocampus may provide an explanation. Newly generated granule cells may be uniquely vulnerable to environmental disruptions relative to their mature neighbors. Here, we determine whether there is a critical period after neuronal birth during which neuronal integration can be disrupted by an epileptogenic insult. By bromodeoxyuridine birthdating cells in green fluorescent protein-expressing transgenic mice, we were able to noninvasively label granule cells born 8 weeks before (mature), 1 week before (immature), or 3 weeks after (newborn) pilocarpine-epileptogenesis. Neuronal morphology was examined 4 and 8 weeks after pilocarpine treatment. Strikingly, almost 50% of immature granule cells exposed to pilocarpine-epileptogenesis exhibited aberrant hilar basal dendrites. In contrast, only 9% of mature granule cells exposed to the identical insult possessed basal dendrites. Moreover, newborn cells were even more severely impacted than immature cells, with 40% exhibiting basal dendrites and an additional 20% exhibiting migration defects. In comparison, <5% of neurons from normal animals exhibited either abnormality, regardless of age. Together, these data demonstrate the existence of a critical period after the birth of adult-generated neurons during which they are vulnerable to being recruited into epileptogenic neuronal circuits. Pathological brain states therefore may pose a significant hurdle for the appropriate integration of newly born endogenous, and exogenous, neurons.
Introduction
The hippocampal dentate gyrus is one of few regions in the adult brain capable of generating new neurons. Elucidating the mechanisms regulating the appropriate integration of these new neurons therefore is of great interest, not only for understanding normal brain function but also for developing therapies to replace neurons lost to disease. Temporal lobe epilepsy may provide significant insights into the importance of neuronal environment in regulating neuronal integration. Specifically, temporal lobe epilepsy is associated with a variety of granule cell abnormalities that may contribute directly to disease development, and it has been hypothesized that many of these abnormal cells may be newborn (Parent and Lowenstein, 2002).
The present study focuses on two abnormalities of granule cells, formation of basal dendrites, and ectopic localization of granule cell bodies. Mature granule cells normally lack basal dendrites. In epileptic animals, however, granule cells with basal dendrites are common (Spigelman et al., 1998; Buckmaster and Dudek, 1999; Yan et al., 2001). Basal dendrites are significant because they project into the dentate hilus and are innervated by granule cell axons in this region, creating recurrent excitatory circuits (Ribak et al., 2000; Austin and Buckmaster, 2004). By destabilizing the dentate gyrus, these circuits may contribute to hyperexcitability and seizures. Similarly, ectopic granule cells, found in the dentate hilus of epileptic brains (Houser, 1990), receive inappropriate innervation and may also contribute to network destabilization (Scharfman et al., 2000; Pierce et al., 2005).
Basal dendrites in epileptic animals are found most frequently on granule cells that, based on their migration pattern, are predicted to be newborn (Ribak et al., 2000; Dashtipour et al., 2001, 2002; 2003; Danzer et al., 2002). Similarly, at least some ectopic granule cells are derived from newborn cells (Jung et al., 2004; Jessberger et al., 2005; Parent et al., 2006). Whether these abnormalities are derived primarily from newborn cells or reflect equivalent disruptions of newly generated and mature cells, however, is not known. Here, we test the hypothesis that adult-generated neurons exhibit an intrinsic critical period after their birth during which they are vulnerable to disruption by pathological brain states. Our findings have important implications for epileptogenesis and for strategies aimed at repairing the damaged brain.
Here, we use a novel combination of approaches to examine the morphological responses of mature, immature, and newborn granule cells during epileptogenesis. Specifically, transgenic mice expressing green fluorescent protein (GFP) in dentate granule cells were given injections of bromodeoxyuridine (BrdU) to “birthdate” granule cells born at different times before or after the development of epilepsy. Epilepsy was induced by treating animals with the cholinergic agonist pilocarpine. Pilocarpine causes intense seizure activity acutely [status epilepticus (SE)], followed by a recovery period and the appearance of brief spontaneous seizures (epilepsy) days to weeks later (Turski et al., 1983, 1984; Shibley and Smith, 2002; Borges et al., 2003). Abnormally connected granule cells appear during this recovery period. Using these approaches, the present study is the first to directly compare the responses of mature and immature adult-generated granule cells to a well characterized brain insult.
Materials and Methods
All procedures conformed to National Institutes of Health and institutional guidelines for the care and use of animals.
BrdU labeling and group designation.
Male and female Thy1-GFP-expressing mice on a C57BL/6 background (provided by Dr. G. Feng, Duke University, Durham, NC) were given once-daily subcutaneous injections of BrdU (100 mg/kg in saline solution) on 3 consecutive days beginning 8 weeks before, 1 week before, or 3 weeks after pilocarpine treatment (see Figs. 2A, 5). Mice were 4, 11, or 15 weeks of age, respectively. Litters were divided so that littermates were used for each time point and treatment. The resulting six animal treatment groups are outlined as follows: group 1, BrdU when the mice were 4 weeks old, no SE, killed at 16 weeks; group 2, BrdU when the mice were 4 weeks old, SE when the mice were 12 weeks old, killed at 16 weeks; group 3, BrdU when the mice were 11 weeks old, no SE, killed at 16 weeks; group 4, BrdU when the mice were 11 weeks old, SE when the mice were 12 weeks old, killed at 16 weeks; group 5, SE when the mice were 12 weeks old, BrdU when the mice were 15 weeks old, killed at 20 weeks; group 6, no SE, BrdU when the mice were 15 weeks old, killed at 20 weeks. Significantly, within each group, two populations of cells were available for analysis: (1) GFP-expressing cells that were 4–16 weeks old (4–20 weeks for groups 5 and 6) and (2) the subpopulation of GFP-expressing cells colabeled with BrdU (their age was determined by the timing of BrdU injections). These populations are also outlined in Figure 2A (hatched color bars, GFP-expressing cells; filled color bars and text, BrdU-labeled, GFP-expressing cells) for groups 1–4. For the purposes of the present study, BrdU-labeled, GFP-expressing cells born 8 weeks before pilocarpine treatment will be referred to as mature (groups 1 and 2), those born 1 week before treatment will be referred to as immature (groups 3 and 4), and those born after treatment will be referred to as newborn (groups 5 and 6).
Pilocarpine treatment.
At 12 weeks of age, mice were given subcutaneous injections of 1 mg/kg methyl scopolamine nitrate in sterile saline. Fifteen minutes later, mice were given subcutaneous injections of 380 mg/kg pilocarpine in saline. All pilocarpine treatments were conducted between 10:00 A.M. and 12:00 P.M. to control for diurnal variations. Mice were observed after the injections for the development of continuous seizure activity (SE), defined behaviorally by continuous tonic/clonic convulsions. Three hours after the onset of SE, mice were given two doses of 10 mg/kg diazepam at 15 min intervals. With this protocol, mice experience a minimum of 3 h of continuous generalized seizure activity (our unpublished observations based on EEG data). Mice were housed in a 32°C incubator for the next 48 h and given 37°C saline subcutaneously to maintain pretreatment weight and improve recovery. Control animals received all drugs and treatments, except they were given saline instead of pilocarpine. Animals that received pilocarpine and did not develop convulsive seizures were also used (groups 1 and 3 only). Importantly, these animals serve as ideal controls because they receive all drug treatments but do not develop spontaneous seizures or brain injury (Mello et al., 1992). Statistical analysis of saline- and drug-treated controls revealed no differences in the percentage of cells with basal dendrites (mean ± SEM; 5 week saline control, 0 ± 0%; 5 week pilocarpine-treated control, 2.6 ± 2.6%; t test, p = 0.495; 12 week control, 6.0 ± 4.2%; 12 week pilocarpine-treated control, 0 ± 0%; t test, p = 0.463), so these groups were binned together.
Four weeks (groups 1–4) or 8 weeks (groups 5 and 6) after pilocarpine or control treatment (when animals were 16 and 20 weeks old, respectively), mice were overdosed with pentobarbital (100 mg/kg) and perfused with PBS plus 1 U/ml heparin, followed by 2.5% paraformaldehyde and 4% sucrose in PBS, pH 7.4. Brains were postfixed for 12 h and cryoprotected in sucrose (10, 20, 30%), and coronal sections were cut on a cryostat at 60 μm. Slide-mounted sections were stored at −80°C until use (Superfrost Plus slides; Fisher Scientific, Pittsburgh, PA).
Immunohistochemistry.
Immunohistochemistry for BrdU and GFP was conducted simultaneously using the following protocol: slide-mounted sections were thawed in Tris buffer (0.02 m Tris-HCl, pH 7.6), incubated for 1 h in 0.5% Igepal in Tris buffer, and incubated for 30 min in 3 μg/ml protease (#P6911; Sigma, St. Louis, MO) with 20 mm CaCl2 at 37°C in Tris buffer. Sections were then incubated for 10 min in ice-cold 0.1 m HCl in distilled H2O, for 20 min in 2 m HCl in 0.05 m PBS at 37°C, and for 10 min in 0.1 m borate buffer, pH 8.5. Sections were incubated for 1 h in blocker [5% normal goat serum (Invitrogen, Carlsbad, CA) plus 0.5% Igepal in 0.1 m PBS, pH 7.4]. Sections were incubated overnight at 4°C in 1:200 mouse monoclonal anti-BrdU antibody (Becton Dickinson, Franklin Lakes, NJ) and 5 μg/ml rabbit polyclonal anti-GFP antibody (AB3080; Chemicon, Temecula, CA) in blocker. Sections were then rinsed in blocker and incubated for 4 h at room temperature in 1:750 Alexa Fluor 488 goat anti-mouse and 1:750 Alexa Fluor 594 goat anti-rabbit antibodies (Invitrogen, Eugene, OR). After rinsing in PBS, sections were dehydrated in alcohols, cleared in xylenes, and mounted with Cytoseal. Antibody specificity was confirmed by including sections from GFP-negative, BrdU-treated animals and GFP-positive, non-BrdU-treated animals.
Immunohistochemistry for the immature neuronal marker doublecortin was conducted using 1:250 goat polyclonal anti-human primary antibodies (sc-8066; Santa Cruz Biotechnology, Santa Cruz, CA) and 1:750 Alexa Fluor 594 donkey anti-goat secondary antibodies (Invitrogen). Doublecortin is expressed by newborn granule cells up until ∼3–4 weeks of age (Brown et al., 2003; Rao and Shetty, 2004; Plümpe et al., 2006).
Microscopy and data collection.
All data analysis was conducted with the investigator blinded to treatment. Slides mounted with up to nine brain sections, located between bregma −1.22 mm and bregma −3.64 mm (Paxinos and Franklin, 2001), were randomly selected for BrdU/GFP immunostaining and analysis. For a given section, all brightly GFP-labeled cells with the soma contained within the tissue section were used. All sections on a slide were screened until 30 BrdU-positive, GFP-expressing dentate granule cells were identified. Once this criterion was reached, no additional sections from the animal were examined (although all additional double-labeled cells in the last section were included to avoid bias). Additional slides, also randomly selected, were immunostained and examined as needed to reach a minimum of three BrdU-positive, GFP-expressing cells per animal. On average, 15.5 ± 1.5 sections per animal were examined (mature/no SE, 20.5 ± 3.6; mature plus SE, 15 ± 5.1; immature and newborn/no SE, 17.5 ± 2.6; immature plus SE, 12.2 ± 3.0; newborn after SE, 9.8 ± 1.2). BrdU-positive, GFP-expressing dentate granule cells were scored to the presence or absence of hilar basal dendrites [63× oil immersion objective; numerical aperture (NA), 1.4]. Any dendrite projecting off the basal pole of the cell (closest to the hilus) and entering the hilus was scored as a hilar basal dendrite. Recurrent basal dendrites, here defined as dendrites that project off the basal pole of the cell but then turn into the molecular layer without entering the hilus, were found infrequently in the present study and were not included in analyses. Granule cell location in the superior blade, inferior blade, or hilus of the dentate gyrus was also noted. Finally, within the granule cell layer, somas were identified as being in the inner (closest to hilus), middle, or outer third.
To determine the baseline number of basal dendrites for each animal, GFP-expressing dentate granule cells located in the inner third of the granule cell layer were analyzed using the same criteria as for BrdU/GFP-labeled cells, except that presence/absence of BrdU labeling was not considered. Cells in the inner third were examined because the majority of BrdU/GFP-labeled cells came from this region. For each animal, 20–30 granule cells were scored. GFP-expressing hilar ectopic granule cells were also scored for the present study (again without regard to BrdU labeling). Four hippocampal sections (eight hilar regions) were examined per animal, and values are presented as the number of hilar ectopic cells per hilus.
Cell loss after pilocarpine-SE was assessed from brain sections Nissl stained with cresyl violet. Sections examined corresponded to Figure 48 in the mouse brain atlas of Paxinos and Franklin (2001). Cell loss was scored in the hilus, dentate granule cell layer, CA3 pyramidal cell layer, and CA1 pyramidal cell layer under 10× magnification using a semiquantitative scale, as follows: (1) no obvious cell loss; (2) <25% cell loss; (3) ∼50% cell loss; and (4) >90% cell loss. Two hippocampi were scored per animal, and the scores were averaged. Finally, we note that the assessment of cell death in the hilus focused only on the large cells typically present in this region (mainly hilar interneurons and mossy cells). No attempt was made to quantify the accumulation of hilar ectopic granule cells, which are typically three to four times smaller than hilar interneurons and mossy cells, using Nissl stain.
Images for figures and neuronal reconstructions were collected using a Leica (Wetzlar, Germany) SP5 confocal microscope set up on a DM IRB2 inverted microscope equipped with a 63× oil immersion objective (NA, 1.4). Using this system, three-dimensional Z-series stacks were captured at 0.5 μm increments for basal dendrites and 0.3 μm increments for axons. Optical zoom was set at two or three times, respectively. Only cells with brightly labeled axons/basal dendrites were used. To quantify imaged processes, confocal image stacks were imported into Neurolucida software for digital reconstruction (version 7.50.4; MicroBrightField, Williston, VT). Axons were reconstructed when they could be followed for a minimum of 40 μm. Most axons quickly left the tissue section containing the soma and thus could only be followed for short distances. Axonal expansions were counted if they were at least two times the diameter of adjacent axonal regions. Basal dendrites were randomly selected from the entire population without regard to length.
Statistics.
For each animal, a minimum of three BrdU-labeled, GFP-expressing neurons was examined. For statistical analysis, values for individual neurons were averaged to provide a mean score for each animal. Statistical analysis was conducted on animal means to avoid pseudo-replication. Individual statistical tests were used as appropriate and are noted in the text. Analysis was conducted using SigmaStat software (version 2.03). Values are presented as means ± SEM.
Figure preparation.
Effectively presenting three-dimensional confocal image stacks is challenging in a two-dimensional format. Specifically, neuronal processes that are easy to follow in the three-dimensional raw data are obscured by processes above and below the structure in two-dimensional maximum projections. Historically, camera lucida drawings avoid this problem because only the target structure is reconstructed. There are obvious advantages, however, to presenting data using confocal images rather than hand (or computer-generated) drawings. To achieve this in a comprehensible two-dimensional format, target neurons have been colorized relative to adjacent neurons. Using this strategy, the target neuron is easily distinguished from adjacent structures, whereas the resulting figure better reflects the raw data. To produce these figures, contrasting pseudocolor images of a confocal image stack were generated in Leica LAS-AF confocal software (1.3.1 build 525). One image stack (red) was transferred to Adobe Photoshop, and all structures, except the target neuron, were deleted from each image. The images were then merged to create a photographic neuronal reconstruction. This reconstruction was then overlaid onto the contrasting (green) maximum projection of the entire image stack. Brightness and contrast of digital images were adjusted to maximize detail. In all cases, identical adjustments were made to images meant for comparison. Finally, we note that although these reconstructions are composed of original confocal images, they should still be considered as reconstructions, with all the associated limitations. For clarity, unmodified maximum projections are also presented with each reconstructed neuron.
Results
GFP is expressed in mature granule cells in the Thy1-GFP mouse line
The present study uses the Thy1-GFP mouse line to characterize granule cell morphology after the development of epilepsy (Feng et al., 2000; Danzer and McNamara, 2004). In this mouse line, granule cells do not express GFP until they are ∼4 weeks old. Specifically, GFP-labeled granule cells do not colocalize the immature neuronal marker doublecortin (0 of 350 GFP-expressing granule cells) (Fig. 1), which is expressed until neurons are ∼3 weeks old, but do colocalize BrdU injected 5 weeks earlier (Fig. 3). This pattern was not altered in epileptic mice, with analysis of animals killed 4 weeks (n = 3), 8 weeks (n = 3), or 11 weeks (n = 2) after pilocarpine-SE exhibiting no doublecortin-positive, GFP-expressing neurons (0 of 832 cells). Notably, 4-week-old granule cells are generally considered to be functionally mature (Markakis and Gage, 1999; Liu et al., 2000; Jones et al., 2003; Ambrogini et al., 2004; Espósito et al., 2005; Overstreet-Wadiche et al., 2006a; Zhao et al., 2006). GFP expression in these animals therefore provides an efficient means to examine the morphology of mature hippocampal dentate granule cells.
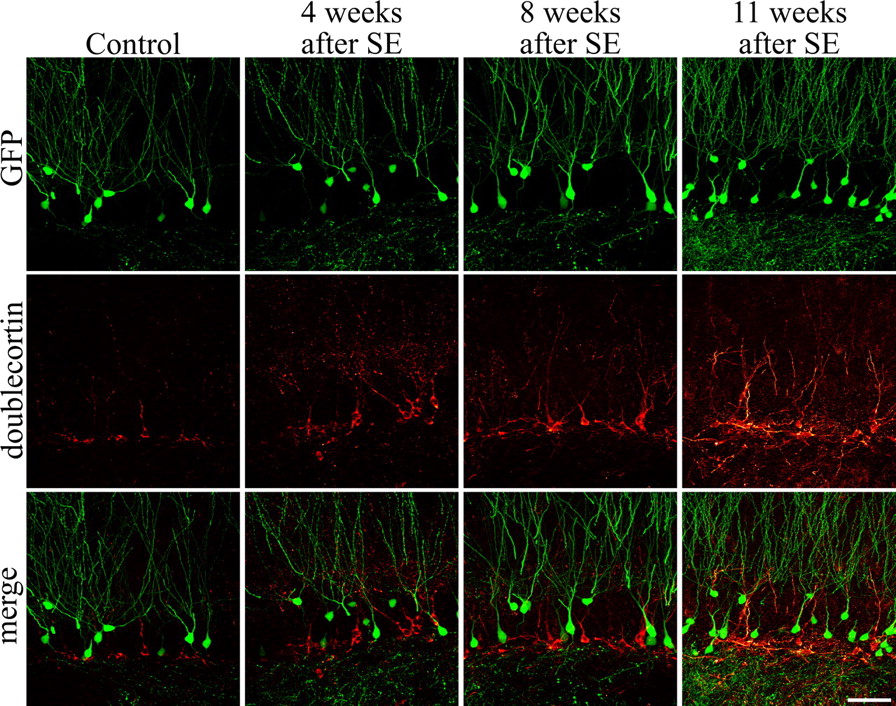
GFP-expressing neurons in the Thy1-GFP mouse line do not colocalize with the immature neuronal marker doublecortin in control animals or animals examined 4, 8, or 11 weeks after SE. Images are confocal optical sections through the dentate gyrus showing GFP (green) and doublecortin (red) immunolabeling. No double-labeled cells were found, indicating that GFP is not expressed until neurons are ∼4 weeks old. Scale bar, 50 μm.
GFP-expressing granule cells from epileptic animals frequently exhibit basal dendrites
Our initial studies focused on the 4- to 16-week-old GFP-expressing granule cells (see Fig. 2A for a description of predicted GFP-expressing granule cell age). Previous studies, using Golgi and biocytin labeling techniques, revealed numerous granule cells with hilar basal dendrites in epileptic animals but not in control animals (Spigelman et al., 1998; Buckmaster and Dudek, 1999; Yan et al., 2001). Analysis of 4- to 16-week-old GFP-expressing granule cells in normal and epileptic animals confirmed the results of these previous studies. Among the two animal groups exposed to pilocarpine-SE (Fig. 2A, groups 2 and 4), 19.6 ± 2.9% of 4- to 16-week-old GFP-expressing granule cells from group 2 and 19.5 ± 2.7% of 4- to 16-week-old cells from group 4 possessed hilar basal dendrites (Fig. 2B, red and blue hatched bars, respectively). Analysis of the two control groups confirmed the typical lack of basal dendrites in normal rodents, with 1.6 ± 2.5% of 4- to 16-week-old cells from group 1 and 1.5 ± 3.2% of 4- to 16-week-old cells from group 3 possessing hilar basal dendrites (Fig. 2B, orange and green hatched bars, respectively). Both pilocarpine-treated groups differed significantly from the two control groups (p < 0.001). The two pilocarpine-treated groups (2 and 4) and the two control groups (1 and 3) did not, however, differ from each other. Because the neuronal populations examined were identical (4- to 16-week-old cells), it is no surprise that they shared identical morphologies.

Schematic detailing the experimental design. The different colors reflect different groups of animals (groups 1–4), whereas hatched versus filled bars within a group reflect different populations of neurons within the same animals. Four groups of animals and eight different populations of neurons are depicted. A, For each group of animals, hatched bars depict the time period over which GFP-expressing cells were generated (0–12 weeks), whereas filled bars depict the period during which the subpopulation of BrdU-labeled, GFP-expressing neurons were generated (either week 4 or 11). Animals received saline or pilocarpine treatment to induce SE when they were 12 weeks old. All animals were killed when they were 16 weeks old. B, Four- to 16-week-old GFP-labeled granule cells exposed to SE when they were 0–12 weeks old (hatched blue and red bars) exhibit basal dendrites significantly more frequently than age-matched neurons from animals that did not experience SE (hatched orange and green bars). Moreover, the two epileptic groups were statistically identical, indicating that pilocarpine treatments were identical between groups receiving injections of BrdU at 4 weeks (blue bar) or 11 weeks (red bar), as expected. C, BrdU-labeled, GFP-expressing dentate granule cells exposed to SE at 1 week of age (red bar) were significantly more likely to possess hilar basal dendrites 4 weeks later relative to age-matched control cells (green bar), cells exposed to SE when they were 8 weeks old (blue bar), and 12-week-old control cells (orange bar). Granule cells exposed to status at 8 weeks of age (blue bar) did not differ from age-matched cells from control animals (orange bar). All values are means ± SEM. ***p < 0.001. P0, Postnatal day 0; DGC, dentate granule cells; HBD, hilar basal dendrites.
Immature, but not mature, granule cells exposed to SE exhibit hilar basal dendrites
A total of 253 BrdU-labeled, GFP-expressing granule cells were obtained from animal groups 1–4 [47 12-week-old cells from group 1 (mature/no SE, n = 8 mice); 75 12-week-old cells born 8 weeks before SE from group 2 (mature plus SE, n = 6); 25 5-week-old cells from group 3 (immature/no SE, n = 5); 106 5-week-old cells born 1 week before SE from group 4 (immature plus SE, n = 7)]. Abbreviations reflect the developmental stage of the neuron at the time of pilocarpine-induced SE. At the end of the experiment, all neurons were mature. Sections were assessed for BrdU-labeled, GFP-expressing cells with basal dendrites projecting into the dentate hilus (hilar basal dendrites) and for BrdU/GFP-labeled cells located within the hilus (ectopic granule cells).
Immature granule cells exposed to SE (immature plus SE) were almost five times more likely to possess a hilar basal dendrite 4 weeks later relative to mature granule cells (mature plus SE) exposed to the identical insult (Fig. 3). Stated differently, almost half of immature granule cells exposed to SE had basal dendrites projecting into the hilus 4 weeks later (Fig. 2C, red bar) (48.4 ± 4.1%). In contrast, significantly fewer mature granule cells exposed to SE had hilar basal dendrites 4 weeks after the insult (Fig. 2C, blue bar) (9.3 ± 3.4%; p < 0.001, ANOVA). A significant interaction was found between treatment and granule cell age (p < 0.001, two-way ANOVA). As expected, immature and mature granule cells exposed to saline treatment or pilocarpine treatment without convulsive SE had typical morphologies 1 month later (Fig. 3), with only 1.5 ± 1.5 and 4.5 ± 3.2% of cells possessing hilar basal dendrites, respectively (Fig. 2C, green and orange bars, respectively). Only immature granule cells exposed to SE were significantly different from their age-matched control group (Fig. 2C, red vs green bar, respectively) (p < 0.001, ANOVA). Mature granule cells exposed to SE did not differ from controls (Fig. 2B, blue vs orange bar, respectively) (p = 0.744). Semiquantitative analysis of the extent of BrdU immunostaining among immature cells exposed to status revealed no differences between cells with and without basal dendrites, suggesting that neurons with basal dendrites do not reflect a distinct population that divided more frequently (thus diluting their BrdU content) relative to cells without basal dendrites (data not shown). Similarly, no systematic bias in neuronal position was observed among groups. Neuronal properties are known to vary with position in the dentate gyrus (Claiborne et al., 1990). The anteroposterior locations of granule cells in the present study, however, were equivalent across groups (data not shown) and between upper and lower blades of the dentate (data not shown). Additionally, the majority of BrdU-labeled, GFP-expressing granule cells were located in the inner third of the dentate granule cell layer, where adult-generated granule cells are typically found. Double-labeled cells were not observed in regions of the brain where adult neurogenesis is limited (data not shown). Combined, these data strongly support the conclusion that the accumulation of basal dendrites in the epileptic brain results from the exposure of immature granule cells to epileptogenic conditions.

A, C, Age-matched granule cells from control animals do not have basal dendrites. B, D, Immature (D), but not mature (B), granule cells exposed to SE exhibit basal dendrites 1 month later. A–D, Digital reconstructions of BrdU-labeled, GFP-expressing granule cells (orange) superimposed on maximum projections showing BrdU-negative, GFP-expressing neighbor cells (green). BrdU labeling is shown in blue. A.1, B.1, C.1, D.1, Confocal maximum projections from confocal z-series stacks used to generate the reconstructions shown in A–D. A.2, B.2, C.2, D.2, Confocal maximum projections showing BrdU labeling. Insets, Merged 90° rotations of GFP (orange) and BrdU (blue) confocal stacks showing colocalization for each neuron. Arrowheads denote axons. Arrows denote basal dendrites. Scale bar: A–D, 20 μm; A.1–D.2, insets, 40 μm.
Persistence of granule cell neurogenesis after SE
To determine whether significant numbers of new cells are generated after pilocarpine-SE, we examined doublecortin immunoreactivity in control and epileptic animals. Doublecortin-immunoreactive cells were present in control animals and animals examined 4, 8, and 11 weeks after pilocarpine-SE (Figs. 1, 4). Gross examination of sections immunostained 4 and 8 weeks after status suggested an increase in labeled cells (Fig. 4), whereas evidence of a decrease was apparent by 11 weeks (although cell numbers were not quantified). Clearly, however, neurogenesis continues for at least 8 weeks after SE, sufficient time for the addition of large numbers of new cells to the dentate gyrus (Cameron and McKay, 2001).

Granule cell neurogenesis persists for at least 2 months after pilocarpine-SE, as demonstrated by labeling of newborn granule cells with the immature neuronal marker doublecortin. Images are single confocal optical sections through the hippocampal dentate gyrus from a control animal and animals examined 4, 8, and 11 weeks after pilocarpine-SE. By 11 weeks, a trend toward declining doublecortin labeling was evident, although labeled cells were still present. Asterisks depict regions of the subgranular zone devoid of doublecortin labeling. Arrowheads depict doublecortin-immunoreactive hilar ectopic granule cells. Scale bar, 200 μm.
Granule cells born after SE develop basal dendrites
To determine the fate of newborn neurons in the seizure-damaged brain, BrdU was given 3 weeks after SE, and animals were killed 5 weeks later (Fig. 5). Eighty-four BrdU-labeled, GFP-expressing neurons from epileptic brains (newborn after SE, n = 6) and 21 neurons from control brains (newborn/no SE, n = 6) were identified for analysis. Just like immature granule cells exposed to SE, almost half of the granule cells born after status possessed hilar basal dendrites, a significant increase over age-matched neurons from control animals (Fig. 6) (newborn/no SE, 1.4 ± 1.4%; newborn after SE, 46.2 ± 6.6%; p = 0.002, Mann–Whitney rank sum test). In comparison, neighboring granule cells, examined without regard to BrdU labeling, in the same pilocarpine-treated animals exhibited hilar basal dendrites only 26.3 ± 5.4% of time (p = 0.041, post hoc t test vs newborn after SE). Finally, only BrdU-labeled newborn granule cells with their cell bodies located in the granule cell layer were included in the analysis of basal dendrites. Significant numbers of granule cells were ectopically located in the hilus in these animals; however, because hilar granule cells invariably have abnormal dendritic morphologies, they were examined separately.

Schematic detailing the experimental design. At 12 weeks of age, animals received pilocarpine to induce SE (group 5). Control animals received saline (group 6). Three weeks after treatment, animals received injections of BrdU to label granule cells. Five weeks later, animals were killed, and BrdU-labeled, GFP-expressing granule cells from epileptic and control animals were analyzed. DGC, Dentate granule cell.

A, B, Newborn granule cells generated after SE possess basal dendrites (B), whereas age-matched granule cells from control animals do not (A). Digital reconstructions of BrdU-labeled, GFP-expressing granule cells (orange) superimposed on maximum projections showing BrdU-negative, GFP-expressing neighbor cells (green) are shown. BrdU labeling is shown in blue/green. A.1, B.1, Maximum projections from confocal z-series stacks used to generate the reconstructions shown in A and B. A.2, B.2, Maximum projections showing BrdU labeling. Insets, Merged 90° rotations of GFP (orange) and BrdU (blue/green) confocal stacks showing colocalization for each neuron. Arrowheads denote axons. Arrows denote basal dendrites. Scale bar: A, B, 20 μm; A.1–B.2, insets, 40 μm.
Granule cells born after SE migrate to ectopic locations in the hilus
Migration of newborn granule cells to ectopic locations in the dentate hilus has been described in the epileptic brain and is thought to contribute to hippocampal hyperexcitability (Scharfman et al., 2000, 2003). Interestingly, 21.4 ± 5.7% of newborn granule cells generated 3 weeks after SE were found in the hilus 5 weeks later (Fig. 7) (p = 0.015, Mann–Whitney rank sum test), whereas none of the age-matched granule cells from control animals were ectopically located (0 of 21 cells). These data contrast with that found for mature and immature granule cells exposed to SE, where negligible numbers were found in the hilus under either control or epileptogenic conditions. Specifically, of the 253 BrdU-positive, GFP-expressing mature and immature granule cells examined in control and epileptic animals, only two were found to be ectopically located in the dentate hilus (data not shown). Both were from epileptic animals (immature plus SE, 1 of 106; mature plus SE, 1 of 75). Combined, these data suggest an increasing vulnerability to disruption with decreasing neuronal age and/or progression of the disease. Consequently, almost 60% of granule cells exposed to abnormal conditions from birth had a basal dendrite, were ectopically located in the hilus, or both.

Granule cells born after SE migrate into the hilus. A, Confocal maximum projection of a control animal showing GFP-labeled granule cells with their axons in the hilus. No BrdU-labeled, GFP-expressing hilar ectopic granule cells were found in control animals. Arrows denote BrdU-positive, GFP-negative cells. B, Digital reconstructions of BrdU-labeled, GFP-expressing granule cells (orange) superimposed on the maximum of the same field showing BrdU-negative, GFP-expressing neighbor cells (green). BrdU labeling is shown in blue. B.1, Maximum projection generated from confocal z-series stacks used to create the reconstruction shown in B. B.2, Maximum projection showing BrdU labeling. Inset, Merged 90° rotation of GFP (orange) and BrdU (blue) confocal stacks showing colocalization within the two hilar ectopic granule cells. DGC, Dentate granule cell. Arrowheads denote hilar ectopic granule cells. Scale bar: A, B, 60 μm; B.1, B.2, inset, 30 μm.
Analysis of ectopic BrdU-negative, GFP-expressing neurons in these same animals paralleled the findings for BrdU-positive, GFP-expressing neurons. No increase in the number of ectopic BrdU-negative, GFP-expressing neurons was observed among animals from groups 1–4, killed 4 weeks after pilocarpine treatment [group 1 (no SE), 0.17 ± 0.17 ectopic granule cells per hilus; group 2 (SE), 0.54 ± 0.22; group 3 (no SE), 0.71 ± 0.29; group 4 (SE), 0.69 ± 0.44; p = 0.490, ANOVA]. Although immature cells appear to be ectopically located in the hilus by 4 weeks after SE (Fig. 4, arrowheads) (see also Parent et al., 2006), these cells are likely too young to express GFP (Fig. 1). Eight weeks after SE (groups 5 and 6), however, BrdU-negative, GFP-expressing neurons were increased in the hilus [group 6 (no SE), 0.33 ± 0.12 ectopic granule cells per hilus; group 5 (SE), 2.4 ± 0.63; p = 0.012, t test]. Presumably, a subset of immature hilar ectopic granule cells not yet expressing GFP 4 weeks after SE now express GFP 8 weeks after status.
Length and branching of basal dendrites
In the present study, basal dendrites were found in large numbers on cells born 1 week before or 3 weeks after SE. To better characterize basal dendrite structure, subsets of BrdU-labeled, GFP-expressing granule cells with basal dendrites were randomly selected from each group for reconstruction (Neurolucida; MicroBrightField). Most basal dendrites were prematurely truncated at the surface of the tissue section, so values underestimate total length. In the present study, mean basal dendrite length was 73.2 ± 12.0 μm for mature/no SE (n = 2 animals, 4 dendrites), 75.0 ± 11.7 for mature plus SE (n = 3 animals, 8 dendrites), 50.6 ± 0.0 for newborn/no SE (based on a single cell), 120.3 ± 40.1 for immature plus SE (n = 5 animals, 13 dendrites), and 115.6 ± 22.1 for newborn after SE (n = 5 animals, 22 dendrites). Although basal dendrites tended to be longer on immature and newborn cells exposed to epileptogenic conditions, the effect was not significant (ANOVA, p = 0.661). Basal dendrites were frequently branched (percentage of branched basal dendrites ± SEM; mature/no SE, 67 ± 33%; mature plus SE, 50 ± 29%; immature plus SE, 95 ± 38%; newborn after SE, 83 ± 27%) regardless of treatment group (ANOVA, p = 0.815). The longest basal dendrites (all immature plus SE or newborn after SE) extended for >250 μm, branching multiple times. For branched basal dendrites, the length of the first-order dendritic segment (distance to first branch) did not differ by treatment group (mature/no SE, 18.4 ± 0.9 μm; mature plus SE, 17.9 ± 10.0 μm; immature plus SE, 21.6 ± 7.9 μm; newborn after SE, 14.2 ± 5.3 μm; ANOVA, p = 0.866). Together, these data indicate that basal dendrites are quite long, frequently projecting well into the dentate hilus (Fig. 3), and are often branched (Fig. 6), similar to apical dendrites.
Mossy fiber axons
Epileptogenesis is associated with aberrant sprouting of granule cell mossy fiber axons to the inner molecular layer of the dentate gyrus (termed “mossy fiber sprouting”). In the present study, most granule cell axons could only be followed a short distance, because they typically left the tissue section after only tens of micrometers. It was therefore not possible to assess mossy fiber sprouting per se in these animals. Only a single cell (mature plus SE) was observed with an axonal projection that could be followed to the inner molecular layer (data not shown). Interestingly, however, mossy fiber sprouts frequently originate close to the soma (Wenzel et al., 2000). Because axons of BrdU-labeled, GFP-expressing neurons could be followed for a short distance, branch frequency was assessed along this proximal axonal region as a potential indicator of increased mossy fiber sprouting. For this analysis, only axons that could be followed for a minimum of 40 μm were used. Most axons could not be followed for >100 μm. To increase statistical power, BrdU-labeled, GFP-expressing neurons from control animals were supplemented with BrdU-negative, GFP-expressing neurons from control animals, and control groups were binned after confirming that they were statistically identical (ANOVA, p = 0.932). The mean axon length examined (before truncation) did not significantly differ among groups [controls, 86.7 ± 8.5 μm (n = 7 animals, 20 axons); mature plus SE, 96.7 ± 16.0 μm (n = 3 animals, 7 axons); immature plus SE, 66.8 ± 5.4 μm (n = 5 animals, 15 axons); newborn after SE, 67.7 ± 7.4 μm (n = 4 animals, 9 axons); ANOVA, p = 0.101]; however, to normalize the data for different axonal lengths, branch frequency was assessed by comparing interbranch interval (total axonal length measured/branch number + 1). Axons that did not branch before truncation received their entire length measured as their score. No significant differences in interbranch interval were observed among groups (control, 63.1 ± 4.3 μm between branches; mature plus SE, 61.5 ± 9.2 μm; immature plus SE, 57.7 ± 4.5 μm; newborn after SE, 52.3 ± 7.7 μm). Indeed, as is evident from these values, many axons did not branch at all in the region examined. Interestingly, however, among the small number of axons that did branch, the distance from the soma to the first branch point was significantly shorter for neurons born after SE relative to controls [controls, 71.6.7 ± 6.7 μm to the first branch (n=5 animals, 6 axons); mature plus SE, 44.3 ± 2.7 μm (n = 2 animals, 4 axons); immature plus SE, 33.5 ± 18.0 μm (n = 2 animals, 3 axons); newborn after SE, 5.7 ± 3.9 μm (n = 2 animals, 4 axons); ANOVA, p = 0.007]. Whether this reflects increased mossy fiber sprouting among these neurons remains to be determined.
Surprisingly, while examining granule cell axons, a previously undescribed change was observed. Granule cell axons from control animals were typically smooth and rapidly tapered as they left the soma and projected into the dentate hilus (Fig. 8, newborn/no SE). Axons from epileptic animals, however, were frequently decorated with bulbous protrusions, most often in close proximity to the soma (Fig. 8, newborn after SE). A quantitative analysis of the number of protrusions located within 20 μm of the soma revealed a significant increase (ANOVA, p < 0.001) among neurons born 8 weeks before (mature plus SE, 2.93 ± 0.81 protrusions/20 μm), 1 week before (immature plus SE, 2.54 ± 0.57), and 3 weeks after (newborn after SE, 3.40 ± 0.61) SE relative to control neurons (0.52 ± 0.23). Binned control groups (mature/no SE; immature/no SE; BrdU-negative, GFP-expressing from control animals) were statistically identical (ANOVA, p = 0.641). No differences were noted among neurons exposed to status at different ages, suggesting that this particular change is not age dependent. Finally, determining the functional significance of these protrusions will require additional ultrastructural and physiological studies; however, the possibility that they are synaptic terminals (al-Hussain and al-Ali, 1995; Acsady et al., 1998) is intriguing given that they were often found within the granule cell layer, and therefore a potential target might be other dentate granule cells.

Composite of confocal maximum projections showing increased numbers of axonal expansions (arrows) on a granule cell born 3 weeks after SE relative to an age-matched granule cell from a control animal. Scale bar, 5 μm.
Neuronal cell loss after SE
Epileptogenesis is associated with neuronal loss in most animal models and is characteristic of the human disease. The extent of neuronal loss among models, however, is highly variable. Even within a model (e.g., pilocarpine), cell loss varies by treatment protocol, age, time after insult, species, genetic background within a species, and even among animals within a study group (Mello et al., 1993; Sankar et al., 1998, 2000; Borges et al., 2003; Yang et al., 2005). Because loss of hippocampal neurons impacts granule cell neurogenesis (Mohapel et al., 2004), cell death was characterized in all animals in the present study. Cell loss was scored in the hilus, dentate granule cell layer, CA3 pyramidal cell layer, and CA1 pyramidal cell layer using a semiquantitative scale. All animals that underwent SE exhibited extensive loss of hilar neurons (Fig. 9, asterisks), variable loss of CA1 (data not shown) and CA3 pyramidal cells (data not shown), and minimal loss of granule cells (Fig. 9). Combined scores for all four regions revealed statistically equivalent loss among epileptic animals (immature plus SE, 8.6 ± 0.6; mature plus SE, 7.6 ± 0.3; newborn after SE, 7.3 ± 0.4; p = 0.25, Kruskal–Wallis ANOVA on ranks). No cell loss was observed in control animals. Perhaps most important, loss of granule cells was minimal and equivalent across status groups (immature + SE, 1.2 ± 0.2, mature + SE, 1.0 ± 0.0; newborn after SE, 1.0 ± 0.0; p = 0.42, Kruskal–Wallis). Finally, within pilocarpine-treated groups, the small amount of variability in cell death that was present did not correlate with either the presence of basal dendrites (Pearson product moment correlation; immature plus SE: R = −0.001, p = 0.998; newborn after SE: R = 0.714, p = 0.111) or the presence of ectopic granule cells (Pearson product moment correlation; newborn after SE: R = −0.104, p = 0.845), supporting the conclusion that variability in cell death was not a confounding factor in the present study.

Bright-field micoroscopy images of cresyl violet-stained sections from control (no SE) and pilocarpine-treated groups. All animals that underwent SE exhibited extensive loss of hilar neurons (asterisks) and minimal loss of granule cells. Cell loss was equivalent among pilocarpine-treated groups. Scale bar, 200 μm.
Discussion
Aberrantly connected dentate granule cells are characteristic of the epileptic brain and may contribute directly to the development of the disease. Only a subset of granule cells, however, exhibits these abnormalities. Exactly why some cells develop abnormalities, and others do not, is unknown. Here, we tested the hypothesis that granule cell age is a critical variable determining whether abnormalities develop. Three principal findings emerge. First, immature granule cells exposed to an epileptogenic insult were significantly more likely to possess abnormal basal dendrites 4 weeks later relative to mature granule cells exposed to the identical insult. Second, the conditions promoting abnormal neuronal integration persisted well after the initial insult, because granule cells born 3 weeks after SE exhibited basal dendrites in equivalent numbers to age-matched granule cells born 1 week before the insult. Finally, in addition to basal dendrites, newborn neurons generated after the insult also migrated to ectopic locations in the dentate hilus. In contrast, negligible numbers of neurons born before the insult were found in the hilus, regardless of developmental stage (mature or immature). The present data provide the first direct evidence that granule cells with basal dendrites in the epileptic brain are newly generated. Moreover, these studies are the first to demonstrate a selective vulnerability of newly generated neurons to disruption in epilepsy, relative to their mature neighbors. Given the predicted destabilizing effect of these abnormalities on hippocampal function, the present data suggest that newly generated neurons may contribute significantly to the development of epilepsy.
Advantages and limitations of the present approach
The main advantage of our approach is that dividing neurons can be birthdated at virtually any time point, whereas the morphology of the birthdated neurons can be assessed months later. Traditional neuroanatomical methods, such as Golgi staining, provide detailed anatomical data but no direct information as to neuronal age (Spigelman et al., 1998; Buckmaster and Dudek, 1999; Yan et al., 2001). Markers of immature neurons, on the other hand, while providing information about the morphology of young cells, provide no information on mature cells (Shapiro et al., 2005; Overstreet-Wadiche et al., 2006b; Shapiro and Ribak, 2006). Viral labeling strategies, to our knowledge, would provide the best alternate means for comparing newborn cells labeled at different time points, although a recent report demonstrating fusion of viral-labeled microglia to mature neurons raises concerns (Ackman et al., 2006). Moreover, infusion of virus invariably leads to brain injury, which can alter the expression of numerous genes, including the neurotrophins (Ballarin et al., 1991; Mudo et al., 1993). Neurotrophins are implicated in epileptogenesis (Danzer et al., 2002; He et al., 2004). The approach used here therefore has the additional advantage of being noninvasive. Finally, although with the present approach double-labeled cells appeared to be more common in epileptic animals, consistent with the neurogenic and survival-promoting effects of seizures (Parent et al., 1997, 1998; Scott and Burnham, 2006), sufficient numbers of control neurons were available without the need for other neurogenic stimuli such as wheel running or environmental enrichment (Kempermann et al., 1998; van Praag et al., 1999). Notably, these stimuli can also alter granule cell morphology (Eadie et al., 2005) and epileptogenesis (Young et al., 1999; Auvergne et al., 2002).
Two concerns of BrdU labeling are worth noting: labeling of cells born well after the BrdU injection and labeling of postmitotic rather than dividing neurons. In the first case, the majority of detectable BrdU-labeled cells appear to be born within 4 d of injection (Dayer et al., 2003), although we cannot exclude the possibility that some small number of labeled granule cells were born later. In the second case, labeling of postmitotic neurons can occur when damaged cells resume DNA synthesis in the presence of BrdU (Kuan et al., 2004). Significantly, however, labeling in the present study occurred either well before or well after SE. Systemic BrdU, therefore, was not present during periods of maximal cell injury. Most importantly, all of the double-labeled cells in the present study were found in neurogenic regions of the hippocampus (data not shown), consistent with their predicted age and with previous studies (Parent et al., 1998, 2006; Rao and Shetty, 2004).
Neuronal critical period for the disruption of adult-generated granule cells
During development, the brain transitions through critical periods during which appropriate stimuli are required for proper formation of axonal and dendritic connections. Visual input, for example, is required for proper maturation of visual cortex. Although the animals examined in the present study were adult, our findings suggest that newborn neurons in these animals go through critical periods similar to those in developing animals, the difference being that the timing of the critical periods is determined by the age of the neuron rather than that of the animal. Specifically, mature granule cells exposed to SE developed neither migration deficits nor basal dendrites (Fig. 2). In contrast, almost half of the immature granule cells exposed to an identical insult developed basal dendrites (Fig. 2). The most severely afflicted neurons, however, were those newborn cells generated after the insult. These neurons were exposed to an abnormal environment from birth and exhibited basal dendrites and ectopic migration (Figs. 6, 7). It is noteworthy that neurons of equivalent ages (5 weeks), born at different times in relation to the insult (1 week before or 3 weeks after), exhibit different phenotypes. This may reflect different critical periods for the disruption of neuronal migration versus dendrite formation (Fig. 10). Typically, migration precedes dendrite formation. The window of time during which migration can be disrupted therefore may end earlier than that for basal dendrite formation. Thus, newborn cells exhibit migration defects and basal dendrites, whereas slightly older, yet still immature, cells exhibit only basal dendrites. Alternatively, changing conditions in the epileptic brain, or the different exposure histories of immature versus newborn cells, might account for the different responses. Interestingly, enhanced plasticity of these young cells (Schmidt-Hieber et al., 2004), although likely important for a variety of physiological processes (Gould et al., 1999; Shors et al., 2001; Cao et al., 2004; Snyder et al., 2005), may render them susceptible to pathological brain states.

Proposed model for the recruitment of adult-generated granule cells into aberrant hippocampal circuits. In this model, only the youngest cells can be induced to migrate into the dentate hilus, whereas the critical period for basal dendrite formation persists for a longer period of time. With neuronal maturity, both critical periods end, and exposure to epileptogenic brain injury induces neither abnormality. DGC, Dentate granule cell.
Mossy fiber plasticity
In contrast to data examining basal dendrites and hilar ectopic granule cells, no significant age-dependent differences in mossy fiber plasticity were observed. It is true that the distance to the first axonal branch point was shorter among neurons born after status relative to controls and neurons exposed to SE/epilepsy had increased numbers of axonal expansions relative to controls, however, neurons from epileptic animals did not differ from each other. These data suggest that neuronal age is not a key factor regulating these axonal changes, but the technical limitations of our analysis (small N and analysis of only proximal axons) should be kept in mind.
Implications for epilepsy
The present study demonstrates a selective vulnerability of newborn and immature neurons to disruption. Abnormal connections formed by these neurons, in turn, may contribute to the development of epilepsy. Whether the net effect of adult neurogenesis is to promote the development of epilepsy, however, is still controversial. Blocking neurogenesis after SE has produced mixed results, for example, with some researchers reporting impaired epileptogenesis and reduced accumulation of ectopic neurons (Jung et al., 2004, 2006), whereas others reported no effect (Parent et al., 1999; Radley and Jacobs, 2003). Notably, however, neurogenesis was blocked either just before or just after the epileptogenic insult in these studies. Because our data indicate that neurons born 1 week before the insult contribute to the pathogenesis of epilepsy, it follows that antimitotic treatments may need to be applied earlier to be truly effective at preventing aberrant neuronal integration.
Recent studies, in contrast, suggest that new neurons could play a protective role in the epileptic brain. In particular, granule cells generated after seizure activity showed reduced excitatory synaptic input and decreased excitability (Jakubs et al., 2006). Notably, however, only 5% of these new neurons exhibited basal dendrites, and none were reported in the hilus. Perhaps significantly, a different model of epileptogenesis was used, and the efficacy at which abnormal newborn neurons were produced appears much lower. Variability across animal models and complex temporal changes within models may account for much of the controversy in the field (Kempermann, 2006). Clearly, additional studies are needed to define the role of newborn neurons in the development of epilepsy.
Implications for neuronal replacement
Neuronal stem cells hold great potential for repairing the damaged brain. It is likely, however, that for any therapy to be effective the new neurons will have to appropriately integrate into existing, and damaged, neuronal circuits. The model system used here provides a unique opportunity to examine the integration of endogenous neural progenitors into a damaged brain. These studies reveal that immature neurons are extremely sensitive to disruptions in the environment and, as a consequence, develop abnormal features that may actually contribute to disease. These findings highlight the need for additional studies aimed at elucidating the factors regulating the appropriate, or maladaptive, integration of newborn neurons into the adult brain.
Footnotes
-
This work was supported by Cincinnati Children's Hospital Medical Center and the Epilepsy Foundation of America. We thank Keri Kaeding, Kenny Campbell, and Chia Yi Kuan for helpful comments on previous versions of this manuscript.
- Correspondence should be addressed to Dr. Steve C. Danzer, 3333 Burnet Avenue, ML 2001, Cincinnati, OH 45229-3039. steve.danzer@cchmc.org




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}