
Abstract
Hsp90 is a specialized molecular chaperone that is capable of buffering the expression of abnormal phenotypes. Inhibition of Hsp90 activity results in the expression of these phenotypes that are otherwise masked. Selection of offspring from the crossing of affected progenies results in inheritance and enrichment of these phenotypes, which can become independent of their original stimuli. The current combined evidence favours a model involving the interplay between genetics and epigenetics. The recent proteomics efforts to characterize the Hsp90 interaction networks provide further clues into the molecular mechanisms behind this complex phenomenon. This review summarizes the most recent experimental observations and briefly discusses the genetic and epigenetic views used in explaining the different observations.
Similar content being viewed by others

Introduction
Hsp90 is an abundant and highly conserved ATP-dependent molecular chaperone that is well characterized with respect to its function 1, its physical and genetic interactors 2, 3, and its influence on phenotypic expression 4. Hsp90 is distinct from other chaperone systems in two main aspects. Unlike the regular chaperone systems, Hsp90 does not act to fold non-native proteins, but rather binds to substrate proteins that are in a near-native state and, thus, which are at a late stage of folding 5. Furthermore, Hsp90 seems to be a specialized chaperone that targets a specific set of client proteins that are mainly involved in signal-transduction pathways. Hsp90 substrates include many transcription factors and protein kinases 6. The in vivo and in vitro activity of Hsp90 depends on its association with cochaperones, such as Hsp40 and Hsp70, and cofactors, such as p23 (yeast Sba1) and Hop (yeast Sti1), that are components of the large Hsp90-multiprotein complexes involved in folding client proteins. Cochaperones regulate the Hsp90 ATPase activity, assist in protein folding, or function as a scaffold for Hsp90 complexes 7. Several small-molecule drugs, such as geldanamycin and radicicol 8, 9, have been discovered that specifically inhibit Hsp90 ATPase; these drugs bind in the ATP-binding pocket at the N-terminal domain of the chaperone 10.
Recently, it has been found that Hsp90 inhibition in several model organisms induces the inheritance and enrichment of abnormal phenotypes 11, 12, 13. By inbreeding the affected progenies for successive generations, the frequencies of the abnormal phenotypes increase in a non-Mendelian fashion. To date, there is no definitive molecular model for explaining this experimental phenomenon. While some evidence currently available would favour a genetically based model 11, 12, other evidence seems to better suit an epigenetically based model 13. In this regard, a recently published integrated proteomic and genomic study of the Hsp90 interaction network in Saccharomyces cerevisiae 3 revealed putative physical and genetic interactions between Hsp90 and several components of the INO80 and SWR-C chromatin remodelling complexes strongly suggesting a functional linkage between Hsp90 and the chromatin remodelling machinery, at least in yeast. Since Hsp90 is highly conserved across many different species, it is reasonable to expect that the general features of a similar Hsp90 interaction network and the possible functional linkage between the chaperone and the chromatin remodelling complexes will also be conserved. This review provides a summary of the most recent research in this area.
How to evolve?
In natural selection, changes in the environment drive the selection for phenotypes that are adaptive, and the fixation of selected phenotypes gives rise to new traits. In classical evolution, phenotypes are determined by the corresponding genotypes that arise from non-deleterious mutations or recombinations of genes, which produce adaptive genotypes that are favoured and, therefore, positively selected (Figure 1A). This selection leads to preservation of 'useful' genes in the population's gene pool, while 'useless' genes are lost in the selection process (Figure 1A). In addition, genetic drift, which is defined as random changes in allele frequencies in the offspring population due to the imperfect genetic representation of the parental population, is also likely to play a part in removing rare genes from the population. The effect of genetic drift would be more pronounced if the population size is small.

A comparison between genetics and epigenetics in trait acquisition. (A) Genetically, a novel trait, i.e., phenotype, arises from non-deleterious mutations and/or gene recombinations, thus altering parts of the genome of the affected progeny. Positive selection for the new trait then results in an increased proportion of affected progenies in the population and the accumulation of novel genes, which give rise to the new trait, in the population's gene pool. (B) Epigenetically, a novel trait might result from the expression of genes that are normally suppressed 4. Environmental stress induces covalent modifications of DNA 17 and histones 17, 18, changes in chromatin structure 16, 18, 19, and/or RNAi 20, which alter the gene expression profile of the affected progeny. Inheritance of the parental epigenome and positive selection for the new trait thus results in an increased proportion of progenies having the altered epigenome, although there have been no changes to the population's genetic makeup.
An alternative mechanism was proposed in CH Waddington's developmental canalization model 14, 15. The model states that, in the wild population, there are masked phenotypes that can be expressed by environmental perturbations. Selection for adaptive phenotypes over several generations results in their fixation, and the resulting phenotypes become the new traits for the population. The model has two important features. (1) The 'new' phenotypes are inducible by environmental stress and are inherent to the population, and they do not entail any changes to the population's genome. (2) Positive selection for these phenotypes over several generations results in their fixation and independence from the original stimuli that was required for their initial expression; in other words, the phenotypes become 'canalized'.
There is evidence that supports the inheritance of different modification states of DNA from one generation to the next 16, 17, 18, 19. Since these modifications lead to changes in gene expression profile, this suggests that epigenetics, which is defined as the inheritance of the changes in gene expression profile without alterations in the genomic sequence, should also be considered as part of the evolutionary equation, at least in terms of phenotypic plasticity. Established epigenetic factors include DNA methylation 17, chromatin remodelling 16, 18, 19, histone modification 17, 18, and RNA interference (RNAi) 20. In other words, changes in the DNA sequence of an organism's genome might not be required to alter its phenotype and the development of a new trait through natural selection might be, in part, the result of epigenetic variations (Figure 1B).
Phenotypic buffering by Hsp90
The buffering of stress-induced phenotypes by Hsp90 has been studied in several different organisms. Rutherford and Lindquist 11 showed that genetic and pharmacological impairment of Hsp90 in Drosophila melanogaster (fruit flies) produces abnormal phenotypes that are enhanced at mildly higher or lower than normal temperatures. Phenotypic selection of a wing or an eye phenotype by crossing affected progenies for several generations resulted in an increase of their frequencies as well as in the divergence of each phenotype. In addition, distribution of affected individuals varied between different inbred lines. Similarly, progenies from wild-type flies crossed to Hsp83 mutants (gene encoding Hsp90 protein in Drosophila) as well as geldanamycin- (an Hsp90 inhibitor) treated flies also showed a similar increase in morphological phenotypes, thus showing that the phenotypic buffering by Hsp90 is naturally occurring 11.
Several important observations were made in these experiments. First, the acquired phenotypes generated by Hsp90 impairment became independent of Hsp90 after successive inbreeding between affected individuals. PCR genotyping revealed that affected progenies from the F6 and F7 generations no longer possessed the original Hsp83 mutation that induced the abnormal phenotype 11. Second, the increase in affected progenies did not occur in a Mendelian fashion. More than 80% of the F6 and F7 generations were affected when only 66% was expected. The authors explained these observations as the result of the involvement of multiple genetic determinants. Third, the phenotypic buffering capacitance of Hsp90 in fruit flies harbouring the Hsp83 mutation was compromised when compared to wild-type strains. Similarly, flies harbouring Hsp83 mutations were more sensitive to environmental perturbations such as abnormal growth temperatures. Moreover, the number of affected individuals expressing abnormal phenotypes upon exposure to geldanamycin was much larger in Hsp83 mutant strains as compared to wild-type strains.
Because Hsp90 is highly conserved across species, it comes as no surprise that phenotypic buffering by Hsp90 was also observed in the plant Arabidopsis thaliana (mouse ear cress). Queitsch et al. 12 observed that pharmacological inhibition of Hsp90 by geldanamycin or radicicol, two structurally and chemically dissimilar drugs 8, 9, produced similar spectra of abnormal phenotypes within a given inbred line. Different lines displayed their own unique spectra of these phenotypes. Similar to Drosophila, plants exposed to higher than normal growth temperature (27 °C) were able to induce the same spectra of abnormal phenotypes in almost all of the inbred lines. Furthermore, it was found that the dark response, which is an adaptation for growing in the dark and an example of phenotypic plasticity, showed varying Hsp90 dependencies among the different inbred lines. One important observation is that the Hsp90-buffered phenotypes in Arabidopsis did not affect the plant's vitality and overall fitness; rather, some of these phenotypes, such as altered leaf morphology and purple pigment accumulation, might actually be beneficial depending on the environment. These findings in Arabidopsis further reinforce the idea that Hsp90 buffers phenotypic variation.
Interplay between genetics and epigenetics
Similar to studies from Lindquist's lab 11, it was recently reported that impairment of Hsp90 activity in flies either by genetic mutation or pharmacological inhibition resulted in inheritance of a specific eye phenotype in an isogenic fly line 13. However, in contrast, an epigenetic basis for capacitor function of Hsp90 was revealed by the finding that depletion of Hsp90 induced an altered chromatin state by affecting global histone H3 acetylation levels, which was suggested to be responsible for epigenetic inheritance of eye phenotype. The eye outgrowth phenotype was found to be reversed by histone deacetylase (HDAC) inhibitors like trichostatin A and sodium butyrate. Interestingly, mutations in some trithorax group genes like verthandi (vtd3) also induced the same eye phenotype in the same sensitized fly line. Importantly, after out crossing the verthandi mutation, the phenotypic selection of individuals with an ectopic eye outgrowth resulted in an increase in the number of affected progenies in successive generations. Upon exposure to geldanamycin, an identical phenotype was also observed in an isogenic fly line that did not harbour the vtd3 mutation. Phenotypic selection for this drug-induced phenotype produced an increase in its frequency in successive generations that were not treated with geldanamycin. Similarly, expression of the phenotype in the geldanamycin-treated sensitized fruit flies became independent of the drug as early as the F1 generation. Evidence that relates the above observations to chromatin acetylation states came from modified selection experiments. In these experiments, F6 fruit flies, selected for the geldanamycin-induced vtd3 phenotype, were fed trichostatin A and sodium butyrate. It was found that both HDAC inhibitors were able to suppress the vtd3 phenotype by two- to three-fold magnitudes. Similar phenotypic suppression was also observed in Hsp83 and original vtd3 mutants. These data provide strong evidence that heritable chromatin acetylation states are associated with Hsp90-induced phenotypic inheritance. However, the exact molecular mechanism and link between Hsp90 and trithorax or Hsp90 and epigentics remains elusive.
Inheritance of phenotypes induced by Hsp90 inhibition
Although there is clear evidence that phenotypes induced by Hsp90 inhibition are heritable and can be fixed by phenotypic selection 11, 12, 13, the exact mechanism of the inheritance process remains unclear. Several lines of evidence suggest that phenotypic variations induced by Hsp90 inhibition become independent of the chaperone after they are inherited across successive generations. In characterizing the inheritance of the deformed eye phenotype in Drosophila, Rutherford and Lindquist 11 mapped the hereditary determinants to at least two variants on the second chromosome, which are in turn dependent on the presence of others on the third chromosome. Hsp90 itself was found not to be the hereditary determinant for the phenotype, as revealed by the lack of Hsp83 mutation in the affected progenies of later generations. In Arabidopsis, Queitsch et al. 12 reported that the dependence of phenotypic plasticity on Hsp90 activity was not the result of an Hsp90 polymorphism. For example, the phenotypic variation in hypocotyl elongation, one of the characteristic traits of dark response, did not map to the cytoplasmic Hsp90 gene cluster.
If Hsp90 is not responsible for phenotypic inheritance, what might be the possible underlying mechanism of the process? Both the studies from the Lindquist laboratory 11, 12 proposed that the phenomenon is genetic in nature. They proposed that the inheritance and increase of the Hsp90-dependent phenotypes is the result of enrichment of multiple genetic determinants. For example, in Drosophila, the second and third chromosomes were required to sustain the deformed eye phenotype since heterozygosity in either one of the chromosomes with a control chromosome resulted in a marked reduction in phenotypic inheritance 11. In Arabidopsis, differences in phenotypes, generated by geldanamycin treatment, are less dramatic between the different recombinant inbred (RI) lines than between the parental lines from which the RI lines were generated 12. Furthermore, the exact spectra of phenotypes displayed upon inhibition of Hsp90 were dependent on the genetic background of the inbred line 12.
At the same time, there is evidence that supports the involvement of epigenetic elements in the inheritance of Hsp90-induced phenotypes. It has been proposed that specific DNA methylation and chromatin modifications, which might be functionally linked to Hsp90, are epigenetic mechanisms by which patterns of gene activity and silencing are inherited without any changes to the genomic sequence 4, 15, 21 (Figure 2). For example, CARM1 (coactivator-associated arginine methyltransferase 1), which participates in histone H3 arginine-17 methylation, associates with estrogen receptor (ER) and plays an important role in transcription regulation. ER is regulated by Hsp90 such that the presence of estrogen induces release of ER from Hsp90, and the ligand-bound ER then activates transcription of specific genes 22. As another example, SMYD3 (SET- and MYND-domain containing 3), the human histone H3 lysine-4 methyltransferase, requires Hsp90 for optimal activity. SMYD3 is in turn required for the activation of WNT genes (coined after the wingless gene in D. melanogaster), which are involved in a complex pathway governing changes in cell behaviour and morphogenesis in normal organ development and in tumours.

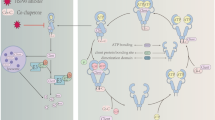
A proposed model for Hsp90-induced chromatin remodelling. Shown is a hypothetical mechanism for the S-phase replication of altered chromatin states in the epigenetic inheritance of Hsp90-induced phenotypes. The residue-specific acetylation (tagged tails) of core histone H3 at specific loci gives rise to localized non-restrictive euchromatin (A), which results in specific gene activation 18, 19. When Hsp90 is inhibited, for example, by geldanamycin (black hexagons) (B) or by mutations, the activity of the core constituents of the chromatin remodelling complex SWR-C (Rvb1/2, Vps71, Swr1, Aor1), which interact physically/genetically with the chaperone 3, could be compromised. This could result in SWR-C inactivity, which would in turn lead to the spread of the neighbouring restrictive heterochromatin into the euchromatin region (D). The modified core histones are also likely to be deacetylated by HDAC 13. On the other hand, if Hsp90 is active (C), then active SWR-C can mediate an exchange of the H2A-H2B core histone dimer for the H2A.Z-H2B dimer, which would prevent the spread of neighbouring heterochromatin into the euchromatin 28 (E). H2A.Z is a modified version of H2A. When DNA is replicated during the S phase (replicates are shown as red and blue lines), the core histones are dissociated from DNA. The histone chaperone complex CAF-1, which is specific for the S-phase core histone H3 and might function with ASF1, binds the H3-H4 dimer upon nucleosome dissociation (F, G) 29, 30. Upon the reconstruction of nucleosomes, CAF-1 is likely to deposit the H3-H4 dimer (H, I), in which H3 has maintained its specific modifications, back to its original binding site on one of the two DNA replicates 28. Through additional mechanisms yet to be identified, each of the daughter cells' chromatin would be reconstructed to resemble the parent cell's chromatin before replication 28. If Hsp90 was inhibited, the resulting chromatin state would be inherited by the two reconstructed chromatins (J), which in turn results in the penetrance of an altered phenotype. Subsequent selection for this phenotype over generations thus maintains these altered chromatin states. On the other hand, maintenance of the original phenotype occurs when the corresponding parental chromatin structure is intact through a putative Hsp90-dependent SWR-C chromatin remodelling activity and is inherited by the two reconstructed chromatins (K).
The inheritance and enrichment of Hsp90-induced phenotypes is indeed a very complex phenomenon. Whether a given phenotype would be inheritable after its induction depends on many different variables: the biochemistry involved in the production of the phenotype, the identities of the genes and gene products involved, the effect of specific genetic backgrounds, the subsequent requirement of the original stimuli that resulted in the phenotype, and others. But perhaps one of the most important determinants is the stability of the inheritance. As pointed out by Sangster et al. 23, the instability of an epigenetic event due to the reversibility of biochemical modifications on DNA and/or histones would present problems in the maintenance of any altered chromatin states from one generation to the next. Thus, a stable inheritance of the phenotype requires the interplay between genetics and epigenetics. While the rapid production of the phenotype upon Hsp90 inhibition could be accounted for by epigenetic events, phenotypic selection would lead to the accumulation of relevant genetic determinants that serve to canalize the phenotype.
The global Hsp90 interaction network: implications for phenotypic inheritance
A recent integrated proteomic and genomic study of the Hsp90 interaction network in S. cerevisiae (budding yeast) revealed that the chaperone interacts physically and/or genetically with 10% of the yeast proteome/genome 3. The study was based on a combination of four complementary experimental strategies. First, systematic genome-wide screens were carried out for protein-protein interactions mediated by Hsp90 using a two-hybrid technique based on ordered strain arrays 24. Second, protein mass spectrometry to identify proteins that copurify with Hsp90 in tandem affinity purification (TAP) procedures were carried out. In this procedure, Hsp90 or yeast ORFs were chromosomally TAP-tagged 25 and the tag was used to pull down Hsp90-containing complexes. Third, synthetic lethal interactions between a temperature-sensitive mutant allele of Hsp90 and members of a panel of ∼4700 single yeast gene deletion strains were identified using the synthetic genetic array technology 26. Lastly, the deletion mutant strains were screened for differential hypersensitivity to the Hsp90 inhibitor geldanamycin in liquid culture using a microarray-based readout 27.
In this study, 198 putative physical interactors and 451 genetic interactors of the chaperone were uncovered. These putative interactors included established Hsp90 cofactors (e.g. Cns1, Cpr6, Cpr7, and Ppt1) and cochaperones (e.g. Ssa1 and Sti1). Several transcription factors (e.g. Adr1, Aft2, Cha4, Gln3, etc.) and protein kinases (e.g. Cdc15, Cka2, Ctk1, Snf1, etc.) were also identified as interacting with the chaperone, suggesting roles for Hsp90 in multiple signalling pathways. Importantly, several components of the INO80 and SWR-C chromatin remodelling complexes were also shown to interact with Hsp90. These include the DNA helicases Rvb1 and Rvb2 (components of both INO80 and SWR-C complexes), Aor1 (component of the SWR-C complex), Swr1 (component of the SWR-C complex, ATPase of the SWI/SNF family), Vps71 (component of the SWR-C complex), Ies1 (subunit of the INO80 chromatin remodelling complex), as well as the H2A histone variant, Htz1/H2A.Z 3. Furthermore, novel cofactors of Hsp90, termed Tah1 and Pih1, that link Hsp90 to the Rvb1 and Rvb2 DNA helicases were identified in this study. Hence, this detailed analysis of global Hsp90 interactors in yeast strongly suggests a functional linkage between Hsp90 and the chromatin remodelling machinery. It is reasonable to speculate that the general features of such a network will be conserved in higher organisms.
Previous studies on SWR-C revealed its specificity in exchanging the core histone dimer H2A-H2B for H2A.Z-H2B (H2A.Z being a variant of H2A) in the nucleosomes of euchromatin (Figure 2C), thereby preventing the spread of neighbouring heterochromatin (Figure 2E) 28. When entire nucleosomes in the parent chromatin are disassembled (Figure 2F and 2G), CAF-1 (chromatin assembly factor 1), which is part of the replication-dependent histone chaperone complex specific for the S-phase H3 histone 29, 30, carries a single H3-H4 dimer 29 to be delivered back to one of the two DNA replicates during re-assembly (Figure 2H and 2I) 28. CAF-1 might function with another H3-H4 chaperone ASF1 (anti-silencing function 1) 30. The redistribution of parental H3 histones, which carry with them specific covalent modifications, serves to transfer part of the parent's epigenome into the chromatin of each daughter cell. Other mechanisms, yet to be identified, might subsequently reconstruct the missing half of the transferred epigenome, using the parental half as the template 28. Thus, as suggested by the proteomics work discussed above, the activity/inactivity of Hsp90 might affect SWR-C-dependent chromatin remodelling and, hence, could affect the chromatin state that is inherited by the chromatin in each daughter cell (Figure 2A-2E). This could be extrapolated to explain the effect of HDAC inhibitors in suppressing phenotypic inheritance as observed by Sollars et al. 13, since inhibiting histone deacetylation prevents the spread of heterochromatin, which allows the maintenance of parental chromatin states and their inheritance (Figure 2B). This scenario, which is similar to a hypothetical mechanism proposed by Sangster et al. 23, allows for an explanation of an epigenetic effect of Hsp90 function. Other scenarios involving different molecular mechanisms have also been proposed 23.
Concluding remarks
Hsp90 functions as a capacitor for phenotypes that are normally suppressed. Their expression via Hsp90 inhibition, genetically, pharmacologically, or by mild environmental perturbations, reveals a molecular mechanism for changing gene expression patterns in response to the environment. Depending on the organism's lifestyle, the expression of abnormal phenotypes can be advantageous in adapting to new environmental conditions.
The Hsp90-induced phenotypes in Drosophila and Arabidopsis discussed here display phenotypic inheritance and enrichment when affected progenies are inbred. The process is likely the result of the interplay between genetics and epigenetics. Considering the stability of phenotypic inheritance associated with the accumulation of genetic determinants, and that different genetic backgrounds harboured different spectra of these phenotypes 11, 12, genetics is definitely one of the major determinants. At the same time, the fact that gene mutations are not required to maintain the abnormal phenotypes 13, the interaction of Hsp90 with chromatin remodelling complexes 3, and the effect of chromatin acetylation states on the stability of phenotypic inheritance 13, all indicate a significant role for epigenetics. Owing to the large number of factors required, the different pathways involved to produce particular phenotypes, and the complex interactions between genetics and epigenetics, it is likely that the inheritance of different Hsp90-induced phenotypes might employ different genetic-epigenetic mechanisms for their expression and subsequent maintenance. Further investigation is required for a better understanding of the complex relationship between Hsp90 activity and the canalization of previously buffered phenotypes.
References
Terasawa K, Minami M, Minami Y . Constantly updated knowledge of Hsp90. J Biochem 2005; 137:443–447.
Pratt WB, Toft DO . Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 2003; 228:111–133.
Zhao R, Davey M, Hsu Y, et al. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the Hsp90 chaperone. Cell 2005; 120:715–727.
Rutherford SL . Between genotype and phenotype: protein chaperone and evolability. Nature 2003; 4:263–274.
Jakob U, Meyer I, Bugl H, et al. Structural organization of procaryotic and eucaryotic Hsp90. Influence of divalent cations on structure and function. J Biol Chem 1995; 270:14412–14419.
Buchner J . Hsp90 & Co. – a holding for folding. Trends Biochem Sci 1999; 24:136–141.
Zhao R, Houry WA . Hsp90: a chaperone for protein folding and gene regulation. Biochem Cell Biol 2005; 83:703–710.
Workman P . Combinatorial attack on multistep oncogenesis by inhibiting the Hsp90 molecular chaperone. Cancer Lett 2004; 206:149–157.
Zhang H, Burrows F . Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med 2004; 82:488–499.
Pearl LH, Prodromou C . Structure, function, and mechanism of the Hsp90 molecular chaperone. Adv Protein Chem 2002; 59:157–186.
Rutherford SL, Lindquist S . Hsp90 as a capacitor for morphological evolution. Nature 1998; 396:336–342.
Queitsch C, Sangster TA, Lindquist S . Hsp90 as a capacitor of phenotypic variation. Nature 2002; 417:618–624.
Sollars V, Lu X, Xiao L, et al. Evidence for an epigenetic mechanism by which Hsp90 acts as a capacitor of morphological evolution. Nat Genet 2003; 33:70–74.
Waddington CH . Canalization of development and the inheritance of acquired characters. Nature 1942; 150:563–565.
Ruden DM, Garfinkel MD, Sollars VE, Lu X . Waddington's widget: Hsp90 and the inheritance of acquired characters. Semin Cell Dev Biol 2003; 14:301–310.
Smith CM, Haimberger ZW, Johnson CO, et al. Heritable chromatin structure: mapping “memory” in histones H3 and H4. Proc Natl Acad Sci USA 2002; 99:16454–16461.
Tariq M, Paszkowski J . DNA and histone methylation in plants. Trends Genet 2004; 20:244–251.
Hake SB, Xiao A, Allis CD . Linking the epigenetic 'language' of covalent histone modifications to cancer. Br J Cancer 2004; 90:761–769.
Dejardin J, Cavalli G . Epigenetic inheritance of chromatin states mediated by polycomb and trithroax group proteins in Drosophila. Prog Mol Subcell Biol 2005; 38:31–63.
Rao M, Sockanathan S . Molecular mechanisms of RNAi: implications for development and disease. Birth Defects Res 2005; 75:28–42.
Ruden DM, Xiao L, Garfinkel MD, Lu X . Hsp90 and environmental impacts on epigenetic states: a model for the trans-generational effects of diethylstibesterol on uterine development and cancer. Hum Mol Genet 2005; 14:R149–R155.
Beato M, Klug J . Steroid hormone receptors: an update. Hum Reprod Update 2000; 6:225–236.
Sangster TA, Queitsch C, Lindquist S . Hsp90 and chromatin: where is the link? Cell Cycle 2003; 2:166–168.
Uetz P, Giot L, Cagney G, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 2000; 403:623–627.
Puig O, Caspary F, Rigaut G, et al. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 2001; 24:218–229.
Tong AH, Evangelista M, Parsons AB, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 2001; 294:2364–2368.
Giaever G, Chu AM, Ni L, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 2002; 418:387–391.
Korber P, Horz W . SWRred not shaken: mixing the histones. Cell 2004; 117:5–7.
Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y . Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 2004; 116:51–61.
Zabaronick SR, Tyler JK . The histone chaperone anti-silencing function 1 is a global regulator of transcription independent of passage through S phase. Mol Cell Biol 2005; 25:652–660.
Acknowledgements
We thank Jamie Snider (Department of Biochemistry, University of Toronto, Toronto, Canada) and Dr Muhammad Tariq (Centre for Molecular Biology, University of Heidelberg, Heidelberg, Germany) for careful reading of the manuscript. Work in the corresponding author's laboratory is supported by research grants from the Canadian Institutes of Health Research, Natural Sciences and Engineering Research Council of Canada, and the National Cancer Institute of Canada.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wong, K., Houry, W. Hsp90 at the crossroads of genetics and epigenetics. Cell Res 16, 742–749 (2006). https://doi.org/10.1038/sj.cr.7310090
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7310090
