


Abstract
Immunotherapy has led to a paradigm shift in the treatment of some malignancies, providing long-term, durable responses for patients with advanced cancers. However, such therapy has benefited only a subset of patients, with some patients failing to respond to treatment at all and others achieving a limited response followed by tumor progression. Understanding factors contributing to an effective response and further elucidating mechanisms of resistance will be crucial as these therapies are applied more broadly. Genomics-based approaches have significantly advanced the study of response and resistance to immunotherapy in general, and to immune checkpoint blockade more specifically. Here, we review how genomic and transcriptomic approaches have identified both somatic and germline positive correlates of response, including high mutational/neoantigen load and low intratumoral heterogeneity, among others. The genomic analysis of resistant tumors has additionally identified crucial factors involved in resistance to immune checkpoint blockade, including loss of PTEN and upregulation of other immune checkpoints. Overall, the continued use of genomic techniques at the point of care, combined with appropriate functional studies, would ideally lead to a better understanding of why certain patients respond to immune-based therapies, allowing clinicians to identify the subset of patients likely to benefit from such therapy, and potentially providing insight into how other therapies may be added in combination to increase the number of patients who may benefit from immunotherapy. Clin Cancer Res; 22(23); 5642–50. ©2016 AACR.
Introduction
Immunotherapy represents a truly exciting therapeutic modality in oncology. Prior attempts to harness the body's immune system to target malignant cells, including the use of high-dose cytokines (IL2) or adoptive cell transfer of tumor-infiltrating lymphocytes, generated a response in a limited subset of patients and carried the potential for significant toxicity (1, 2).
More recently, the field of cancer immunotherapy has grown tremendously, including the use of therapeutic cancer vaccines and novel adoptive cell transfer with chimeric antigen receptor (CAR) T-cell therapy (3, 4). The discovery of natural “brakes” or checkpoints within the immune system, including CTL-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1), led to the development of antibodies that inhibit these checkpoints—in a manner, “cutting the brakes” to allow T-cell activation and effector functions (5–7). The potential benefit was first demonstrated with the anti–CTLA-4 antibody ipilimumab in patients with advanced melanoma (8, 9) and soon followed by antibodies inhibiting the PD-1 axis, including nivolumab and pembrolizumab, either alone or in combination with ipilimumab (10, 11). These therapies are now FDA-approved in many advanced malignancies, including melanoma, non–small cell lung cancer, renal cell cancer, Hodgkin lymphoma, bladder cancer, and head and neck cancer (8, 10, 12–19).
Unfortunately, only a minority of patients respond to these agents, and fewer still achieve a durable response. For instance, the 3-year survival rate for patients with advanced melanoma treated with ipilimumab is under 30% (9). Here, we discuss genomic and transcriptomic approaches to understanding response and resistance to immune checkpoint therapy, review how such techniques might lead to appropriate selection of “responders” likely to benefit from immunotherapy, and provide a framework for understanding (and ideally overcoming) resistance.
Understanding Response to Immunotherapy
Genomic and transcriptomic approaches have both been employed to better understand the correlates of response to immune check point blockade (Fig. 1).

Genomic correlates of response. Genomics-based approaches and other investigations have led to the identification of multiple mechanisms of response to immune checkpoint blockade, including high mutational and neoantigen load, low intratumoral heterogeneity, infiltration with a clonal T-cell popular, and deficiencies in DNA repair machinery.
Genomic correlates of response. Genomics-based approaches and other investigations have led to the identification of multiple mechanisms of response to immune checkpoint blockade, including high mutational and neoantigen load, low intratumoral heterogeneity, infiltration with a clonal T-cell popular, and deficiencies in DNA repair machinery.
DNA-based approaches
Analysis of mutational and neoantigen load.
The number of somatic mutations varies widely both between and within cancer types (20). Nonsynonymous mutations that are transcribed and translated into a polypeptide may generate a neoepitope, with presentation on the major histocompatibility complex (MHC) molecules and recognition by the adaptive immune system (Fig. 2). Snyder and colleagues identified a relationship between mutational load (nonsynonymous mutations) and response to immune checkpoint therapy in patients with advanced melanoma (21) using whole-exome sequencing on tumor samples from 64 patients treated with CTLA-4 blocking agents (mostly ipilimumab, with 5 patients treated with tremelimumab). A higher mutational load correlated with long-term clinical benefit from CTLA-4 blockade (defined as freedom from disease or stable disease for at least 6 months). Importantly, they experimentally validated the immunogenicity of predicted mutated peptides, demonstrating that they generated T-cell responses in vitro using peripheral blood mononuclear cells from patients (22). These results are concordant with other studies demonstrating that in silico prediction of MHC class I–peptide binding corresponds to experimentally identified T-cell immune responses (23).
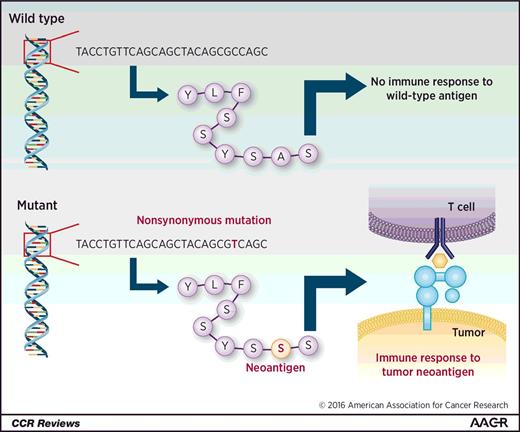
Generation of neoantigens. Wild-type antigens are recognized as “self” and do not generate an immune response. Nonsynonymous mutations may lead to an altered peptide sequence that is ultimately presented on MHC molecules. This altered peptide sequence, therefore, produces a new or “neoantigen,” which may then be recognized by the host immune system, leading to an antitumor immune response.
Generation of neoantigens. Wild-type antigens are recognized as “self” and do not generate an immune response. Nonsynonymous mutations may lead to an altered peptide sequence that is ultimately presented on MHC molecules. This altered peptide sequence, therefore, produces a new or “neoantigen,” which may then be recognized by the host immune system, leading to an antitumor immune response.
This association between mutational load and clinical benefit from CTLA-4 blockade in advanced melanoma was confirmed in work by Van Allen and colleagues using whole-exome sequencing of 110 pretreatment melanoma samples with matched germline tissue (22). Putative neoantigens were identified by generating all possible 9- and 10-mer peptide sequences resulting from a mutation, and were filtered for neoantigens computationally predicted to have a high HLA class I binding affinity. Neoantigen load positively correlated with response to CTLA-4 blockade. Unfortunately, no truly predictive biomarker or recurring neoantigen was identified.
A similar relationship between mutational load and response to PD-1–axis blockade has also identified (24, 25). Rizvi and colleagues performed whole-exome sequencing on non–small cell lung cancer samples from 34 patients treated with the anti–PD-1 antibody pembrolizumab, comprising two independent cohorts (discovery and validation sets). In both cohorts, patients with a durable clinical benefit from pembrolizumab therapy carried a significantly higher nonsynonymous mutational load as compared with patients with no durable benefit. Furthermore, a high mutational load positively correlated with a significant improvement in progression-free survival. Here too, candidate neoantigens were experimentally validated using a high-throughput multimer screen to identify neoantigen-specific reactive T cells that recognized mutant but not wild-type peptide. In some responders to pembrolizumab, such neoantigen-specific T-cell responses appeared to temporally correlate with clinical response.
Rizvi and colleagues further employed a previously validated molecular smoking signature (based on high transversion rates) in an attempt to identify other correlates of response. Interestingly, although presence of the molecular smoking signature did significantly correlate with an improved progression-free survival, self-reported smoking history did not. Prospective, clinically based studies that explore whether this genomic signature may be used to stratify patients above self-reported smoking history are necessary.
The significant association between mutation (and neoantigen) load and response to immune checkpoint blockade has focused attention on tumors with typically high mutational loads (i.e., melanoma, non–small cell lung cancer, and renal cancer). However, this association is not absolute, a number of responders carry a low mutation load, and nonresponders may carry a high mutation load. This discordance holds true for tumor types (such as colorectal cancer) that, on average, have a relatively high number of somatic mutations but do not typically respond to immune checkpoint blockade (20, 26). One possible explanation is the degree of intratumoral heterogeneity (ITH), as bulk sequencing of a tumor may not fully capture the spatial complexity of the mutational landscape (27). In a hypothetical tumor comprised of a single clone with a high mutational load, an immune response to a single neoantigen could target the entire population of tumor cells. Conversely, in another tumor with the same total number of mutations but comprised of a large and heterogeneous population of subclones, each with only a few mutations, an immune response generated against a neoantigen would target a subclonal population, thereby leaving the remainder of tumor cells unaffected.
The importance of ITH was explored by McGranahan and colleagues (28). The authors established heterogeneity by multiregion sequence analysis of a small number of non–small cell lung cancer tumor samples and computationally calculated the proportion of cancer cells harboring a specific mutation. A mutation identified in all spatially distinct regions of a tumor was deemed a clonal neoantigen, and, conversely, a mutation present in only a subset of regions was categorized as a subclonal mutation. By applying this definition to both the Rizvi and Snyder cohorts, the authors demonstrated the importance of clonal architecture. For example, in one patient with non–small cell lung cancer who had a high neoantigen load but no durable clinical benefit with PD-1 blockade, analysis revealed that the tumor was very heterogeneous, with most (over 80%) of the mutations being subclonal. Overall, a low neoantigen subclonal fraction was significantly associated with response to immune checkpoint blockade, suggesting that the spatial organization and depth of mutations may also play a role.
Taken together, these studies support the hypothesis that the number of nonsynonymous mutations (and ultimately neoantigens) significantly correlates with clinical response to immune checkpoint blockade, but with certain limitations. First, the clonality of a neoantigen appears to be important. Second, current immunogenomic analyses typically infer a patient's specific HLA type to identify putative neoantigens. However, some studies have demonstrated that, perhaps unexpectedly, clinical benefit appears to correlate with neoantigen load, even when the HLA type is randomized (22). Such results are compatible with degenerate binding of neopeptides across HLA types, but to further support the validity of these computational predictions, experimental confirmation is needed to demonstrate that these neopeptides do, in fact, bind different MHC molecules, ultimately generating an immune response.
Generating high mutational loads.
Deficiencies in mismatch repair are expected to generate a high mutational load. Le and colleagues explored anti–PD-1 therapy with pembrolizumab in patients with (primarily) chemorefractory colorectal cancer and defects in DNA mismatch repair using whole-exome sequencing (26). Patients with mismatch repair–deficient tumors had, on average, a higher mutational load (1,782 mutations in the mismatch repair deficient compared with 73 in the mismatch repair proficient), which correlated to a significantly higher objective response rate, immune-related progression-free survival, and overall survival (of note, in this series, zero mismatch repair–proficient patients had an objective response to pembrolizumab). Additional data presented at the 2016 American Society of Clinical Oncology Annual Meeting further suggest that response to pembrolizumab may not depend on tumor histology (29). In a basket trial of 29 patients with mismatch repair–deficient tumors (including histologies assumed to be less responsive to immune checkpoint blockade, such as prostate cancer and sarcoma), treatment with pembrolizumab led to an impressive disease control rate of 72%. Emerging data suggest that the prevalence of mismatch repair deficiency across different tumor histologies may be higher than previously thought, implying a potential role for checkpoint blockade in tumors once thought to be resistant to such therapy (29, 30).
Mutations in genes related to DNA repair and replication may also influence response to immunotherapy. Hugo and colleagues examined whole-exome sequencing of melanomas from patients treated with pembrolizumab (24) and found BRCA2 mutations enriched in patients who were responsive to PD-1 blockade (28% of responding tumors but only 6% of nonresponding tumors). These predicted loss-of-function mutations in BRCA2 may affect homologous recombination and double-stranded DNA break repair. Interestingly, Rizvi and colleagues also identified mutations in DNA repair and replication genes in responders to PD-1 blockade, which, in turn, often correlated with a higher mutational load (25). Thus, even with cancer types that on average have few somatic mutations, such genomic analysis may identify a subset of patients across tumor types whose tumors harbor mutations in DNA replication or repair genes, leading to a higher mutational load and the potential for response to immune checkpoint blockade.
Augmenting immunotherapy with radiotherapy or chemotherapy has also been postulated. The abscopal effect, where distant tumor sites outside of a radiation field decrease in size following radiotherapy, may, in part, be related to effects on the number of and release of tumor antigens. Case reports of the abscopal effect (including complete responses) in patients with metastatic melanoma treated with ipilimumab and local radiotherapy (31, 32) have, in part, prompted a number of prospective clinical trials combining immunotherapy and radiotherapy (33). Furthermore, preclinical studies suggest that hypomethylating agents might increase tumor antigen exposure (and, more generally, tumor cell visibility), leading to improved response to immune-based therapies (34, 35). These and other preclinical and early clinical studies provide initial evidence that chemotherapy or radiotherapy may affect tumor neoantigen load or trigger an immunogenic cell death through release of damage-associated molecular patterns (such as calreticulin or HMGB1, among others; ref. 36). Further study is needed to determine whether these adjunctive therapies increase the number of clonal neoantigens without increasing tumor clonal diversity, which could reduce the effectiveness of the immune response.
Beyond mutational load.
Response to checkpoint blockade is not purely tumor cell autonomous and depends on a complex cancer–immunity cycle. The cancer–immunity cycle, as eloquently described by Chen and Mellman, requires the release of tumor antigens, presentation of these antigens leading to effector cell activation, successful trafficking, and infiltration of such cells into the tumor microenvironment as well as overcoming an often immunosuppressive microenvironment to recognize and ultimately kill tumor cells (37). Here too, genomics-based approaches have identified host immune factors, including germline changes that might play a role in response to immune therapy.
Tumeh and colleagues analyzed the T-cell infiltrate in patients with melanoma who were successfully treated with pembrolizumab (38). Using quantitative IHC and multiplex immunofluorescence, they demonstrated that CD8 T-cell infiltration into the tumor and at the invasive tumor margin positively correlated with response to pembrolizumab. Next-generation sequencing of the variable T-cell receptor beta chain to assess T-cell clonality demonstrated a positive correlation between a more clonally restricted (i.e., less diverse) infiltrative T-cell population and response to pembrolizumab. Furthermore, in a posttreatment analysis, responders to pembrolizumab demonstrated a clonal expansion of these T cells (when compared with the nonresponding group). This work highlights the value of T-cell infiltration and suggests that the quality of the T-cell response (i.e., clonality) may underlie effective responses to PD-1 blockade.
Genomic analysis of exceptional responders to immunotherapy may also provide insight into unique mechanisms of response. Analysis of tumor and germline samples from a patient with chemorefractory, metastatic lung adenocarcinoma with an exceptional response to anti–PD-L1 therapy identified both a somatic and a germline mutation in JAK3 (39). These activating mutations led to increased tumor expression of PD-L1, and, because of the germline mutation, increased tissue macrophage expression of PD-L1. Abrogation of PD-L1 activity with atezolizumab in vitro led to enhanced T-cell activity and correlated with an in vivo clinical response for this patient. Overall, this analysis suggested a mechanism for the individual's exceptional response to therapy.
In addition to checkpoint blockade, genomic approaches may guide other immune-based therapeutics, such as therapeutic vaccines. Rajasagi and colleagues (40) used whole-exome sequencing of chronic lymphocytic leukemia (CLL) samples from 91 patients to identify tumor neoantigens based on computational prediction of HLA class I–binding peptides, which were then experimentally confirmed to elicit CD8 T-cell responses in patients who achieved long-term remission following allogeneic stem cell transplantation. On the basis of this approach, clinical trials are exploring the benefit of identifying tumor-specific neoantigens and administering them via therapeutic peptide vaccines tailored to the individual patient, (i.e., NCT01970358 for patients with melanoma and NCT02287428 for patients with glioblastoma). Recent work by Kranz and colleagues (41) has further shown that neoantigen delivery may be possible using RNA encapsulated in a lipid carrier (RNA-lipoplexes). Given that, at least in melanoma, recurrent mutations and corresponding neoantigens have not consistently been identified (22, 24), such personalized therapeutic vaccine approaches may generate an effective antitumor immune response.
RNA-based approaches
Transcriptomics allows for another layer of investigation into mechanisms of response to immune checkpoint blockade. Such gene expression analysis could be used for biomarker discovery, including identification of potentially predictive gene signatures, and may hint at functional mechanisms underlying response to therapy.
Rooney and colleagues used a relatively simple RNA-based approach to measure immune cytolytic activity [defined as expression of granzyme A (GZMA) and perforin (PRF1)] for a variety of cancer types (42). Indeed, the presence of this cytolytic activity signature was significantly associated with response to CTLA-4 blockade in metastatic melanoma (22). Similarly, in PD-L1 blockade, the presence of effector T cells in the tumor microenvironment was associated with clinical response. Herbst and colleagues used a Fluidigm gene expression assay (“immunochip”) to investigate transcriptional differences between responders and nonresponders to the anti–PD-L1 antibody atezolizumab in patients across multiple solid malignancies (43). Expression of IFNγ (IFNG) and IFNγ-inducible genes in pretreated tumors positively correlated with response to therapy in patients with melanoma, albeit with weaker associations in renal and non–small cell lung cancers. Furthermore, gene expression analysis performed on atezolizumab-responsive tumors demonstrated upregulation of markers of CD8 T-cell activation and Th cell type 1 response (granzyme A, perforin 1, among others).
Transcriptomic analysis may also identify individual genes or signatures that correlate with response to immune checkpoint blockade. The expression of CTLA-4 and PD-L2 (but not PD-L1) correlated with response to CTLA-4 blockade in advanced melanoma (22). In a pretreatment transcriptomic analysis of melanoma patients who went on to receive anti–PD-1 therapy, responders had lower expression of immunosuppressive genes, macrophage chemotactic genes, and mesenchymal transition genes (24). As technology for RNA analysis further develops, including single-cell RNA-sequencing, such approaches may helpful in exploring complexities of the tumor microenvironment (44).
Genomic Approaches to Understanding Resistance
Elegant work in mouse models and cell lines have elucidated pathways through which the tumor has increased PD-L1 expression through JAK/STAT signaling (45); dysregulated chromosomal alterations (46); and increased expression of multiple coinhibitory molecules including CTLA-4, PD-L1, TIM-3, TIGIT, and OX-40, among others (47, 48). Genomic screens may further illuminate methods by which tumors have evolved mechanisms of immune evasion and how resistance might evolve in the presence of checkpoint blockade (Fig. 3).

Mechanisms of resistance. Genomic techniques have contributed to the identification of multiple mechanisms of resistance to immune checkpoint blockade, including alterations in signaling pathways involved in cell proliferation of and apoptosis, stabilization of immune checkpoints, and alterations in MHC peptide presentation. TCR, T-cell receptor.
Mechanisms of resistance. Genomic techniques have contributed to the identification of multiple mechanisms of resistance to immune checkpoint blockade, including alterations in signaling pathways involved in cell proliferation of and apoptosis, stabilization of immune checkpoints, and alterations in MHC peptide presentation. TCR, T-cell receptor.
Genomic techniques identify the intersecting pathways between cell growth and antiapoptosis with those of immune surveillance
Genomic profiling has identified pathways associated with tumor cell differentiation, proliferation, and apoptosis (49), with recent studies linking these pathways to immune surveillance. In melanoma cell lines, BRAF, which is mutated in approximately half of all melanoma, functionally modulates the T-cell microenvironment through upregulation of IL1A and IL1B, leading to expression of PD-L1 and PD-1 in tumor-associated fibroblasts (50). Spranger and colleagues classified cutaneous melanoma from The Cancer Genome Atlas (TCGA) as “T-cell enriched” versus “T-cell poor” based on a 13-gene subset and correlated β-catenin signaling targets (i.e., APC, SOX11, WNT7B) with a lack of CD8 T-cell infiltrate. In a mouse model lacking BRAF and PTEN, they then demonstrated that β-catenin signaling was necessary for effective dendritic cell recruitment as well as initial T-cell priming and infiltration (51). Notably, deletion of PTEN leads to upregulation of the PI3K–AKT pathway and increased expression of PD-L1 in glioblastoma (52)—an effect not seen across melanoma cell lines (53). Peng and colleagues queried TCGA data for a relationship between PTEN copy number and T-cell activity and infiltration. Copy number served as a proxy for gene expression, as gene deletion is a common mechanism for PTEN loss in melanoma. Tumors with low copy number had lower cytolytic scores and lower histologically detectable lymphocyte infiltration (54). Peng and colleagues demonstrated that PTEN-mutated tumors in mice shrank and had greater T-cell infiltration with inhibition of both PI3K and PD-L1, suggesting a future role for combination therapy. Modulation of either the β-catenin/WNT signaling pathway or PTEN may thus improve T-cell infiltration into tumors. Genomic approaches, particularly when paired with functional studies, might identify additional therapeutic targets in pathways traditionally associated with cell differentiation and apoptosis that further augment immune recognition or infiltration.
Genomic techniques identify mechanisms of immune evasion
Genomic screens might offer novel insight into shared mechanisms of immune evasion across tumor types. Kataoka and colleagues used whole-genome sequencing of patients with adult T-cell leukemia to identify structural variations within the 3'-untranslated region of the PD-L1 locus (55). They further identified 31 structural variants of PD-L1 in 10,210 tumors in TCGA, with the highest prevalence found in diffuse large B-cell lymphoma and stomach adenocarcinoma. These variants were validated in functional studies in murine models and human cell lines, demonstrating increased stability and selection of PD-L1 within the tumor. Intriguingly, addition of anti–PD-1 therapy abrogated the effect in murine models, possibly by circumventing the PD-1–PD-L1 axis. Stabilization of these inhibitory checkpoint ligands through increased transcription or delayed degradation may thus present a novel target. This mechanism further highlights the importance of PD-L1 in immune evasion.
Genomic techniques offer insight into why certain tumors may fail in response to immunotherapy
Tumor antigens must go through the antigen-processing machinery and be presented on MHC molecules for recognition by T cells. Genomic screens of tumors have identified mutations within this pathway, including the HLA genes (42, 56) and B2M (42). Until recently, the significant polymorphisms within the HLA loci have limited the ability to sequence the germline, much less understand the impact of somatic mutations (57, 58). Recently, Shukla and colleagues (59) generated a novel algorithm based on whole-exome sequencing of tumor and normal tissue to infer HLA type and then to identify loss-of-function mutations (i.e., nonsense, frameshift, indels, splice site). Somatic mutations in the MHC were enriched in colon adenocarcinoma, head and neck, lung squamous, and stomach cancers—tumors that carry a higher neoantigen burden. Functionally, mutations lay more commonly in the domain that binds the CD8 coreceptor and the peptide-binding groove domain, potentially abrogating T-cell recognition of the MHC–peptide complex.
Previous efforts have identified mutations in tumors not treated with immune checkpoint blockade agents. Zaretsky and colleagues selected four individuals treated with pembrolizumab whose tumors initially regressed and then relapsed (60). In two individuals, homozygous truncating mutations in either the JAK1 or JAK2 protein, when studied in cell lines, correlated with decreased JAK/STAT signaling, MHC expression, and PD-L1 expression. IFNγ signaling has been linked to increased PD-L1 signaling in multiple cell lines (45), with upregulation of the JAK2 pathway identified in selective lymphomas (46). Decreased reliance on IFN signaling cascades may result in increased dependence on other mechanisms of evasion. A third patient had a mutation in B2M, abolishing MHC expression on the cell surface and abrogating T-cell recognition, a phenomenon previously described in patients undergoing other forms of immunotherapy and also de novo (61, 62). Of note, a fourth patient had no identifiable mutation, suggesting that novel mechanisms of evasion might be at play. The small sample size limits the generalizability of these mechanisms, but certainly future sequencing will determine whether predominant mutation patterns arise. Tumors either de novo or in the presence of immunotherapy might face selective pressure to develop mutations in the antigen processing and presentation pathway that lead to immune evasion.
Conclusions
Integration of genomic mutations, transcriptomic analysis, and neoantigen prediction might ultimately allow for a more robust prediction of response to immune checkpoint blockade. For instance, an integrated approach might identify tumors with a high mutational load but with significant ITH and no clonal CD8 T-cell infiltration, suggesting that they are unlikely to respond to immune checkpoint blockade. Furthermore, although the focus thus far has been on identifying predictors of response, given the potential for severe autoimmune adverse effects with these therapies (63–65), future work should also aim to find predictors of toxicity.
Notable limitations to current approaches exist. The computational identification of neoantigens typically involves the use of in silico algorithms, such as NetMHCpan (66) and others (67, 68), that attempt to predict the binding affinity of peptides to MHC class I molecules. Such predictions require experimental confirmation of MHC class I–peptide binding, which can be challenging and requires integrating techniques such as mass spectrometry (69, 70). Furthermore, these algorithms do not necessarily predict whether a given peptide will generate an in vivo immune response, although the immunogenicity of a number of predicted peptides has been validated experimentally (23, 40, 71, 72). Additional computational tools may be needed to predict which neoantigens have the capacity to bind MHC class I molecules and generate an immune response that is recognized by the T-cell repertoire (73). Although NetMHCpan and other algorithms might predict peptide binding to MHC class I molecules, the development of additional tools is needed to predict peptide binding to MHC class II molecules, which are ultimately recognized by the CD4+ T-cell compartment and may well have clinical relevance (74, 75). With further refinement and experimental validation, however, genomics-based approaches will hopefully allow for the selection of patients likely to achieve long-term, durable responses to such therapies.
Disclosure of Potential Conflicts of Interest
E.M. Van Allen reports receiving a commercial research grant from Bristol-Myers Squibb. No potential conflicts of interest were disclosed by the other authors.
Grant Support
This work supported by the Kure It-AACR Research Grant for Immunotherapy for Kidney Cancer, BroadIgnite, and BroadNext10.