
Abstract
The Blood-Brain Barrier (BBB) restricts access of large molecules to the brain. The low endocytic activity of brain endothelial cells (BECs) is believed to limit delivery of immunoglobulins (IgG) to the brain parenchyma. Here, we report that endogenous mouse IgG are localized within intracellular vesicles at steady state in BECs in vivo. Using high-resolution quantitative microscopy, we found a fraction of endocytosed IgG in lysosomes. We observed that loss of pericytes (key components of the BBB) in pdgf-bret/ret mice affects the intracellular distribution of endogenous mouse IgG in BECs. In these mice, endogenous IgG was not detected within lysosomes but instead accumulate at the basement membrane and brain parenchyma. Such IgG accumulation could be due to reduced lysosomal clearance and increased sorting to the abluminal membrane of BECs. Our results suggest that, in addition to low uptake from circulation, IgG lysosomal degradation may be a downstream mechanism by which BECs further restrict IgG access to the brain.
Similar content being viewed by others


Introduction
Therapeutic antibodies hold considerable potential in both diagnosis and treatment of diseases1,2. However, their use for passive or active immunotherapy in the central nervous system (CNS) is limited by the blood–brain barrier (BBB). It is estimated that the BBB prevents over 95% of drugs, including large molecules such as immunoglobulins (IgG), from accessing the brain3. In mice, less than 0.1% of peripherally administered IgG reaches the brain parenchyma4. This function of the BBB is critical for maintenance of brain homeostasis and results from the unique properties of brain endothelial cells (BECs). These cells are distinguished from peripheral endothelial cells by the presence of particularly tight intercellular junctions that prevent paracellular transport, by the expression of specialized molecular transporters and receptors at the apical and basolateral membranes and by a higher pericyte coverage. Furthermore, they interact with CNS-specific cell types, such as astrocytes, microglia and neurons, which together form the functional neurovascular unit (NVU)5,6,7. The precise role of BECs in protecting the brain from peripheral protein influx has been extensively studied. However, intracellular sorting and transport through the transcytosis pathway in BECs remains largely unexplored8.
Morphological studies of the BBB using transmission electron microscopy (TEM) showed that exogenous horseradish peroxidase (HRP) was poorly internalized within BECs9. This observation led to the widely held view that a low rate of endocytosis is a hallmark of the BBB3,5,6. Specifically, it is believed that “minimal vesicular trafficking”10 may be responsible for minimizing the amount of IgG that reaches the brain parenchyma11. However, additional mechanisms downstream of uptake may be involved. Despite extensive research on the delivery of therapeutic antibodies to the brain, surprisingly little is known about transcytosis of IgG4,12,13,14. Most studies focusing on uptake and sorting of IgG have been performed in cultured cells and data showing that IgG is present within BECs in situ in the NVU is limited15.
In this study, we investigated the distribution of IgG at the BBB and in BECs. By using quantitative high-resolution confocal microscopy, we show for the first time that endogenous mouse IgG (mIgG), one of the main components of plasma16, is present in intracellular vesicles within BECs. At steady state, a fraction of mIgG is found in lysosomes. We observed that loss of pericytes in pdgf-bret/ret mice17 affects the intracellular distribution of endogenous mIgG and of a peripherally administered antibody in BECs. Our data suggest that pericytes modulate IgG trafficking by reducing their lysosomal transport in BECs. Overall, our results suggest that, in addition to a low basal rate of uptake, lysosomal degradation of IgG is a downstream mechanism by which BECs may limit the amount of IgG that enters the brain.
Results
We first applied a confocal light-microscopy protocol to image different cell types of the NVU. Our aim was to visualize intracellular structures that could thus far be detected only by electron microscopy (Fig. 1a). We reconstructed a 3D model of the NVU by immunofluorescent-labelling of BECs, pericytes and basal lamina markers (Fig. 1b,c; Table 1). Next, we examined the distribution of endogenous mIgG within the NVU. Under physiological conditions, it is believed that the low endocytosis rate of BECs is sufficient to exclude mIgG from the brain parenchyma11. Unexpectedly, we detected numerous mIgG puncta within capillaries (Fig. 1d–f; Supplementary Video 1). This distribution of mIgG was not an artefact caused by unspecific antibody binding since (i) we observed the same pattern using three different anti-mouse antibodies (Fig. 1d,g–j, Supplementary Fig. 1), (ii) no signal was observed using secondary antibodies against goat or human IgGs (Supplementary Figs 1 and 5), and (iii) the signal was restricted to the intracellular space in capillaries delineated by CollagenIV (Fig. 1d–f). We found that the distribution of mIgG was widespread along the vasculature in the cerebral cortex. However, the punctate pattern of mIgG was only evident at high-resolution (Supplementary Fig. 2). The majority of these puncta occurred within BECs and not pericytes, as shown by staining with CD31 (Fig. 1g,h) or CD13 (Fig. 1i,j).

(a) Representative TEM cross-section of a brain cortical microvessel. The arrow points to a non-coated vesicle budding from the luminal membrane, the arrowhead points to a clathrin-coated-vesicle and the asterisk marks an intracellular vesicle. BEC, Brain Endothelial Cell; TJ, Tight Junction; BL, Basal Lamina; Per, Pericyte. (b,c) Representative 3D reconstruction of the NVU showing a BEC (marked by CD31 expression, green), Pericyte (marked by CD13 expression, red) within the Basal Lamina (CollagenIV, grey). The cross-section in the right panel (c) is a single optical section to highlight the vascular lumen. (d–f) Representative 3D reconstruction of a microvessel (marked by CollagenIV, green) with mIgG (red) in punctate structures (d). (e) shows a high magnification 3D reconstruction of the boxed area. Cross-sections in (f) show that vesicles are within the Basal Lamina and not in the parenchymal space. (g–j) Representative single optical sections of mIgG puncta localizing specifically within BECs marked with CD31 in green (g,h) but not in pericytes marked with CD13 in green (i,j). (k,l) Histograms for the radius of mIgG-positive intracellular vesicles (k) and normalized mIgG intensity per vesicle (l) in semi-logarithmic scale. Points show the mean value ± SEM of each size or intensity bin for 30 microvessels from 3 different C57BL/6 mice. The continuous line shows a log-normal fit of the experimental data. In all images, DAPI-stained nuclei are shown in blue.
The mIgG puncta had a radius between 0.2 and 0.4 μm with the majority below 0.5 μm, in agreement with the size of endosomes (Fig. 1k, Supplementary Fig. 3). We also estimated the amount of mIgG contained in individual puncta by quantifying the fluorescence intensity per puncta. Intensity values corresponding to individual puncta spanned nearly three orders of magnitude (Fig. 1l) suggesting that some puncta accumulate mIgG18.
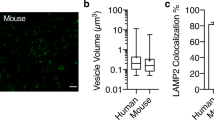
Next, we analyzed the localization of mIgG within BECs. Extensive antibody screening was performed to identify suitable endosomal markers compatible with our ex vivo technique. However, many antibodies gave non-specific punctate staining and were not considered for further analysis. As an alternative we exploited two recently described “Brain Shuttle” antibodies as markers of different endosomal populations19. Monovalent Brain Shuttle (BS-sFab) binds to the transferrin receptor (TfR) and is trafficked through the transferrin receptor-mediated transcytosis pathway, which includes both early and recycling endosomes, ultimately reaching the brain parenchymal space. Divalent Brain Shuttle (BS-dFab) also binds to the transferrin receptor but is trafficked for degradation in lysosomes19. The Brain Shuttles were detected using a secondary antibody against human IgG. Mice were administered with the different BS constructs (6 mg/kg) and sacrificed either 30 minutes (BS-sFab) or 8 hours (BS-dFab) post-injection. After 8 hours, BS-sFab is localized in the brain parenchyma with only minimal signal in capillaries and was thus not suitable for this analysis19. We found BS-sFab in vesicular structures within capillaries (Fig. 2a). Interestingly, we observed colocalization between BS-sFab and mIgG in vesicles close to the luminal membrane (Fig. 2a, asterisk). In contrast, there was little overlap between BS-sFab- and large mIgG-positive vesicles (Fig. 2a, arrows), suggesting that BS-sFab and mIgG are initially transported through a common endosomal compartment and that mIgG is subsequently sorted away from the canonical TfR transcytosis pathway. On the other hand, we observed extensive colocalization between BS-dFab and mIgG (Fig. 2b). Since BS-dFab is sorted to lysosomes this suggested that mIgG-positive vesicles are also transported to lysosomes19. To confirm this hypothesis, we performed a double immunostaining using an antibody against LAMP2, a lysosomal marker, and mIgG. We found that 20% of the total intracellular mIgG colocalized with lysosomes at steady state (Fig. 2c,d). In BECs, therefore, a substantial amount of mIgG is transported to lysosomes for degradation.

(a,b) Representative 3D reconstruction of the intracellular distribution of the Brain Shuttle sFab (a) or dFab (b) (both in green) and endogenous mIgG (red) within a single brain microvessel. The right panels show high magnification image of the boxed area. Arrows highlight the minimal overlap between mIgG and BS-sFab vesicles in (a) and extensive colocalization between mIgG and BS-dFab in (b). Asterisks show Brain Shuttle-positive vesicles overlapping with faint mIgG vesicles. (c) Representative 3D reconstruction of the intracellular distribution of LAMP2-positive lysosomes (green) and endogenous mIgG (red) within a single brain microvessel. The right panel shows a high magnification image of the boxed area and highlights the colocalization between mIgG and LAMP2-positive lysosomes. In all images, DAPI-stained nuclei are shown in blue. (d) Colocalization of mIgG to LAMP2 expressed as the fraction of the total vesicular mIgG intensity colocalized with LAMP2 vesicles. Points show measurements from individual microvessels. Each symbol corresponds to a different animal. Lines show the mean ± SD for 33 microvessels from 3 different animals.
Pericytes are key components of the NVU that are known to control the permeability of the BBB. Previous work showed that reduction of pericyte coverage of brain microcapillaries in pdgf-bret/ret mice results in the acute extravasation of injected tracers due to increased transcytosis17. However, the specific changes in the transcytosis pathway (for example uptake, sorting or fusion with the plasma membrane) were not explored. Therefore, we applied our imaging and quantification protocol to analyze the changes in endogenous mIgG transport in pdgf-bret/ret mice in comparison to control mice (Fig. 3a–c). We used CollagenIV, a marker of the basal lamina, to delineate capillaries in 3D and then quantified the amount of mIgG that accumulated within the basal lamina, the amount of mIgG that accumulated in the brain parenchymal space and the number of mIgG-positive vesicles contained within microcapillaries (Fig. 3d–f, respectively). The amount of endogenous mIgG increased 100-fold within the basal lamina and 50-fold in the brain parenchymal space of pdgf-bret/ret mice compared to both C57BL/6 and pdgf-bret/wt mice (Fig. 3a–e, Supplementary Fig. 4). The enlargement of capillaries in pdgf-bret/ret was previously reported (Fig. 3c)17. Strikingly, the number of mIgG-positive intracellular vesicular structures was significantly reduced in pdgf-bret/ret mice compared to C57BL/6 or pdgf-bret/wt mice (Fig. 3f). Moreover, the few remaining vesicular structures did not colocalize with LAMP2-positive lysosomes (Fig. 3g, inset and arrows). Therefore, the reduced number of intracellular mIgG-positive vesicles in pdgf-bret/ret mice could result from the increased transport and delivery of mIgG to the abluminal membrane.

(a–c) Representative maximum projection images of brain microvessels (marked by CollagenIV in green) showing the localization of mIgG (red) in C57BL/6 (a) pdgf-bret/wt (b) and pdgf-bret/ret (c) mice. DAPI-stained nuclei are shown in blue. (d–f) Graphs showing the quantification of mIgG intensity per μm2 of basal lamina (d) mIgG intensity per μm3 of brain parenchyma (e) and number of vesicles per μm3 of microvessel volume (f) in C57BL/6 (blue), pdgf-bret/wt (red), and pdgf-bret/ret mice (green). Points show measurements from individual microvessels. Each symbol corresponds to a different animal. Lines show the mean ± SD for 30 microvessels from 3 different animals for each phenotype. **p < 0.0001 by Fisher’s LSD test. (g) Representative maximum projection of a brain microvessel in pdgf-bret/ret mice showing the localization of mIgG (red) and lysosomes marked by LAMP2 (green). The right panel shows a high magnification of a single optical slice image of the boxed area and highlights the reduced colocalization between LAMP2 and mIgG. Individual mIgG and LAMP2 vesicles are marked with arrowheads and arrows, respectively.
To test this hypothesis, we used TEM to analyze the distribution of the total pool of intracellular vesicles in pdgf-bret/ret mice (Fig. 4a). Vesicles were classified as (i) luminal, including coated and non-coated vesicles docked or budding from the luminal membrane (Fig. 4b), (ii) intracellular, including tubules and vesicles within the cytoplasm (Fig. 4c) or (iii) abluminal i.e. vesicles docked to the abluminal membrane (Fig. 4d). There was no significant difference between luminal vesicles in pdgf-bret/ret and pdgf-bret/wt mice (Fig. 4e). However, both the number of intracellular and abluminal vesicles were significantly increased in pdgf-bret/ret BECs (Fig. 4f,g). These observations suggest that the increased delivery of endogenous mIgG to the brain parenchyma upon pericyte depletion results from an increased intracellular transport to the abluminal membrane.

(a) Scheme representing the different vesicle populations identified by TEM in BECs. (b–d) representative TEM cross-sections of brain microvessels in pdgf-bret/wt (left panel), and pdgf-bret/ret mice (right panel) showing coated and non-coated luminal vesicles (b) intracellular vesicles and tubules (c) and abluminal vesicles (d). (e–g) Graphs showing the quantification of the number of vesicles per capillary identified by TEM in pdgf-bret/wt (red), and pdgf-bret/ret mice (green). Points show measurements from individual microvessels. Each symbol corresponds to a different animal. Columns represent the median and error bars the interquartile range for at least 18 microvessels from at least 3 different animals per phenotype. **p < 0.005 by Mann-Whitney U test.
To further confirm these data, we investigated the intracellular distribution in BECs of a human IgG following peripheral administration to pdgf-bret/ret and pdgf-bret/wt mice. Mab86 is a humanized antibody that binds specifically to phosphorylated tau20. One hour post-injection, Mab86 was localized within vesicular structures in pdgf-bret/wt BECs where it colocalized with endogenous mIgG (Fig. 5a, Supplementary Fig. 5). On the contrary, we did not detect Mab86-containing vesicular structures in pdgf-bret/ret BECs (Fig. 5b).

(a,b) Representative maximum intensity projections of microcapillaries comparing the intracellular distribution of Mab86 (green) after acute injection and endogenous mIgG (red) in pdgf-bret/wt (a), and pdgf-bret/ret (b) mice. (c,d) High-resolution images of neurons on the hippocampus of 3Tg x pdgf-bret/wt (c) or 3Tg x pdgf-bret/ret (d) mice. In all images, DAPI-stained nuclei are shown in blue. (e,f) Representative low-magnification images of the hippocampus of 3Tg x pdgf-bret/wt (e) or 3Tg x pdgf-bret/ret (f) mice showing phosphotau positive neurons (red) and Mab86 (green). (g,h) High magnification images of the boxed area in 3Tg x pdgf-bret/wt (g) or 3Tg x pdgf-bret/ret (h) mice highlighting the accumulation of Mab86 in phosphotau positive neurons in 3Tg x pdgf-bret/ret mice.
Finally, we generated a new mouse model by crossing pdgf-bret/ret animals with the TauPS2APP triple transgenic mouse model (3Tg) of Alzheimer’s Disease (AD)21 to test whether pericyte loss would increase transcytosis and delivery of Mab86 to phosphotau-containing hippocampal neurons. We injected 13 months-old mice i.p. with 30 mg/kg of Mab86 and analyzed brain sections 48 hours post-injection. Neither Mab86 nor mIgG were detectable in the brain parenchyma of 3Tg x pdgf-bret/wt mice (Fig. 5c). In 3Tg x pdgf-bret/ret mice, Mab86 and mIgG diffusely localized in the parenchymal space (Fig. 5d). However, Mab86 specifically accumulated in hippocampal neurons (Fig. 5d). We then examined tau pathology in the hippocampus of 3Tg x pdgf-bret/wt and 3Tg x pdgf-bret/ret mice and found that phosphotau localized predominantly at the tip of the CA1 region and the subiculum as previously described (Fig. 5e,f)20,21. In 3Tg.x pdgf-bret/wtmice, very few Mab86-positive cells were detected (Fig. 5e,g). However, Mab86 target engagement was strongly enhanced in phosphotau positive neurons of 3Tg x pdgf-bret/ret mice (Fig. 5f–h). These data confirm that pericyte depletion increases the delivery of a functional IgG to the brain. Overall, our results support the hypothesis that increased IgG transcytosis to the brain in pdgf-bret/ret mice results from an increased intracellular trafficking to the abluminal membrane.
Discussion
Earlier studies using electron microscopy showed that vesicular transport is extremely low in BECs, providing one possible explanation for how the BBB limits entry of peripheral proteins to the brain3,5,9. However, although TEM allows detailed characterization of the morphological properties of BECs and the NVU, it has limitations for the study of transcytosis. In particular, the number of intracellular vesicles within endothelial cells observed by TEM gives no information on the rates of endosome transport. A recent review on TEM studies found only a weak correlation between intracellular vesicles and transport capacity in different types of vascular bed22. This consideration prompted us to further investigate the mechanisms that restrict protein transport into the brain parenchyma.
We used TEM to quantify the total number of vesicular structures contained within BECs in pdgf-bret/ret and pdgf-bret/wtmice. Whereas luminal vesicle numbers were similar in pdgf-bret/ret and pdgf-bret/wtmice, pericyte loss in pdgf-bret/ret mice leads to increased numbers of intracellular vesicles. Docking and fusion of these vesicles with the abluminal membrane probably contributes to the increased protein permeability of the BBB as previously described17. To complement this analysis, we tried to identify the specific location of endogenous IgG at the NVU using quantitative high-resolution confocal imaging. In control mice, BECs contained numerous intracellular vesicles filled with varying amounts of mIgG. The steady state vesicular distribution observed ex vivo reflects the balance of uptake, endosome fusion, recycling, and degradation that has previously been described in in vitro models for cargo(s) such as LDL18. Importantly, our observations do not challenge the fact that BECs have a low basal uptake from circulation compared with peripheral endothelial cells. However, despite such a low uptake there are abundant mIgG-positive vesicles in BECs. Therefore, mechanisms downstream of uptake must exist to prevent the delivery of this intracellular mIgG pool to the brain parenchyma. First, mIgG may interact with FcRn and get recycled to the plasma membrane and released in the blood stream. FcRn is the only Fc receptor known to be expressed by BECs23. In vitro, FcRn-expressing endothelial cells can recycle IgG via Rab11-positive endosomes15. Therefore, binding of mIgG to FcRn within BEC endosomes could prevent their sorting to the abluminal membrane. Additional experiments using specific markers of FcRn recycling pathway would be required to confirm this hypothesis. Second, mIgG may be prevented from reaching the parenchyma through trafficking to lysosomes for degradation. In a series of in vivo experiments, Broadwell et al showed that circulatory proteins are endocytosed and eventually degraded in lysosomes24. Interestingly, we observed significant colocalization of mIgG with both LAMP2 (a lysosomal marker) and BS-dFab (gets trafficked to lysosomes) indicating that lysosomal degradation prevents delivery of mIgG to the brain. Together, these data suggest that lysosomal clearance of proteins in BECs may limit protein access to the brain parenchyma. Additional work using quantitative analysis of transport kinetics across multiple endosomal compartments in vivo will be required to fully characterize the sorting pathway for IgG across the BBB.
Previous work showed that pericytes are key regulators of BBB permeability17,25. For example, the transcytosis flux is increased in pericyte-deficient mice such as pdgf-bret/ret17. We observed a significant reduction in the number of intracellular mIgG-positive vesicles in pdgf-bret/ret mice compared to control mice. Conversely, mIgG delivery to the brain was significantly increased. This could be due to an unchanged, saturated IgG uptake together with an increased release of mIgG to the brain in pdgf-bret/ret BECs13. Supporting this hypothesis we found (i) no difference in the number of luminal coated and non-coated vesicles and (ii) a significant increase in the number of abluminal vesicles in pdgf-bret/ret mice compared to control mice. These data suggest that pericyte loss may lead to increased vesicular flux to the abluminal membrane of BECs. The findings do not exclude that other fluid-phase uptake pathways26 are altered upon pericyte depletion25,27 but suggest that pericytes act on specific endocytosis pathways.
The BBB poses a formidable obstacle for delivery of therapeutic antibodies since less than 0.1% of peripherally administered IgG reaches the brain parenchyma4. We show for the first time that a systemically administered antibody, Mab86, can be detected in vesicular structures within BECs. The low brain exposure of Mab86 and its colocalization with endogenous mIgG in BECs suggests that lysosomal clearance may limit Mab86 transport to the brain parenchyma. Loss of pericytes significantly enhanced Mab86 brain delivery as shown by substantially increased target engagement in neurons and, similar to endogenous mIgG, did not increase the number of intracellular Mab86-positive vesicles in BECs. Overall our results suggest that pericytes regulate IgG transcytosis in part by controlling trafficking to lysosomes for degradation. Our data suggest that strategies to reduce IgG lysosomal clearance in BECs may enhance delivery of antibodies across the BBB and into the brain.
Methods
Mice
TauPS2APP (3Tg) and pdgf-bret/ret mice were described previously17,21. 3Tg mice were crossed with pdgf-bret/wtmice to generate 3Tgx pdgf-bret/ret and 3Tg x pdgf-bret/wt littermates. All animal experiments were approved by the Swiss Veterinary Office Basel-Stadt and were carried out in accordance with the approved guidelines described in the Swiss animal permission #1902.
Brain sectioning and immunofluorescence
Brain processing was performed as previously described19. Briefly, 19–20 months-old C57BL/6, pdgf-bret/wt or pdgf-bret/ret mice were euthanized with CO2 and transcardially perfused with PBS at 37 °C, followed by perfusion with 2% PFA. The brain was then removed and incubated overnight in 2% PFA at 4 °C before sectioning. Brains were included in 2% agarose and 100 μm sagittal sections were cut using a Leica VT1000M vibratome. Sections were stored at −20 °C in 1:1 PBS/Glycerol. Sections were processed for immunofluorescence by washing with PBS and permeabilization with PBS 0.3% Triton X-100 and 10% donkey serum for blocking. Primary antibodies were diluted in 5% donkey serum in PBS and incubated with sections for 72 hours at 4 °C, followed by washing with PBS and 1 hour incubation at room temperature with appropriate fluorescently-labelled secondary antibodies (Donkey anti-goat, donkey anti-rabbit, or donkey anti-rat IgG coupled to AlexaFluor488, 555, or 647, from LifeTechnologies) in 5% PBS. Finally, sections were washed with PBS, stained with 1 μg/ml DAPI and mounted using DAKO Fluorescent Mounting medium on glass slides with a 0.17 mm coverslip. Table 1 shows the list of antibodies used for the study.
Electron microscopy
Brain samples were fixed by immersion in 2.5% glutaraldehyde and 2.5% paraformaldehyde in cacodylate buffer (0.1 M, pH 7.4) and washed in cacodylate buffer for 30 minutes. The samples were post-fixed in 1% osmium tetroxide in 0.1 M cacodylate buffer for 1 hour at 4 °C and dehydrated through graded alcohol (50, 70, 90, and 100%) and propylene oxide for 30 minutes each. Next, samples were oriented longitudinally and embedded in Epon 812. Ultrathin 70 nm sections were contrasted with uranyl acetate and lead citrate and examined at 70 kv with a Morgagni 268D electron microscope. Digital images were acquired with a Mega View III camera (Soft Imaging System). Cortical capillaries were selected randomly for quantification of intracellular vesicles.
Imaging of intracellular mIgG at the NVU
Mounted brain sections were imaged using the Leica TCS SP8 microscope using a HC PL APO 63x/1.4 oil objective. Images were 12-bit and either 1024 × 1024 with 90 nm pixel size or 512 × 512 with 46.46 pixel size. Laser intensity and detector gain settings were optimized to minimize pixel saturation and maximize dynamic range. Between 15 and 20 optical sections were acquired per vessel covering a z-distance of 7–12.5 μm. Deconvolution of confocal images was performed using the Leica LAS-AF 3D deconvolution tool. Movies and 3D reconstructions of vessels were performed using Imaris. Cross-sections were performed using FIJI.
Quantification of intracellular mIgG
Two different methods were used to quantify intracellular mIgG within BECs. Since the density of mIgG-positive structures was low and the z-stack acquired was only 7 μm thick, we could analyze the maximum intensity projection of each microvessel. Quantitative multiparametric image analysis (QMPIA) was performed using Kalaimoscope MotionTracking as previously described18,28,29. Object-based colocalization was estimated after substraction of random colocalization as previously described30. To quantify and compare the accumulation of mIgG in intracellular vesicles, basal lamina and parenchymal space, quantification were performed using Imaris. 3D-reconstructed images were segmented using an absolute intensity threshold mask on CollagenIV to identify intracellular structures (within CollagenIV mask), Basal Lamina (colocalized with CollagenIV mask) and parenchymal signal (outside of CollagenIV mask). Intracellular vesicles were segmented using Imaris spot detection algorithm with default parameters and an estimated diameter of 0.8 μm. mIgG intensity values were normalized by the surface area of the mask or the parenchymal volume. The total number of vesicles was normalized by the volume of CollagenIV mask. Quantifications were obtained from 10 microvessels per animal using at least 3 animals per genotype. Multiple statistical comparisons were performed using Fisher’s LSD test in GraphPad.
Detection of BS or Mab86
14-months old TAuPS2APP mice were injected with 6 mg/kg BS i.v. and euthanized by pentobarbital 30 minutes after injection. Then, animals were transcardially perfused with PBS, followed by perfusion with 2% PFA. The brains were removed and incubated overnight in 2% PFA at 4 °C before sectioning. For Mab86 detection, 13 months old 3Tg x pdgf-bret/wt or 3Tg x pdgf-bret/ret mice were injected with 30 mg/kg Mab86 and euthanized by pentobarbital 1 or 48 hours after injection. Whole brain images were acquired from frozen sections prepared and immunostained with an anti-human PHF-TAU AT8 antibody and a goat anti-human IgG (H + L) AlexaFluor555 (LifeTechnologies) to detect Mab86 (Pierce, MN1020) as previously described20. Imaging was performed using a Metafer 4 slide scanning system (MetaSystems).
Additional Information
How to cite this article: Villaseñor, R. et al. Trafficking of Endogenous Immunoglobulins by Endothelial Cells at the Blood-Brain Barrier. Sci. Rep. 6, 25658; doi: 10.1038/srep25658 (2016).
References
Wu, A. M. & Senter, P. D. Arming antibodies: prospects and challenges for immunoconjugates, Nat Biotechnol 23, 1137–1146, 10.1038/nbt1141 (2005).
Adams, G. P. & Weiner, L. M. Monoclonal antibody therapy of cancer, Nat Biotechnol 23, 1147–1157, 10.1038/nbt1137 (2005).
Pardridge, W. M. Drug transport across the blood-brain barrier, J Cereb Blood Flow Metab 32, 1959–1972, 10.1038/jcbfm.2012.126 (2012).
St-Amour, I. et al. Brain bioavailability of human intravenous immunoglobulin and its transport through the murine blood-brain barrier, J Cereb Blood Flow Metab 33, 1983–1992, 10.1038/jcbfm.2013.160 (2013).
Rubin, L. L. & Staddon, J. M. The cell biology of the blood-brain barrier, Annu Rev Neurosci 22, 11–28, 10.1146/annurev.neuro.22.1.11 (1999).
Blanchette, M. & Daneman, R. Formation and maintenance of the BBB, Mech Dev 138 Pt 1, 8–16, 10.1016/j.mod.2015.07.007 (2015).
Abbott, N. J., Patabendige, A. A., Dolman, D. E., Yusof, S. R. & Begley, D. J. Structure and function of the blood-brain barrier, Neurobiol Dis 37, 13–25, 10.1016/j.nbd.2009.07.030 (2010).
Preston, J. E., Joan Abbott, N. & Begley, D. J. Transcytosis of macromolecules at the blood-brain barrier, Adv Pharmacol 71, 147–163, 10.1016/bs.apha.2014.06.001 (2014).
Reese, T. S. & Karnovsky, M. J. Fine structural localization of a blood-brain barrier to exogenous peroxidase, J Cell Biol 34, 207–217 (1967).
Chow, B. W. & Gu, C. The Molecular Constituents of the Blood-Brain Barrier, Trends Neurosci 38, 598–608, 10.1016/j.tins.2015.08.003 (2015).
Triguero, D., Buciak, J. B., Yang, J. & Pardridge, W. M. Blood-brain barrier transport of cationized immunoglobulin G: enhanced delivery compared to native protein, Proc Natl Acad Sci USA 86, 4761–4765 (1989).
Bard, F. et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease, Nat Med 6, 916–919, 10.1038/78682 (2000).
Zlokovic, B. V. et al. A saturable mechanism for transport of immunoglobulin G across the blood-brain barrier of the guinea pig, Exp Neurol 107, 263–270 (1990).
Tuma, P. & Hubbard, A. L. Transcytosis: crossing cellular barriers, Physiol Rev 83, 871–932, 10.1152/physrev.00001.2003 (2003).
Ober, R. J., Martinez, C., Vaccaro, C., Zhou, J. & Ward, E. S. Visualizing the site and dynamics of IgG salvage by the MHC class I-related receptor, FcRn, J Immunol 172, 2021–2029 (2004).
Waldmann, T. A. & Strober, W. Metabolism of immunoglobulins, Prog Allergy 13, 1–110 (1969).
Armulik, A. et al. Pericytes regulate the blood-brain barrier, Nature 468, 557–561, 10.1038/nature09522 (2010).
Foret, L. et al. A general theoretical framework to infer endosomal network dynamics from quantitative image analysis, Curr Biol 22, 1381–1390, 10.1016/j.cub.2012.06.021 (2012).
Niewoehner, J. et al. Increased Brain Penetration and Potency of a Therapeutic Antibody Using a Monovalent Molecular Shuttle, Neuron 81, 49–60, 10.1016/j.neuron.2013.10.061 (2014).
Collin, L. et al. Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer’s disease, Brain 137, 2834–2846, 10.1093/brain/awu213 (2014).
Grueninger, F. et al. Phosphorylation of Tau at S422 is enhanced by Abeta in TauPS2APP triple transgenic mice, Neurobiol Dis 37, 294–306, 10.1016/j.nbd.2009.09.004 (2010).
Stewart, P. A. Endothelial vesicles in the blood-brain barrier: are they related to permeability? Cell Mol Neurobiol 20, 149–163 (2000).
Schlachetzki, F., Zhu, C. N. & Pardridge, W. M. Expression of the neonatal Fc receptor (FcRn) at the blood-brain barrier, J Neurochem 81, 203–206, 10.1046/j.1471-4159.2002.00840.x (2002).
Broadwell, R. D. & Salcman, M. Expanding the definition of the blood-brain barrier to protein, Proc Natl Acad Sci USA 78, 7820–7824 (1981).
Daneman, R., Zhou, L., Kebede, A. A. & Barres, B. A. Pericytes are required for blood-brain barrier integrity during embryogenesis, Nature 468, 562–566, 10.1038/nature09513 (2010).
Mayor, S., Parton, R. G. & Donaldson, J. G. Clathrin-independent pathways of endocytosis, Cold Spring Harb Perspect Biol 6, 10.1101/cshperspect.a016758 (2014).
Ben-Zvi, A. et al. Mfsd2a is critical for the formation and function of the blood-brain barrier, Nature 509, 507–511, 10.1038/nature13324 (2014).
Rink, J., Ghigo, E., Kalaidzidis, Y. & Zerial, M. Rab conversion as a mechanism of progression from early to late endosomes, Cell 122, 735–749, 10.1016/j.cell.2005.06.043 (2005).
Collinet, C. et al. Systems survey of endocytosis by multiparametric image analysis, Nature 464, 243–249, 10.1038/nature08779 (2010).
Kalaidzidis, Y., Kalaidzidis, I. & Zerial, M. A Probabilistic Method to Quantify the Colocalization of Markers on Intracellular Vesicular Structures Visualized by Light Microscopy, Aip Conf Proc 1641, 580–587, 10.1063/1.4906025 (2015).
Acknowledgements
We thank Annie Girardeau, Christof Kugler and Michelle Ammann for excellent technical assistance. R.V is supported by the Roche Postdoctoral Fellowship (RPF) program (2014_2016). We thank Yves Lutz, the IGBMC Imaging platform, Francoise Gerber, Juerg Messer and Bernd Bohrmann for their support.
Author information
Authors and Affiliations
Contributions
The project was designed by L.C. The manuscript was prepared by R.V., L.O., F.G., H.L., P.-O.F., L.C. and R.V. carried out the experimental work. L.O. carried out the in vivo work. N.M. performed the electron microscopy. F.G. provided the Mab86 antibody. A.K. and C.B. provided the pdgf-ret mice. R.V. and L.C. analysed the experimental data. Correspondence to be addressed to L.C.
Corresponding author
Ethics declarations
Competing interests
R.V., L.O., F.G., H.L., P.-O.F. and L.C. are all are under paid employment by the company F. Hoffmann-La Roche. R.V. is a Roche post-doctoral fellow.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Villaseñor, R., Ozmen, L., Messaddeq, N. et al. Trafficking of Endogenous Immunoglobulins by Endothelial Cells at the Blood-Brain Barrier. Sci Rep 6, 25658 (2016). https://doi.org/10.1038/srep25658
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep25658
This article is cited by
-
Nano-immunotherapy: overcoming delivery challenge of immune checkpoint therapy
Journal of Nanobiotechnology (2023)
-
Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours
Nature Reviews Clinical Oncology (2023)
-
The Association of Serum Immunoglobulins with Risk of Cardiovascular Disease and Mortality: the Rotterdam Study
Journal of Clinical Immunology (2023)
-
CD98hc is a target for brain delivery of biotherapeutics
Nature Communications (2023)
-
Activation of NMDA receptors in brain endothelial cells increases transcellular permeability
Fluids and Barriers of the CNS (2022)
