Article Text

Abstract
Background Non-small cell lung cancer (NSCLC) patients bearing targetable oncogene alterations typically derive limited benefit from immune checkpoint blockade (ICB), which has been attributed to low tumor mutation burden (TMB) and/or PD-L1 levels. We investigated oncogene-specific differences in these markers and clinical outcome.
Methods Three cohorts of NSCLC patients with oncogene alterations (n=4189 total) were analyzed. Two clinical cohorts of advanced NSCLC patients treated with ICB monotherapy [MD Anderson (MDACC; n=172) and Flatiron Health-Foundation Medicine Clinico-Genomic Database (CGDB; n=894 patients)] were analyzed for clinical outcome. The FMI biomarker cohort (n=4017) was used to assess the association of oncogene alterations with TMB and PD-L1 expression.
Results High PD-L1 expression (PD-L1 ≥50%) rate was 19%–20% in classic EGFR, EGFR exon 20 and HER2-mutant tumors, and 34%–55% in tumors with ALK, BRAF V600E, ROS1, RET, or MET alterations. Compared with KRAS-mutant tumors, BRAF non-V600E group had higher TMB (9.6 vs KRAS 7.8 mutations/Mb, p=0.003), while all other oncogene groups had lower TMB (p<0.001). In the two clinical cohorts treated with ICB, molecular groups with EGFR, HER2, ALK, ROS1, RET, or MET alterations had short progression-free survival (PFS; 1.8–3.7 months), while BRAF V600E group was associated with greater clinical benefit from ICB (CGDB cohort: PFS 9.8 months vs KRAS 3.7 months, HR 0.66, p=0.099; MDACC cohort: response rate 62% vs KRAS 24%; PFS 7.4 vs KRAS 2.8 months, HR 0.36, p=0.026). KRAS G12C and non-G12C subgroups had similar clinical benefit from ICB in both cohorts. In a multivariable analysis, BRAF V600E mutation (HR 0.58, p=0.041), PD-L1 expression (HR 0.57, p=0.022), and high TMB (HR 0.66, p<0.001) were associated with longer PFS.
Conclusions High TMB and PD-L1 expression are predictive for benefit from ICB treatment in oncogene-driven NSCLCs. NSCLC harboring BRAF mutations demonstrated superior benefit from ICB that may be attributed to higher TMB and higher PD-L1 expression in these tumors. Meanwhile EGFR and HER2 mutations and ALK, ROS1, RET, and MET fusions define NSCLC subsets with minimal benefit from ICB despite high PD-L1 expression in NSCLC harboring oncogene fusions. These findings indicate a TMB/PD-L1-independent impact on sensitivity to ICB for certain oncogene alterations.
- immunotherapy
- lung neoplasms
- tumor biomarkers
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
PD-1/PD-L1 immune checkpoint blockade (ICB) has revolutionized the treatment of many cancer types, including non-small cell lung cancer (NSCLC). ICB has the potential to induce durable responses; however, the response rate from single agent ICB is only 15%–20%.1–3 High PD-L1 expression4–6 and high tumor mutational burden (TMB)7–10 have been well documented to be associated with superior benefit from ICB in NSCLC. However, driver alterations in EGFR and ALK oncogenes have been associated with cold immune microenvironment11 and poor outcomes from ICB therapy.1 9 12–14 More recently, a study on the IMMUNOTARGET registry demonstrated distinct patterns of responses to ICB in NSCLC patients carrying rare canonical oncogene alterations.15 These data highlighted the critical impact of oncogene alterations on response to ICB. However, due to the scarcity of these rare oncogene-altered NSCLCs treated with ICB, the numbers of patients for certain oncogene groups were relatively small precluding definitive conclusions. Furthermore, the mechanisms underlying these distinct responses, and the impact of oncogene alterations on TMB, PD-L1 expression and on their predictive values are largely unknown, primarily due to lack of appropriate molecular and clinical data from rare oncogene-altered NSCLC patients treated with ICB.
To fill this void, we leveraged a large NSCLC cohort of 4189 patients, including 4017 NSCLC patients with oncogene alterations, TMB and PD-L1 data as well as two large registry cohorts consisting of 1066 NSCLC patients treated with ICB monotherapy to assess the impact of oncogene alterations on TMB, PD-L1 expression and clinical outcomes from ICB therapy.
Methods
Biomarker cohort
FMI biomarker cohort
A total of 4017 NSCLC patients with molecular data including oncogene alterations, TMB and PD-L1 expression from the FMI FoundationCORE database were analyzed to assess the impact of oncogene mutations on TMB and PD-L1 (hereafter referred as FMI biomarker cohort).
Clinical cohorts
MDACC cohort
GEMINI, a MD Anderson lung cancer database was queried. A total of 172 patients with advanced NSCLC whose tumor harbored oncogene alterations (see ‘Oncogene alterations’ section) and treated with single-agent ICB (pembrolizumab, nivolumab, atezolizumab, and durvalumab) between January 2014 and May 2018 were included. Data were collected through chart review, and dataset was locked on April 5, 2019. The GEMINI database is approved by the MD Anderson Cancer Center (MDACC) Institutional Review Board (IRB), and all patients signed informed consent.
Clinico-Genomic database (CGDB) immunotherapy cohort
The Flatiron-Foundation Medicine Inc CGDB, which is a clinically annotated subset of the FMI biomarker cohort, was queried and a total of 894 patients with advanced NSCLC harboring oncogene alterations (as defined in the ‘Oncogene alterations’ section) and treated with single-agent ICB between January 2011 and December 2018 were included (hereafter referred as CGDB immunotherapy cohort).
CGDB chemotherapy cohort
To understand whether the impact of oncogene alterations was specific to ICB, we also queried the Flatiron-Foundation Medicine Inc CGDB for patients with advanced NSCLC with oncogene alterations (as defined in the ‘Oncogene alterations’ section) and treated with combination or single-agent chemotherapy (hereafter referred as CGDB chemotherapy cohort). The study was IRB approved and included a waiver of informed consent. Data were collected from database repository, and dataset was locked on December 31, 2018.
This study was conducted in accordance with ethical guidelines including Declaration of Helsinki and US Common Rule.
Oncogene alterations
For the MDACC Cohort, genomic profiling information was obtained through Clinical Laboratory Improvement Amendments certified laboratory assay results annotated in the GEMINI database. For the CGDB and FMI biomarker cohorts, the information was obtained from respective data repositories and sequencing was performed by FoundationOne assay.
Oncogene alterations included in this study are: BRAF: codons 457–723, including V600E and non-V600E; EGFR exon 20: codons 762–823 (T790M mutation not included); HER2: codons 755 and 770–785; KRAS: codons 12, 13, 33, and 61; classic EGFR: exon 19 deletions and exon 21 p.L858R mutation (±T790M mutation); MET: exon 14 skipping mutations; and ALK, ROS1, and RET: gene fusion/rearrangements.
Tumor mutational burden
For the CGDB cohorts and for the FMI biomarker cohort, TMB was determined through tissue-based next-generation sequencing (FoundationOne). TMB was defined as all mutations (including all non-synonymous and synonymous mutations with germline variants and driver mutations excluded) divided by the total covered exonic regions16 and presented as number of mutations/megabase (Mb). High TMB is defined as ≥10 mutations/Mb, corresponding to FMI diagnostic test definition of high TMB or ≥16 mutations/Mb based on OAK and POPLAR trials.17 TMB data was not available for the MDACC cohort.
PD-L1 staining
For the MDACC cohort, PD-L1 staining was obtained from pathology reports. For the CGDB cohorts, PD-L1 status was obtained from data repository. For the FMI biomarker cohort, PD-L1 staining was assessed through immunohistochemistry using the 22C3 PharmDx assay (Dako North America). PD-L1 expression was quantified by tumor proportional score (TPS) and defined as positive (≥1%) or negative (<1%), and high (≥50%) or low (1%–49%).5 6 18
Statistical analysis
Progression-free survival (PFS) was defined as time from starting treatment until disease progression or death. Overall survival (OS) was defined as time from starting treatment until death. Patients without an event were censored at last follow-up. Left truncation method was applied to real-world PFS and OS analysis to adjust for situations where molecular profiling occurred after the start of treatment.19
Kaplan-Meier method was used to estimate PFS and OS, and differences were assessed by log-rank test. HRs and 95% CIs were determined through Cox proportional hazards model. Multivariable analysis was performed using the Cox regression method. Best response to ICB treatment determined through RECIST V.1.1 was available only for MDACC Cohort. The KRAS group was used as the reference comparator because: (1) it was the most commonly mutated oncogene in these cohorts of NSCLCs; and (2) KRAS-mutant NSCLCs constitute a heterogeneous patient population that have been shown to have similar prognosis compared with KRAS wild-type patients20–23 and to derive similar benefit from ICB therapy when compared with KRAS wild-type NSCLC patients.1 24
Differences in categorical variables were assessed through χ2 or Fisher’s exact test with Bonferroni method for between group comparisons. Continuous variables were tested for normality using Shapiro-Wilk test, and Dunn’s test with Benjamini-Hochberg correction was used for between group comparisons. Significance was established at p value ≤0.05. Statistical analysis was performed on GraphPad Prism V.7.0 (La Jolla, California, USA), IBM SPSS Statistics V.24.0 (Armonk, New York, USA), SAS (Cary, North Carolina, USA), and RStudio V.3.5.5 (Boston, Massachusetts, USA).
Results
TMB and PD-L1 are associated with clinical benefit from immunotherapy in oncogene-driven NSCLC
We first analyzed the CGDB immunotherapy cohort (median follow-up 17.8 months (95% CI 15.6 to 19.8)) to assess the predictive performance of TMB or PD-L1 for benefit from ICB in oncogene-driven NSCLC. We observed that higher TMB using either 10 or 16 mut/Mb as the cut-off was associated with longer PFS (TMB ≥10 vs <10 mut/Mb: HR 0.61, 95% CI 0.50 to 0.75, p<0.001; TMB ≥16 vs <16 mut/Mb: HR 0.52, 95% CI 0.39 to 0.69, p<0.001), and longer OS (TMB ≥10 vs <10 mut/Mb: HR 0.79, 95% CI 0.64 to 0.97, p<0.001; TMB ≥16 vs <16 mut/Mb: HR 0.69, 95% CI 0.51 to 0.92, p<0.001) consistent with previous findings.7 8 10 17 Positive PD-L1 expression was associated with longer PFS (PD-L1 positive vs negative: HR 0.48, 95% CI 0.30 to 0.77, p<0.001), but the difference did not reach statistical significance for OS (PD-L1 positive vs negative: HR 0.66, 95% CI 0.39 to 1.10, p=0.095). In a multivariable analysis, higher TMB was significantly associated with longer PFS (TMB ≥10 vs <10 mut/Mb: HR 0.66, 95% CI 0.53 to 0.82, p<0.001; TMB ≥16 vs <16 mut/Mb: HR 0.56, 95% CI 0.42 to 0.76, p<0.001) (online supplemental table 1A,B), and longer OS (TMB ≥10 vs <10 mut/Mb: HR 0.76, 95% CI 0.61 to 0.96, p=0.019; TMB ≥16 vs <16 mut/Mb: HR 0.66, 95% CI 0.49 to 0.89, p=0.006) (online supplemental table 2A,B). PD-L1 expression was also associated with longer PFS (TMB cut-off of 10: PD-L1 positive vs negative: HR 0.57, 95% CI 0.35 to 0.92, p=0.022) (online supplemental table 1A).
Supplemental material
Oncogene alterations were associated with distinct clinical outcomes from ICB therapy
We next assessed the impact of oncogene alterations on clinical outcome from ICB treatment using 1066 oncogene-altered advanced NSCLC patients (n=172 from MDACC cohort: median follow-up 29.1 months (95% CI 26.2 to 33.6); and n=894 from CGDB immunotherapy cohort: median follow-up 17.8 months (95% CI 15.6 to 19.8)) (online supplemental figure 1A,B). As outlined in online supplemental table 3, BRAF and KRAS groups were enriched for smokers (p<0.001) in both MDACC cohort and CGDB immunotherapy cohort, while patients in the classic EGFR group received more prior treatment lines (p<0.001) compared with other genomic groups, likely due to higher likelihood of prior exposure to TKIs (p<0.001). Since KRAS-mutant NSCLC constituted the largest population in both MDACC cohort (n=87) and CGDB immunotherapy cohort (n=601) and have been previously shown to have similar prognosis compared with KRAS wild-type patients20–23 and to derive similar benefit from ICB therapy when compared with KRAS wild-type NSCLC patients,1 24 we used the KRAS group as the reference for outcome analysis.
In the MDACC cohort, the BRAF group had the longest PFS among all genomic groups (BRAF 7.4 vs KRAS 2.8 months, HR 0.36, 95% CI 0.14 to 0.88, p=0.026) (figure 1A), while classic EGFR and HER2 groups had the shortest PFS (1.8 and 1.9 months, respectively) (figure 1A). The same trend remained in subsequent multivariable analysis adjusting for PD-L1 expression and smoking status (TMB data were not available for MDACC cohort) (p=0.032 for BRAF group, p=0.022 for Classic EGFR group, and p=0.116 for HER-2 group, online supplemental table 4). Compared with the KRAS group, OS was numerically longer in BRAF group, but the difference was not statistically significant (35.6 vs 16.8 months, HR 0.65, 95% CI (0.26 to 1.63), p=0.363). Meanwhile patients with classic EGFR mutations had the shortest OS (11.3 months, 95% CI 5.8 to 16.9, HR 2.01, p=0.006) (figure 1B). Similar trends were observed in a multivariable analysis adjusting for PD-L1 expression, although the difference was not significant (BRAF vs KRAS: HR 0.69, p=0.454; classic EGFR vs KRAS: HR 1.49, p=0.263; online supplemental table 5). Furthermore, the BRAF group also had highest objective response rate (ORR) (62%) among all oncogene groups (Fisher’s exact test p<0.001). Compared with the KRAS group, the common comparator, this difference was not statistically significant (62% vs 24%, p=0.364) (figure 1C). Importantly, two of three patients with BRAF V600E mutations had recently progressed on vemurafenib and achieved durable responses at 35+ and 20+ months post treatment on ICB. There were seven patients harboring ALK fusion (n=2), RET fusion (n=2) or MET exon 14 skipping mutations (n=3) in the MDACC cohort. Except for two patients with MET mutations who had stable disease, all remaining five patients had progressive disease as best response. Results for PFS, OS and ORR are summarized in table 1.

Clinical outcomes for oncogene-driven non-small cell lung cancers on treatment with single-agent PD-1/PD-L1 immune checkpoint inhibitor. (A) PFS in MDACC cohort; (B) OS in MDACC cohort; (C) waterfall plot for ORR in MDACC cohort for patients with measurable disease; (D) PFS in CGDB immunotherapy cohort; (E) OS in CGDB immunotherapy cohort. *Patients harboring V600E alteration in the BRAF group. CGDB, Clinico-Genomic database; PFS, progression-free survival; OS, overall survival.
Clinical outcomes according to treatment group for the MDACC and CGDB immunotherapy cohorts
Consistent with the results from the MDACC Cohort, the BRAF groups in the CGDB immunotherapy cohort had the longest PFS (V600E 9.8 and non-V600E 5.4 months) (figure 1D and table 1) and OS (V600E 20.8 and non-V600E 14.9 months) (figure 1E and table 1) among all genomic groups although the difference did not reach statistical difference. Meanwhile, patients with classic EGFR mutations had significantly shorter PFS (figure 1D and table 1). Similar to classic EGFR group, patients with oncogene fusions (ALK: n=19; ROS1: n=3; RET: n=14) had poor outcomes with a trend for shorter PFS compared with the KRAS group. The MET exon 14 mutation group (n=34) also had a short PFS of 2.7 months, but the difference was not significant compared with the KRAS group (HR 1.13, 95% CI 0.69 to 1.84, p=0.59) (figure 1D and table 1). In a multivariable analysis adjusting for TMB, PD-L1 expression, and smoking status, BRAF V600E had longer PFS compared with the KRAS group (TMB cut-off of 10: HR 0.58, 95% CI 0.35 to 0.98, p=0.041; TMB cut-off of 16: HR 0.63, 95% CI 0.38 to 1.05, p=0.074) (online supplemental table 1A,B).
KRAS-mutant group is the largest group in our cohorts, and we therefore assessed whether different KRAS-mutant alleles are associated with distinct clinical outcome from ICB treatment. As shown in online supplemental figure 2A–E, ORR and survival were similar between G12C, G12D and G12V subgroups.
To understand whether the impact of these oncogene alterations was only present on clinical outcome from ICB, we analyzed the CGDB chemotherapy cohort (n=933) for clinical outcomes on treatment with chemotherapy across the same NSCLC oncogene groups (online supplemental figure 1C, online supplemental table 6). These analyses demonstrated that the fusion group (ALK: n=39; ROS1: n=12; RET: n=14) had the longest OS with chemotherapy (27.2 months in fusion group vs 11.7 months in KRAS group, HR 0.50, 95% CI 0.34 to 0.74, p<0.001) (online supplemental figure 3A). This difference remained significant in a multivariable analysis adjusting for prior TKI therapy (HR 0.48, 95% CI 0.33 to 0.71, p<0.001). Otherwise, PFS or OS was not different between oncogene groups (online supplemental figure 3A,B).
Oncogene-driven lung cancers show distinct patterns of PD-L1 expression and TMB
We leveraged the 4017 NSCLC patients from FMI biomarker cohort to assess the impact of oncogene alterations on PD-L1 expression and TMB. High prevalence of PD-L1 positivity (TPS ≥1%) was observed in MET (115/145, 79.3%), RET (42/54, 77.8%), BRAF V600E (89/118, 75.4%), ROS1 (40/55, 72.7%) and ALK (136/194, 70.1%) groups, and low prevalence was observed in tumors harboring EGFR exon 20 (75/166, 45.2%), HER2 (50/105, 47.6%), classic EGFR (exon 19 deletion and exon 21 L858R) (368/732, 50.3%), and BRAF non-V600E (116/208, 55.8%). Compared with KRAS-mutant NSCLCs, MET-mutant tumors had higher PD-L1 positivity rate (TPS ≥1%), while EGFR-mutant tumors had significantly lower PD-L1 positivity rate (figure 2A) consistent with our previous findings.25 Furthermore, high PD-L1 expression (TPS ≥50%) was common in tumors harboring alterations in MET (80/145, 55.2%), BRAF V600E (57/118, 48.3%), ROS1 fusion (23/55, 41.8%) and RET fusion (20/54, 37.0%). Among these, tumors harboring MET alterations had significantly higher prevalence of high PD-L1 expression (TPS ≥50%), while classic EGFR, EGFR exon 20, and HER2 groups had lower prevalence of high PD-L1 compared with KRAS group (pairwise comparison vs KRAS p<0.050) (figure 2B).
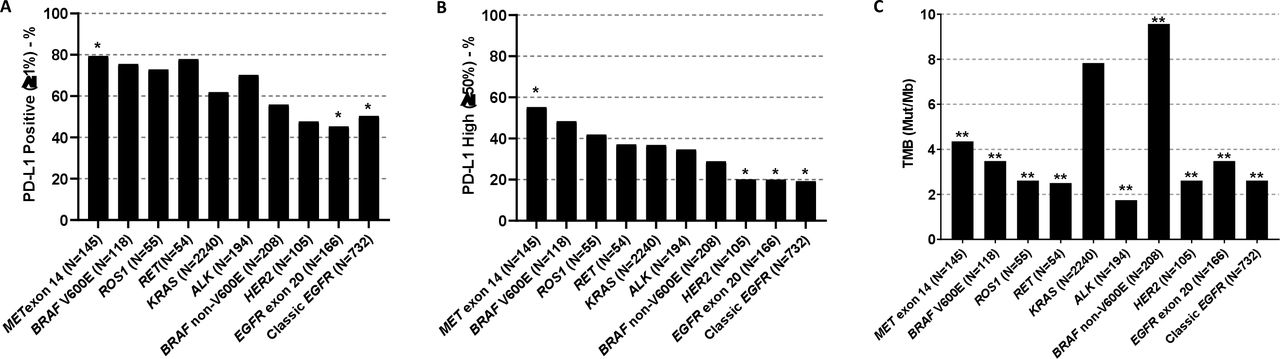
Oncogene-driven non-small cell lung cancers have distinct patterns of PD-L1 expression and TMB – FMI lung cancer database. (A) PD-L1 positivity rates; (B) high PD-L1 expression rates; (C) TMB. *Adjusted p<0.05 versus KRAS group; **adjusted p<0.01 versus KRAS group. TMB, tumor mutational burden.
Compared with the KRAS group, TMB was higher for BRAF non-V600E group (9.6 mut/Mb (n=208) vs 7.8 mut/Mb (n=2240); adjusted p=0.003), while all other oncogene groups had significantly lower TMB (adjusted p<0.001). Of note, ALK (n=194), classic EGFR (n=732), HER2 (n=105), RET (n=54), and ROS1 (n=55) groups had the lowest TMB with median <3 mut/Mb (figure 2C and online supplemental table 7).
Discussion
Oncogene drivers have been long known to determine the cancer biology and clinical behavior of NSCLC. In addition to the classic targetable genomic alterations such as activating EGFR mutations and ALK fusions, more and more oncogene alterations defining distinct lung cancer groups are being discovered.26 In the era of immuno-oncology, emerging evidence has demonstrated the profound impact of oncogene drivers on cancer immune microenvironment11 27 and benefit from ICB treatment.13 15 28 29 However, most of these intriguing findings are based on small cohorts because of the scarcity of data on certain genomic groups of NSCLC treated with single-agent ICB.13 15 28 29 Furthermore, the association between oncogene drivers and established predictive markers for benefit from ICB such as PD-L1 and TMB has not been systemically studied. To fill this void, we leveraged the clinical and molecular data from over 4000 oncogene driven NSCLC patients and analyzed the association of oncogene drivers and TMB/PD-L1 expression as well as the benefit from ICB treatment.
Our study is consistent with previous reports demonstrating the impact of TMB and PD-L1 expression in clinical outcomes from ICB therapy in NSCLC but more specifically in a population of oncogene addicted NSCLC. Furthermore, BRAF-mutant NSCLC had high TMB and PD-L1 expression (figure 2A–C), which may make this group more sensitive to ICB therapy. These results suggest that ICB is a reasonable choice in the frontline setting for BRAF-mutant, PD-L1+ NSCLC given its potential to induce durable clinical benefit. It remains unclear why the outcomes reported for the BRAF groups in the MDACC and CGDB immunotherapy cohorts are superior when compared with two previously published cohorts of BRAF-mutant NSCLC treated with ICB.15 28 Unfortunately, the unavailability of both TMB and PD-L1 data across the four cohorts precludes more definitive conclusions. Of note, BRAF+MEK inhibition enhances immune responses in preclinical models30 31 and increases duration of response and PFS in combination with ICB in melanoma patients.32 33 Although it remains unknown if these combinations have activity in NSCLC, these findings suggest that ICB-TKI combinations could be explored in BRAF-mutant NSCLC.
KRAS G12C and non-G12C mutant NSCLC patients had similar clinical outcome in both analyzed immunotherapy cohorts highlighting that mutant allele was not associated with clinical outcome on treatment with ICB in KRAS-mutant NSCLC. These findings are consistent with prior work from our group and others highlighting that comutations (eg, STK11, KEAP1, and TP53), but not specific KRAS alleles, were associated with distinct subgroups with differing biological phenotypes and response to immunotherapy in KRAS-mutant NSCLC.15 34–37 These results suggest that further therapeutic intervention is warranted to increase immunogenicity of KRAS-mutant NSCLC. The KRAS G12C inhibitor AMG510 resulted in a more inflamed tumor microenvironment and increased the efficacy of ICB in an immune competent mouse model.38 Consequently, clinical investigation of KRAS G12C inhibitors and ICB combinations are ongoing.
Consistent with prior studies, our results also demonstrated that NSCLC patients harboring classic EGFR mutations had inferior outcomes from ICB.1 9 12 13 39–41 Additionally, we observed EGFR exon 20 and HER2 mutations were also associated with less benefit from single-agent ICB. This could be related to low TMB and low PD-L1 expression in these tumors. However, EGFR-mutant NSCLC tumors have been reported to have decreased proliferating and activated CD8+ T cell infiltration,13 increased adenosine pathway signaling, as demonstrated by elevated expression of CD73 and adenosine A1 receptor, increased TGF-β signaling, and lower IFN-γ expression.27 Furthermore, our group has shown significantly lower T cell receptor (TCR) clonality implying less reactive TCR repertoire in EGFR-mutant NSCLC independent of TMB.11 27 These findings indicate molecular mechanisms, other than low TMB, contribute to a cold immune microenvironment in EGFR-mutant NSCLC. Delineating these underlying mechanisms will allow development of effective immunotherapies for EGFR-mutant NSCLC. Contrary to EGFR and HER2, NSCLC harboring ALK/ROS1/RET fusions or MET exon 14 skipping mutations were found to have high PD-L1 expression although with low TMB. However, this did not translate to better clinical outcomes on ICB therapy as demonstrated by the short PFS and low RR in these groups. This suggests that oncogene-specific factors other than TMB and PD-L1 expression also impact clinical outcome from ICB therapy.
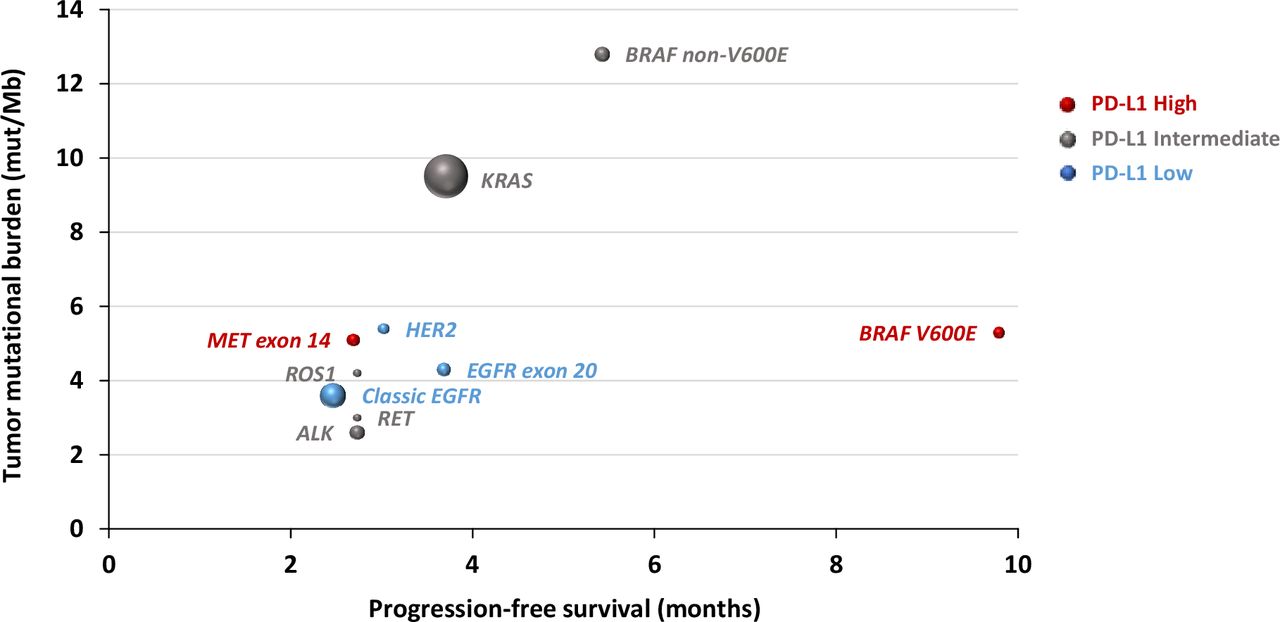
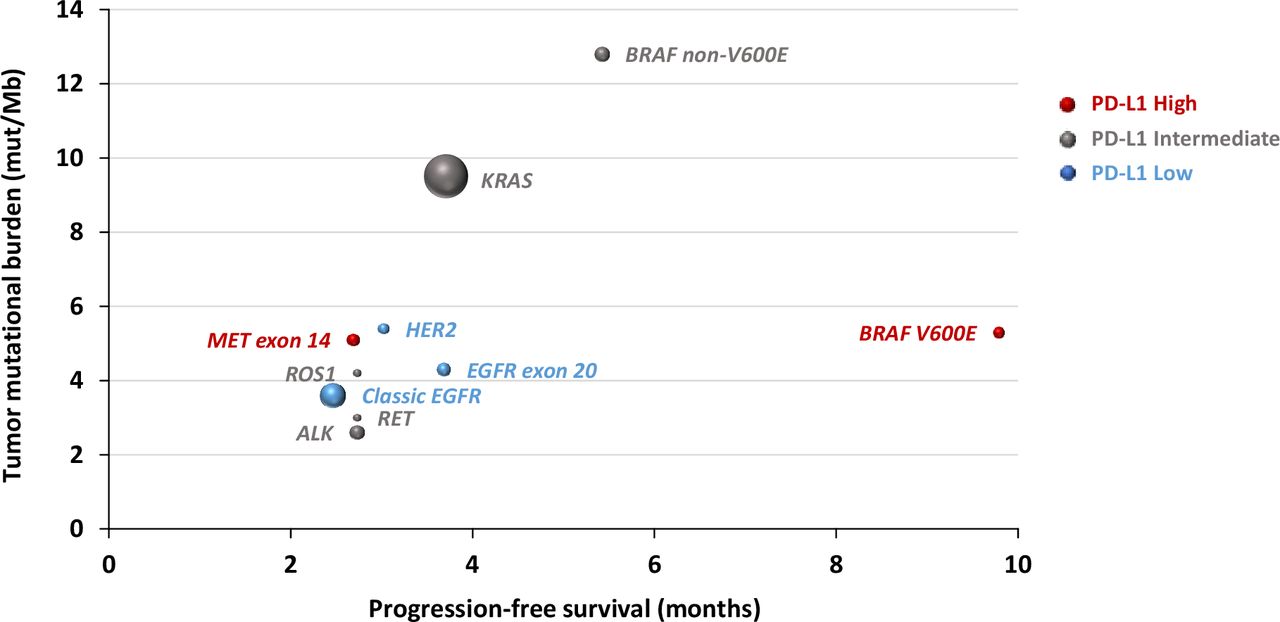
As a real-world study, our study has unavoidable limitations. First, the data are not consistent across different datasets, and many important data were missing. For example, tumor measurement was not available from the CGDB cohorts, which made estimation of ORR not feasible so that we were unable to validate the ORR from the MDACC cohort. In addition, the MDACC cohort did not have TMB data available, which limited further validation of the impact of TMB in predicting ICB outcome for oncogene-driven NSCLC. Second, we focused on NSCLC patients with oncogene drivers who received single-agent ICB. How these oncogene alterations impact the response and survival from ICB in combination with chemotherapy remains to be determined in future studies. Third, although we have over 4000 patients included, the sample size was still small, particularly for patients of certain genomic groups. For example, the number of patients in the BRAF-mutant group in the MDACC cohort was small, and we therefore had to combine V600E and non-V600E mutations for formal analysis, which limited us to validate the findings from the CGDB immunotherapy cohort and hampered further comparisons between V600E and non-V600E groups. Fourth, in the CGDB immunotherapy cohorts, PD-L1 status was missing in 76%–92% of patients and very few patients (n<5) had high TMB (≥10 or ≥16) across all oncogene groups with the exception of KRAS and BRAF non-V600E groups. This impaired our ability to analyze the predictive performance of PD-L1 or TMB in each oncogene group. Despite these limitations, our study creates a framework of groups of oncogene-driven lung cancers with distinct patterns of TMB and PD-L1 and different clinical outcomes on treatment with ICB that can be used for treatment decision in the clinic and for clinical trial design (figure 3).
{kind=link}
{kind=link}
{kind=link}
Correlation between oncogene drivers and tumor mutational burden, progression-free survival and clinical outcome on immune checkpoint blockade therapy. Dot sizes are proportional to sample size; red: high PD-L1 expression; gray: intermediate PD-L1 expression; blue: low PD-L1 expression.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
This study was IRB approved and conducted in accordance with ethical guidelines including Declaration of Helsinki and US Common Rule.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Correction notice This paper has been updated since first published to amend corresponding author details.
Contributors Conception and design: MVN, KS, IB, SH, XL, JPR, AR, JL, LAA, DS, JZ, JVH. Development of methodology: MVN, FS, MM, KS, IB, VS, HX, SH, DS, MEG, KM, C-JW, JZ, DSB, JPR, AR, TC, CMG, KGM, LH, WR, JL, LAA, DS, JZ, JVH. Acquisition of Data: MVN, FS, MM, KS, IB, VS, HX, SH, DS, YYE, XL, MEG, KM, C-JW, JZ, DSB, TC, CMG, KGM, LH, WR, GS, VAM, BMA, GF. Analysis and interpretation of data all authors. Writing, review and or revision of the manuscript: all authors. Administrative, technical, or material support: MVN, FS, MM, KS, IB, VS, HX, SH, DS, YYE, XL, MEG, KM, C-JW, JZ, DSB, JPR, AR, TC, CMG, KGM, LH, WR, GS, JL, AST, VP, DLG, BSG, GS, VAM, BMA, GMF, LAA, DSS, JZ, JVH.
Funding The authors would like to acknowledge funding support from University of Texas MD Anderson Lung Moon Shot Program and the MD Anderson Cancer Center Support Grant P30 CA01667, NIH R01 CA190628, NIH/NCI R01 CA205150-01, Cancer Prevention & Research Institute of Texas RP160652, UT Lung SPORE P50 CA70907, Rexanna's Foundation for Fighting Lung Cancer, Bruton Endowed Chair in Tumor Biology, Standing Fund for EGFR inhibitor resistance, the Hallman fund, Fox Lung EGFR Inhibitor Fund, and The Gil and Dody Weaver Foundation (to JVH).
Competing interests MVN is a consultant for Mirati and Merck/MSD, and reports funding to the institution from Mirati, Novartis, Pfizer, Ziopharm, AstraZeneca, and Checkmate; MM is an employee of Foundation Medicine, and owns stock in Roche; KS is an employee of Genentech, and owns stock in Roche; IB is an employee of Genentech, owns stock in Roche; VS is an employee of Roche, and owns stock in Roche; HX is an employee of Roche, and owns stock in Roche; HS is an employee of Genentech, and owns stock in Roche; XL is a Consultant for Eli Lilly, AstraZeneca, and EMD Serono; MEG is a former employee of Foundation Medicine; KM is an employee of Foundation Medicine, and owns stock in Roche; JPR reports licensing/royalties from Spectrum Pharmaceuticals; TC has received speaker’s fees from the Society for Immunotherapy of Cancer and Bristol-Myers Squibb, receives consulting fees from MedImmune and Bristol-Myers Squibb, and reports research funding to MD Anderson Cancer Center from Boehringer Ingelheim, MedImmune and Bristol-Myers Squibb; AT reports research grants from Eli Lilly, Millennium, Polaris, Genentech, Merck, Boehringer-Ingelheim, BMS, Ariad, Epizyme, Seattle Genetics, Takeda, and EMD Serono; advisory board member for BMS, Eli Lilly, Genentech, Roche, Novartis, Ariad, EMD Serono, Merck, Seattle Genetics, Astra-Zeneca, Boehringer-Ingelheim, Sellas Life Science, Takeda, Epizyme, and Huron; VP is an employee of Pfizer; DLG reports research grants from AstraZeneca, Jenssen R & D, Takeda, Ribun Therapeutics; and honoraria member of Senofi, AstraZeneca, Ribun Therapeutics; GS is an employee of Foundation Medicine, and owns stock in Roche; VAM is an employee of Foundation Medicine, and owns stock in Roche; BA is an employee of Foundation Medicine, and owns stock in Roche; GF is an employee of Foundation Medicine, and owns stock in Roche; LAA is an employee of Foundation Medicine, and owns stock in Roche; DS is an employee of Genentech, and owns stock in Roche; JZ reports research funding from Merck, Johnson and Johnson, and consultant fees from BMS, Johnson and Johnson, AstraZeneca, Geneplus, OrigMed, Innovent outside the submitted work; JVH reports research grants from NIH/NCI, American Cancer Society, Cancer Prevention & Research Institute of Texas, AACR Johnson & Johnson Lung Cancer, AZ, Spectrum, Checkmate Pharmaceuticals; Advisory Committees – AZ, BMS, GSK, Guardant Health, Kairos Venture Investments, BrightPath Biotherapeutics Hengrui Therapeutics, Eli Lilly, Spectrum, EMD Serono, Roche, Foundation One Medicine; Royalties & Licensing – Spectrum & Bio-Tree Systems, Inc.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.