
Abstract
We investigated the mechanisms by which TAp73β and dominant-negative p73 (ΔNp73) regulate apoptosis. TAp73β transactivated the CD95 gene via the p53-binding site in the first intron. In addition, TAp73β induced expression of proapoptotic Bcl-2 family members and led to apoptosis via the mitochondrial pathway. Endogenous TAp73 was upregulated in response to DNA damage by chemotherapeutic drugs. On the contrary, ΔNp73 conferred resistance to chemotherapy. Inhibition of CD95 gene transactivation was one mechanism by which ΔNp73 functionally inactivated the tumor suppressor action of p53 and TAp73β. Concomitantly, ΔNp73 inhibited apoptosis emanating from mitochondria. Thus, ΔNp73 expression in tumors selects against both the death receptor and the mitochondrial apoptosis activity of TAp73β. The importance of these data is evidenced by our finding that upregulation of ΔNp73 in hepatocellular carcinoma patients correlates with reduced survival. Our data indicate that ΔNp73 is an important gene in hepatocarcinogenesis and a relevant prognostic factor.
Similar content being viewed by others

Introduction
The discovery of a ΔNp73 (dominant-negative p73)-based p53–p73 interference network suggests that the p53 status of a tumor should no longer be regarded as the sole predictor of clinical outcome and therapeutic responsiveness.
TP53 is the prototype tumor suppressor gene in human cancer due to its proapoptotic and antiproliferative function in response to oncogenic stress. The p53 pathway is inactivated in the majority of human malignancies.1 TP73, despite significant homology to p53, is not a classic Knudson-type tumor suppressor gene.2, 3, 4 TP73-deficient mice lack a tumor phenotype 5 and inactivating mutations in patients suffering from cancer are extremely rare.3
The expression of the TP73 gene is complicated by the presence of several splicing isoforms at the C-terminus (p73α–ζ) 2, 3, 6, 7 and of two distinct promoters, driving the expression of p53-like proteins containing the transactivation domain (TAp73), and inhibitory proteins lacking TA, called ΔTAp73 (the collective name for four different p73 TA-deficient forms, mainly ΔNp73). ΔNp73 acts as a potent transdominant inhibitor of TAp73 and wild-type p53.3, 8 Thus, the TP73 locus encodes both a tumor suppressor (TAp73) and a putative oncogene (ΔNp73).
The finding that a significant percentage of tumors specifically select for dominant-negative p73 isoforms strongly argues for their oncogenic role during tumorigenesis.8, 9 ΔNp73 cooperates with oncogenic Ras in transforming primary mouse embryo fibroblasts (MEFs) in vitro and in inducing MEF-derived fibrosarcomas in nude mice in vivo.10 Furthermore, mice heterozygous for mutations in both p53 and p63 or p63 and p73 displayed higher tumor burden and metastasis compared to p53+/− mice.11
Endogenous TAp73 is activated in response to a variety of chemotherapeutic drugs and gamma-irradiation in a pathway that depends on the non-receptor tyrosine kinase c-Abl.12, 13 We have recently described a role for TAp63α in the induction of apoptosis and chemosensitivity.14
The involvement of p63 and p73 in p53-mediated apoptosis is controversial. Whereas on the one hand the presence of p63 and p73 has been shown to be essential for p53 to induce apoptosis in fibroblasts following DNA damage,15 results of a recent study indicate on the other hand, that at least in thymocytes, p53-dependent apoptosis occurs independently of p63 and p73.16
Given the possible role of TAp73 and ΔNp73 isoforms in cancer, it is of great interest to determine how they regulate apoptosis, as the critical process which links tumor development, treatment sensitivity and clinical outcome.
We have analyzed the mechanisms of TAp73/ΔNp73-regulated apoptosis and their relevance for chemosensitivity and prognosis in hepatocellular carcinoma (HCC).
Our data suggest possible signaling pathways through which the balance between TAp73 and ΔNp73 regulates the apoptosis response of cancer cells, thereby playing a decisive role in the choice between elimination of cancer-prone cells versus hepatocarcinogenesis as well as between treatment sensitivity versus drug resistance. Of clinical importance, we show that ΔNp73 is a predictor of adverse outcome and a new prognostic factor in HCC.
Results
TAp73β-induced apoptosis involves activation of caspases
Adenoviral transfer of the TAp73β gene into Hep3B cells induced apoptosis in a dose-dependent manner (Figure 1a). TAp73β-mediated apoptosis was strongly inhibited by the caspase inhibitors ZVAD-FMK, DEVD-FMK, Z-IETD-FMK and Z-LEHD-FMK (Figure 1b and c). Involvement of caspases was confirmed by fluorometric determination of the increased enzymatic activity of the caspase-3, -8 and -9 class of proteases (Figure 1d).

Apoptosis induced by TAp73β involves caspase activation. (a) TAp73β induces dose-dependent apoptosis of hepatoma cells (Hep3B) within 72 h. Data obtained in two separate experiments were averaged, values are mean±S.D., n=6. (b) TAp73β-dependent apoptosis was blocked by the broad-spectrum caspase inhibitor zVAD (50 μM), indicating that TAp73β-dependent cell death involves caspase-activation. A FACScan® analysis of propidium iodide-stained nuclei 20 of Hep3B cells (72 h) is shown. Data obtained in five separate experiments were averaged, values are mean±S.D., n=15. *P<0.0001 compared to TAp73β, 10 MOI, Wilcoxon test. (c) The caspase inhibitors DEVD-FMK (C3Inh), Z-IETD-FMK (C8Inh) and Z-LEHD-FMK (C9Inh) significantly reduced TAp73β-dependent apoptosis. Three independent experiments were performed, and a representative result is shown. Mean±S.D., n=3. *P<0.05 compared to TAp73β, 10 MOI, Wilcoxon test. (d) Determination of the increased enzymatic activity of the caspase-3, -8 and -9 class of proteases was performed by fluorometric assays 48 h following rAd-TAp73β transfer (10 MOI) into Hep3B cells. Fold activation represents the DEVDase- (consistent with caspase 3-), IETDase- (consistent with caspase 8-) and LEHDase- (consistent with caspase 9-) activity of Hep3B cells transduced by rAd-TAp73β compared to cells transduced by rAd-GFP. Three independent experiments were performed, and a representative result is shown. Mean±S.D., n=3. *P<0.05, Wilcoxon test
Microarray analysis of TAp73β-mediated apoptosis
Since TAp73β was able to induce classical apoptosis (Figure 1) and since TAp73 is a transcription factor, we decided to investigate the underlying molecular mechanisms by performing a gene array analysis. Following adenoviral TAp73β transfer the genes encoding for the death receptors CD95, TNF-R1, TRAIL-R1 and -R2 were found to be upregulated (Table 1). Additional evidence for the involvement of receptor-mediated signaling in TAp73β-induced apoptosis was provided by the fact that the pro-caspase-8-binding adapter protein Fas-associated death domain (FADD) was found to be upregulated.
Activation of caspases is clearly involved in mediating TAp73β-induced downstream apoptosis signaling. Caspase-1, -2, -3, -4, -6, -8, -9 and -10 were found to be upregulated in response to TAp73β (Table 1).
Furthermore, we identified the genes encoding the proapoptotic Bcl-2 family members BAD and BIK and the genes encoding BNIP3, HRK and RAD9 as targets for transcriptional upregulation in cells overexpressing TAp73β (Table 1). Thus, microarray analyses provide evidence that TAp73β stimulates both, genes that regulate the extrinsic apoptosis pathway initiated by ligation of death receptors and genes that regulate the intrinsic/mitochondrial apoptosis pathway.
TAp73β triggers the extrinsic apoptosis pathway via activation of death receptors
We have previously shown that the CD95 gene is a transcriptional target of wt p53, whose expression is induced through binding of wt p53 to a regulatory region residing within its first intron.17, 18 Based on these observations, we next investigated if TAp73β, like p53, induces CD95-dependent apoptosis by upregulating the expression of the CD95 receptor itself. Semiquantitative RT–PCR showed an increase in the amount of CD95 mRNA in Hep3B cells following adenoviral TAp73β transfer (Figure 2a).

TAp73β induces upregulation of the CD95 receptor and sensitizes towards CD95-mediated apoptosis. (a) TAp73β, like p53, induces upregulation of CD95 mRNA. Semiquantitative PCR-analysis of CD95 mRNA expression following adenoviral transfer (24 h) of either rAd-GFP, rAd-p53 or rAd-TAp73β (10 MOI each) into Hep3B cells (p53−/−). (b) FACScan® analysis of CD95 receptor expression in Hep3B cells following transduction by either rAd-GFP, rAd-p53 or rAd-TAp73β (5 MOI each). Transfer of rAd-TAp73β (72 h) as well as restitution of a wt p53 status restores the ability of the p53−/− cell line Hep3B to increase the CD95 receptor. Presented are representative diagrams of FL-2 (CD95) for rAd-p53 and rAd-TAp73β. (c+d), TAp73β induces upregulation of the CD95 receptor in Hep3B (dose dependent) and in Saos2 cells (50 MOI). FACScan® analyses were performed in triplicates, six independent experiments were performed, a representative result is shown (mean±S.D., n=3). *P<0.05 compared to GFP, Wilcoxon test. (e) TAp73β sensitizes Hep3B cells towards CD95-mediated apoptosis. Only Hep3B cells transduced by rAd-TAp73β exhibited a significantly increased responsiveness towards induction of apoptosis by anti-APO-1. Data are expressed as % increase in specific apoptosis due to addition of anti-APO-1. FACScan® analysis following Nicoletti staining. A representative result of three independent experiments is shown, presented is mean±S.D., n=4. *P<0.0001, MANOVA, between-subject effect
Importantly, FACS analysis revealed that overexpression of TAp73β led to an increase in the amount of CD95 displayed on the cell surface in Hep3B (Figure 2b and c). This increase in CD95 expression was also observed in a second cellular model, in Saos2 cells (Figure 2d). Saos2 cells are p53 negative and show no detectable levels of p63 and p73 at either mRNA or protein level.
The increased number of death receptors on the cell surface is likely to increase sensitivity of cancer cells towards apoptosis. We tested this hypothesis by treating Hep3B cells with agonistic anti-APO-1 antibody. Figure 2e shows that the antibody is in fact able to trigger cell death in TAp73β-overexpressing Hep3B cells, but not in cells infected with a control green fluorescent protein (GFP)-adenovirus. This indicates that TAp73β induces the expression of a functional CD95 death receptor on the cell surface.
TAp73 is a transcriptional activator of the CD95 gene
Upregulation of CD95 mRNA and protein by TAp73β might be due to direct transcriptional activation of the CD95 gene. Since most of the TAp73β-driven promoters seem to share p53-responsive elements, we investigated the possibility that TAp73β transactivates the CD95 gene via binding to the intronic p53-binding site 17, 18 (Figure 3a). This was carried out by transient transfection assays, employing plasmid p1142CD95-luc.

TAp73 transactivates the CD95 gene via the intronic p53-binding site. Map of the human CD95 gene. Exons 1–9 are numbered according to Wada et al.38 and represented by black boxes. The striped box indicates the intronic p53-binding site (p53-IBS) in the first intron of the CD95 gene.18 (a) Wild-type (wt) sequence of the CD95 intronic p53-binding site.18 This sequence (top line) is compared with the consensus p53-binding site (bottom line) according to El-Deiry et al.39 Missing vertical bars indicate deviations in the wt sequence from the consensus. R=purine, Y=pyrimidine, W=A or T. (b) In plasmid mt p1142CD95-luc, essential nucleotides for the binding of wt p53 protein have been mutated in the intronic p53-responsive element. (c) The TAp73 isoforms α, β, γ and Δ transactivated the CD95 gene via binding to the p53-binding site in its first intron. Mutation of this binding site (mt p1142CD95-luc) completely abrogated binding and transactivation by TAp73-isoforms. Cells were transfected with 1 μg of plasmid p1142CD95-luc together with 100 ng of either a wt p53-plasmid, or a TAp73α, β, γ or Δ-plasmid, or an equivalent amount of empty vector. Presented is the fold p53- or TAp73-dependent activation of the p1142CD95-luc reporter plasmid, calculated relative to the value obtained with the same reporter in the absence of p53 or the TAp73-isoforms. Assays were performed in triplicates in five independent experiments. One representative experiment is shown (mean±S.D., n=3)
Figure 3c shows that cotransfection of the TAp73 isoforms α, β, γ and δ significantly increased p1142CD95-luc activity. This supports the hypothesis that TAp73 can induce CD95 gene expression. The TAp73-dependent transactivation of the CD95 gene strictly depends on the intronic p53-binding site, as it is totally abrogated when using a CD95 luciferase construct (Figure 3c) with a mutated intronic p53-binding site (Figure 3b). This argues strongly in favor of the conclusion that the CD95 gene is a direct transcriptional target of TAp73.
ΔNp73 is an efficient inhibitor of the transcriptional activity of TAp73 and inhibits both transactivation of the CD95 gene and apoptosis
As shown in Figure 4a, ΔNp73 is a strong inhibitor of TAp73-mediated CD95 gene transactivation. ΔNp73 is not able to transactivate the CD95 gene by itself. In addition, Figure 4b shows the capacity of the Δ84p73β isoform, which is functionally equivalent to the Δ2p73, Δ3p73 and Δ23p73 isoforms,2 to abrogate TAp73-mediated transactivation of the CD95 gene. Furthermore, Δ84p73β blocks p53-mediated transactivation of the CD95 promoter (Figure 4c). Thus, interference with the CD95 gene transactivation-capacity of TAp73 and that of p53 constitutes a molecular mechanism by which N terminus-deleted p73-isoforms exert their antiapoptotic action. This implies that the presence of the ΔNp73 (or Δ84p73β) protein constitutes a dominant-negative effect controlling apoptosis in hepatoma cells. To test this hypothesis, we evaluated the regulation of apoptosis by ΔNp73. Hep3B cells undergo TAp73β-dependent cell death as assessed by FACS analysis. This apoptotic activity is completely abolished by the coexpression of Δ84p73β (Figure 4d).

Dominant-negative (ΔN) p73β and delta (Δ) 84p73β inhibit TAp73- and p53-mediated transactivation of the CD95 gene and counteract apoptosis induced by TAp73β. (a–c), analysis of TAp73-dependent luciferase activity. Hep3B cells were transfected with 1 μg of plasmid p1142CD95-luc together with 100 ng of either a TAp73α, β, γ or Δ-plasmid, a p53-plasmid, or an equivalent amount of empty vector alone or together with the respective ΔNp73- or Δ84-isoform. Presented is the fold TAp73- or p53-dependent activation of each reporter plasmid, calculated relative to the value obtained with the same reporter in the absence of the respective TAp73-isoform or of p53. ΔNp73 (a) and Δ84p73β (a) were not able to transactivate the CD95 gene. Moreover, they act as dominant-negative being able to repress the ability of TAp73 to drive the CD95 gene. In addition to their ability to inhibit TAp73-mediated transactivation of the CD95 gene, the ΔN- and Δ84 isoforms of p73 inhibit wt p53-induced transactivation of the CD95 gene (c). Assays were performed in triplicates in five independent experiments. One representative experiment is shown (mean±S.D., n=3). (d) ΔN- and Δ84 isoforms of p73 inhibit specific apoptosis induced by TAp73β. Hep3B cells were transduced for 72 h either with rAd TAp73β (10 MOI) or rAd Δ84p73β (10 MOI). Data obtained in three separate experiments were averaged, presented is mean±S.D., n=9
TAp73β engages mitochondrial apoptosis pathways
In order to further characterize the molecular mechanisms of TAp73-mediated apoptosis, we investigated the influence of TAp73β on mitochondrial apoptosis signaling. FACS analysis following JC-1 staining revealed an alteration of the mitochondrial membrane potential following adenoviral TAp73β transfer in Hep3B cells (Figure 5a).

TAp73β engages mitochondrial apoptosis pathways. (a) FACScan® analysis of Hep3B cells following JC-1 staining showed alteration of the mitochondrial membrane potential. After 72 h, there was a significant increase in J-aggregates due to TAp73β (between-subject effect), which was significantly dose-dependent (within-subject effect). Data obtained in three separate experiments were averaged, presented is mean±S.D., n=9. *P<0.001, MANOVA, between-subject effect; GFP versus TAp73β, and P<0.05, MANOVA, within-subject effect; for 48 h: P=0.06, MANOVA, between-subject effect. (b) Overview of the Bax-luciferase gene construct used in (c) containing a luciferase construct of the Bax promoter with its four p53 promoter binding-sites (white boxes).32 C, TAp73β transactivates the Bax gene. Hep3B cells were transfected with 1 μg of the reporter plasmid presented in (b) together with 10 MOI rAd-TAp73β. The fold TAp73β-dependent activation of the Bax-Pr/luc reporter plasmid, calculated relative to the value obtained with the same reporter in the absence of TAp73β is shown. TAp73β induced a significant time-dependent increase of the transactivation of the Bax gene (ANOVA). A representative result of five independent experiments is shown. Presented is mean±S.D., n=3. *P<0.001, ANOVA. (d) Immunoblot of endogenous Bax expression following rAd-TAp73β transfer. (e) ΔNp73 and Δ84p73β act as dominant-negative being able to repress the ability of p53 and TAp73β to transactivate the Bax gene. Presented is the fold TAp73β- or p53-dependent (10 MOI) activation of the Bax-Pr/luc reporter plasmid (1 μg), calculated relative to the value obtained with the same reporter in the absence of TAp73β or p53. Assays were performed in triplicates in three independent experiments. One representative experiment is shown (mean±S.D., n=3)
To investigate the possible involvement of Bax in p73-induced apoptosis, we performed a transient transfection assay using a reporter plasmid (Bax-Pr/luc) containing the full-length Bax promoter placed upstream of a luciferase cDNA (Figure 5b). Figure 5c shows that cotransfection of TAp73β significantly increased Bax-Pr/luc activity. Western blot analysis confirmed induction of endogenous Bax protein following rAd-TAp73β transfer (Figure 5d). In addition, as shown by microarray analysis, BAD, BIK, BNIP3, HRK and RAD9 were induced (Table 1). Thus, TAp73β contributes to apoptosis by inducing the expression of several proapoptotic genes involved in mitochondrial signaling.
Blocking TAp73β by rAd-Δ84p73β significantly decreased Bax-Pr/luc activity. Furthermore, Δ84p73β did also block p53-mediated transactivation of the Bax promoter (Figure 5e). Thus, in addition to interference with the death receptor-apoptosis activity of TAp73β, N terminus-deleted p73-isoforms exert their antiapoptotic action also on mitochondrial apoptosis signaling pathways.
TAp73β sensitizes cancer cells towards chemotherapy
p53 is frequently mutated in human cancer. This has been implicated in resistance towards chemotherapy. We therefore next investigated if TAp73β restores chemosensitivity of cancer cells.
First, we found that chemotherapeutic agents (bleomycin, mitoxantrone and doxorubicin) induce endogenous TAp73 expression in Hep3B cells (Figure 7a). This is in accordance with data obtained in other cell lines.19 We then tested if there is a synergistic effect of TAp73β and chemotherapeutic agents on the induction of cell death. We infected Hep3B (p53-/-), HepG2 (wt p53) AGS (wt p53) and Saos2 (p53-/-) cells with an adenoviral TAp73β (5 and 10 MOI). Then we exposed the cells to different doses of bleomycin. Figure 6a shows that rAd-TAp73β enhances cell killing by bleomycin. All cancer cell lines tested displayed an increased responsiveness towards cell death after adenoviral TAp73β transfer, independent of their p53 status. A balanced two-way analysis of variance (ANOVA) revealed a highly significant synergistic interaction (P<0.0001) between TAp73β and bleomycin in induction of cell death.

Blocking TAp73 function with a dominant-negative isoform, Δ84p73β, confers chemotherapy resistance. (a, b) Western blot analysis showed upregulation of endogenous TAp73 and endogenous ΔNp73 following treatment of Hep3B cells with bleomycin, mitoxantrone or doxorubicin in clinically relevant concentrations. (c) Blocking TAp73β function with Δ84p73β inhibits chemotherapy-induced apoptosis. Hep3B cells transduced with 10 MOI rAd-Δ84p73β were treated with bleomycin for 72 h. FACScan® analysis of propidium iodide-stained nuclei. One representative out of three experiments performed is shown, mean±S.D., n=3. * P<0.05, Wilcoxon test, compared to the (next) left column. (d) Hep3B cells transduced with 10 MOI rAd-Δ84p73β or 10 MOI rAd-GFP were treated with bleomycin and determination of the increased enzymatic activity of the caspase-3, -8 and -9 class of proteases was performed by fluorometric assays 48 h later. Fold activation represents the DEVDase- (consistent with caspase 3-), IETDase- (consistent with caspase 8-) and LEHDase- (consistent with caspase 9-) activity compared to control cells, transduced with rAd-GFP, only. One representative out of three experiments performed is shown, mean±S.D., n=3. *P<0.05, Wilcoxon test. (e) Blocking endogenous TAp73 with Δ84p73β inhibits chemotherapy-induced apoptosis at the mitochondrial level. FACS analysis following JC-1 staining of Hep3B cells treated for 72 h revealed a significant change of the mitochondrial membrane potential following rAd-TAp73β transfer and/or bleomycin treatment, which was inhibited by Δ84p73β. One representative out of three experiments performed is shown, mean±S.D., n=3. * P<0.05, Wilcoxon test, compared to the (next) left column

TAp73β sensitizes cancer cells towards chemotherapy. (a) TAp73β and chemotherapeutic agents synergize in the induction of cell death. All cell lines tested, independent of their p53 status, displayed an increased responsiveness towards cell death after adenoviral TAp73β transfer. A balanced two-way ANOVA revealed a synergistic interaction between TAp73β- and bleomycin-induced cell death for all cell lines tested (P<0.0001). Data are expressed as the fraction of living cells, not treated with rAd-GFP or rAd-p73β or bleomycin (mean±S.D., n=6 wells). * By MANOVA, between-subject effect P<0.0001 compared to GFP 10 MOI. (b) TAp73β significantly increased drug sensitivity of Hep3B and Saos2 cells. Thus, the increase in cell death shown in (a) is due to an increase in specific apoptosis. Data obtained in two separate experiments were averaged. * By MANOVA, between-subject effect P<0.0001, mean±S.D., n=6. (c) DAPI staining shows the synergistic effect of rAd-TAp73β (10 MOI) and chemotherapeutic agents (bleomycin 3 μg/ml) on the induction of cell death in Hep3B cells. (d) Enhanced chemosensitivity is partially due to a cooperative activation of the CD95 receptor/ligand system by TAp73 and anticancer drugs. Cytostatic treatment of Hep3B cells with bleomycin increased CD95 ligand mRNA expression. In contrast, rAd-TAp73β (10 MOI) transfer alone did not upregulate CD95 ligand mRNA. Moreover, Hep3B cells transduced with TAp73β responded with enhanced expression of CD95 receptor mRNA. (e) Apoptosis following adenoviral TAp73β-transfer and bleomycin treatment was partially blocked by an antagonistic anti-CD95L Ab (NOK-1) in Hep3B cells. Data obtained in two separate experiments were averaged, presented is mean±S.D., n=6. *P<0.05, Wilcoxon test, compared to the (next) left column. (f) The cooperative action of TAp73β-transfer and chemotherapy leads to enhancement of Bax gene transactivation. Combined treatment with 3 μg/ml bleomycin led to a further increase of the transactivation of the Bax promoter. Treatment conditions are as detailed in Figure 5c. One representative out of three experiments performed is shown, mean±S.D., n=3. *P<0.001, MANOVA, within-subject effect (time-effect), **P<0.05, MANOVA, between-subject effect (bleomycin-effect). (g) Western blot analysis confirms the cooperative induction of endogenous Bax protein by rAd-TAp73β and bleomycin in Hep3B cells
The observed synergy of TAp73β and chemotherapeutic drugs was due to a cooperative effect on induction of specific apoptosis (Figure 6b). FACScan® analysis20 revealed that TAp73β significantly increased drug-induced apoptosis of Hep3B and Saos2 cells (P<0.0001). DAPI staining also illustrated the cooperative effect on the induction of cell death (Figure 6c).
We further analyzed the mechanisms contributing to the enhanced chemosensitivity conferred by TAp73β. The synergy of TAp73β and bleomycin turned out to be due to a cooperative action at different levels of apoptosis signaling, namely at the death receptor and the mitochondrial level, enhancing apoptosis mediated by the CD95 system (Figure 6d and e) as well as apoptosis mediated by mitochondrial activation (Figures 6f and g and 7e). CD95 ligand mRNA was induced upon bleomycin treatment (Figure 6d and reported previously,17, 18, 21) but not by TAp73β, whereas TAp73β stimulated upregulation of CD95 receptor mRNA. Apoptosis following adenoviral TAp73β-transfer and bleomycin treatment was partially blocked by an antagonistic anti-CD95L Ab (NOK-1), indicating a role of a CD95L/CD95 interaction in the death process (Figure 6e). Thus, the observed synergy of TAp73β and chemotherapy may in part result from enhanced CD95 expression (due to TAp73β) triggered by upregulated CD95 ligand (due to chemotherapy). In addition, we show a cooperative action of TAp73β and bleomycin on mitochondrial apoptosis. FACScan® analysis showed that the change of the mitochondrial membrane potential caused by TAp73β was significantly increased by addition of bleomycin (Figure 7e). The cooperative action of TAp73β and bleomycin on mitochondrial apoptosis is further evidenced by the fact that combined treatment led to a significant increase of the transactivation of the Bax gene (Figure 6f). This was validated on protein level; combined treatment led to a further increase of endogenous Bax protein (Figure 6g).
ΔNp73 confers drug resistance to p53-deficient tumor cells
Since TAp73 and ΔNp73 show opposite effects on apoptosis induction, we investigated if ΔNp73 interferes with chemotherapy-induced apoptosis.
Both, endogenous TAp73 and ΔNp73 are upregulated in response to DNA damage by bleomycin, mitoxantrone and doxorubicin in Hep3B cells (Figure 7a and b). Δ84p73β counteracts specific apoptosis induced by TAp73β and bleomycin (Figure 7c). Interference of ΔNp73 with apoptosis sensitivity can take place at several levels of apoptosis signaling. As shown in Figure 4, ΔNp73 can directly interfere with the transcriptional activation function of p53 and TAp73 and consequently inhibit transactivation of the CD95 gene by p53 or TAp73. Furthermore, resistance towards chemotherapy by ΔNp73 can be imposed at the caspase-, and the mitochondrial level of apoptosis signaling. Figure 7d shows that rAd-Δ84p73β inhibits bleomycin-induced activation of caspase-3, -8, and -9 class of proteases. Adenoviral transfer of Δ84p73β strongly reduces mitochondrial activation induced by bleomycin and TAp73β as measured by Bax reporter assays and FACScan® analysis following JC-1 staining (Figure 5e and 7e). Repression of TAp73β-mediated transactivation of the Bax promoter was one mechanism by which ΔNp73 acts as dominant-negative on mitochondrial apoptosis signaling (Figure 5e). Even though we cannot exclude a direct activity of ΔNp73 on the mitochondria, it is more likely that the effect of ΔNp73 on mitochondria is also due to its interference with TAp73-mediated transactivation, albeit other genes are involved (Table 1). This supports the hypothesis that the ratio of the two isoforms TAp73/ΔNp73 is an important determinant of clinical response to chemotherapy. This prompted us to further analyze the clinical significance of ΔNp73 expression in patients with HCC.
Clinical impact of ΔNp73 overexpression: ΔNp73 is a prognostic marker in patients with HCC
Western blot analysis revealed that TAp73 and ΔNp73 are specifically expressed in tumor tissue, but not in non-neoplastic liver tissue. Hereby, TAp73 and ΔNp73 seemed to be inversely regulated, that is, high expression of TAp73 corresponded to low expression of ΔNp73 and vice versa (Figure 8).

Western Blot analysis of ΔNp73 and TAp73 expression in six HCCs (‘T’) and corresponding non-neoplastic liver tissue (‘N’) using our specific antibodies. TAp73 and ΔNp73 are distinctively expressed in tumor tissue, but not in non-neoplastic liver tissue
By immunostaining, our antibody raised specifically against ΔNp73, identified 31 out of 84 (37%) of the HCCs to overexpress ΔNp73 (Table 2, Figure 9a and b). Within these tumors, we observed a strong immunoreactivity of the tumor cell nuclei. In the case of surrounding, non-neoplastic cirrhotic liver tissue, normal hepatocytes were seen to be occasionally positive for ΔNp73. In contrast to the corresponding tumors, however, in nontumorous tissue only very few nuclei (<1%) expressed ΔNp73 (Figure 9c).

Immunohistochemical demonstration of ΔNp73 protein in hepatocellular carcinoma and in non-neoplastic liver tissue using our specific antibody.30 (a) ΔNp73 staining signal is localized within tumor cell nuclei (red reaction product; original magnification × 20). (b) Positive tumor cell nuclei with a slight perinuclear staining during mitosis (arrowhead): ΔNp73 immunoreactivity occurs also in the cytoplasm. Sinus endothelial cells are ΔNp73-negative (asterisk) (original magnification × 100; oil immersion). (c) Surrounding liver tissue is negative for ΔNp73 (original magnification × 20)
p53 mutational analysis revealed that 46% (39/84) of these HCC patients carried p53 mutations. There was no correlation between ΔNp73 overexpression and the p53 mutational status (Table 3).
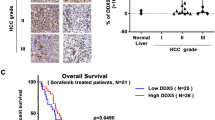
The survival analysis took into account ΔNp73 positivity (defined by immunohistochemistry), UICC tumor stage and Edmondson grade. Consistent with published data, UICC stage and Edmondson grade were valuable prognostic parameters (Table 2). Of clinical importance is our finding of a significantly shorter survival time of patients with tumors overexpressing ΔNp73 compared to patients whose tumors were ΔNp73 negative (P<0.005, log-rank test) (Table 2).
Discussion
Findings obtained in the present study allow us to propose a model that links the regulation of apoptosis by the relative expression of TAp73 and ΔNp73 to chemosensitivity and clinical outcome in HCC (Figure 10). Our data provide three new findings: (1) the identification of new target genes of TAp73/ΔNp73-regulated apoptosis pathways, (2) the molecular mechanisms of TAp73/ΔNp73 in chemosensitivity and (3) the characterization of ΔNp73 as a new prognostic marker for patients with HCC.

Model of TAp73/ΔNp73-regulated apoptosis. Upon DNA damage, TAp73 activates both, the death receptor- (pathway 1) and the mitochondrial- (pathway 2) apoptosis pathway. ΔNp73 is a strong inhibitor of both pathways
Our results support a two-pathway model for the TAp73-apoptotic response in hepatoma cells. Here, we show that TAp73β is involved in the activation of both, the extrinsic/death receptor-mediated apoptosis pathway as well as the intrinsic/mitochondria-mediated apoptosis pathway, pathways 1 and 2, respectively (Table 1 and Figure 10).
TAp73β can trigger each of the major death receptor systems, the CD95-, the TNF-R- and the TRAIL-R-system. We identified the CD95 gene as a transcriptional target of TAp73β via the intronic p53 enhancer. We show that TAp73-dependent transactivation of the CD95 gene depends on the intronic p53 binding site, as it was totally abrogated when we mutated the intronic p53-binding site of the CD95 gene. The TRAIL-R1 and -R2 genes are other death receptor genes that harbor p53-binding elements.22, 23 Thus, TAp73β-mediated transcriptional activation of the TRAIL-R1 and -R2 genes might also be exerted via binding to p53-responsive elements. Furthermore, we found TNF-R1 and TNF to be upregulated by TAp73β. This is in line with recent findings that c-ABL and p73 contribute to apoptosis induced by TNFα, in addition to their role in promoting DNA damage-associated cell death.24 As for the CD95 system, CD95 ligand mRNA was induced upon concomitant treatment with cytostatic drugs but not by TAp73β alone. Thus, TAp73-mediated increases in CD95 expression may lead to apoptosis in a CD95 ligand-independent fashion. Spontaneous multimerizing of overexpressed CD95 and TNFR1 has been reported previously to result in the induction of CD95- and TNF-like effects, independent of the respective ligand, suggesting that the self-association of the death domain suffices to trigger downstream signaling and apoptosis.25, 26
In addition to its direct effect on death receptor-mediated apoptosis, TAp73β can contribute to apoptosis by inducing the expression of several pro-apoptotic proteins acting at the mitochondrial level. Using microarray analysis we identified BAD, BIK, BNIP3, HRK and RAD9 to be upregulated in cells overexpressing TAp73β. Similar to p53, TAp73 was able to transactivate the Bax promoter in hepatoma cells and to induce an increase of endogenous Bax protein levels. This is in line with previous findings from our group in a different cellular system (Saos2 cells).27 Thus, TAp73, like p53,1 engages the major apoptotic pathways in the cell stimulating both death receptor signaling and apoptosis emanating from mitochondria.
Furthermore, our results show a relevant role for TP73 in chemosensitivity. Both, endogenous TAp73 and ΔNp73, are upregulated in response to DNA damage by bleomycin, mitoxantrone and doxorubicin. TAp73β significantly enhances the apoptosis-inducing effect of chemotherapeutic drugs in a variety of solid cancer cell lines. On the contrary, the N terminus-deleted p73 isoform predominantly detected in cancer, Δ84p73, as well as the isoform produced by the second promoter, ΔNp73, confers drug resistance to hepatoma cells. Interference of ΔNp73 with death by apoptosis in tumor cells after treatment with anticancer agents can take place at several levels of apoptosis signaling. Resistance towards chemotherapy imposed by ΔNp73 occurred at least in part by downregulation of the CD95 gene. Our data show that direct interference with the CD95 gene transactivation function of TAp73 and of p53 is one molecular mechanism by which ΔNp73 exerts its antiapoptotic and oncogenic function.8, 10, 28 This inhibitory effect of ΔNp73 is not restricted to the CD95 gene. There may exist additional proapoptotic genes (harboring p53 binding elements) whose expression is downregulated by ΔNp73, contributing to tumor progression and a worse patient outcome. We identified the Bax promoter to be repressed by ΔNp73. Thus, resistance towards apoptosis by ΔNp73 is also imposed by inhibition of mitochondrial apoptosis pathways. Furthermore, ΔNp73 strongly inhibited chemotherapy-induced activation of caspase-3, -8 and -9. Of note, our data couple the disruption of apoptosis at the death receptor- and mitochondrial level by ΔNp73 with intrinsic drug resistance. We show that the functional status of TAp73/ΔNp73 is an important determinant of cellular response to chemotherapeutic drugs. While the expression of TAp73 synergizes with chemotherapeutic drugs, the protein isoform ΔNp73 confers drug resistance. This implies that the natural, or pharmacologically regulated, relative balance of these two isoforms may influence the clinical outcome.
Emerging evidence from the analysis of primary human tumors shows that deregulated ΔNp73 expression is rather frequent.8, 9 In neuroblastoma, which is almost exclusively wild-type for p53, a correlation of ΔNp73 status with the clinical outcome was seen.9
In a series of 193 patients with HCC, we have previously shown detectable (high) p73 by in situ hybridization and immunohistochemistry in 32% of the tumors, whereas all normal tissue had undetectable p73 levels.29 Many of the (early) p73 overexpression studies in human cancers determined total p73 levels, because the antibodies used could not distinguish between TA and ΔNp73. We have generated highly specific antibodies 30 and we show here for the first time that it is the ΔNp73 isoform, which is upregulated in HCC. Our antibody raised specifically against ΔNp73, identified 31 out of 84 (37%) of patients with HCC to overexpress ΔNp73 in their tumor tissue. Importantly, we provide the first evidence that ΔNp73 upregulation in HCC is correlated with a poor prognosis. Patients with tumors overexpressing ΔNp73 exhibited a significantly shorter survival time compared with patients whose tumors were ΔNp73 negative (P<0.005 log-rank test). This is an important and clinically relevant finding, which suggests the use of ΔNp73 status as a prognostic marker for patients with HCC.
Our finding that a significant percentage of HCCs select for dominant-negative p73 isoforms strongly argues for their oncogenic role during hepatocarcinogenesis. Preferential upregulation of ΔNp73 in HCCs might impose oncogenic activity that specifically interferes with the tumor suppressor function of wild-type p53 and TAp73 disabling major apoptosis pathways (death receptor- and mitochondrial pathways), as we have shown in vitro.
Molecular links between apoptosis, tumorigenesis and drug resistance provide the foundation for new therapeutic approaches and for a targeted cancer therapy. Our results show that p53 is not the only component in predicting prognosis and response to chemotherapy, but instead the status of a network that contains p53, p73 and p63.14 Therapeutic modulation of TAp73/ΔNp73 and mutant p53 levels might be used to target the large percentage of human tumors that harbor p53 mutations and/or overexpress ΔNp73. One might predict that interfering with the expression or function of ΔNp73 and/or mutant p53 and/or ΔNp63 in tumor cells may render such tumors more responsive to therapy and reduce their aggressiveness and metastatic capacity.
Materials and Methods
Cell lines
We used Hep3B (human liver carcinoma, deficient in p53 [p53−/−]), HepG2 (human hepatoblastoma, wild-type (wt) p53), AGS (human gastric adenocarcinoma, wt p53) and Saos2 (human osteosarcoma, p53−/−). Hep3B cells were maintained in MEM (Invitrogen, Karlsruhe, Germany), HepG2 and AGS in DMEM (Gibco BRL, Eggenstein, Germany) and Saos2 in a 1:1 mixture of Ham's F-12/DMEM (Biochrom, Berlin, Germany). Medium was supplemented with 10% FCS (Biochrom), 10 mM HEPES pH 7.3 (Invitrogen), 2 mM L-Glutamine (Invitrogen) and 100 μg/ml Gentamycin (Invitrogen).
Treatment with cytostatic drugs
Cells were treated with bleomycin at a dose range of 0.3–300 μg/ml or with doxorubicin at a dose range of 0.005–0.5 μg/ml or with mitoxantrone at a dose range of 0.1–1 μg/ml for 6 h up to 72 h. The concentrations relevant for therapy are 1.5–3 μg/ml for bleomycin, 0.001–0.02 μg/ml for doxorubicin, and 0.03–0.5 μg/ml for mitoxantrone in patients' sera.
Treatment with IgG3 anti-APO-1
The CD95 (Apo-1/Fas) receptor was stimulated with the monoclonal antibody anti-APO-1 IgG3, κ, at a concentration of 1 μg/ml as described.18
Adenoviral constructs and transduction
Replication deficient adenoviral vectors were generated according to the method of He et al.31 The vectors either encoded the complete human wt p53 cDNA (rAd-p53), TAp73β cDNA (rAd-TAp73β) or Δ84p73β cDNA (rAd-Δ84p73β), together with the GFP, or the GFP alone (rAd-GFP), each under the control of the cytomegalovirus immediate/early gene (CMV) promoter. At a MOI of 10, an infection rate of 80–90% of the cells was obtained. Cells were seeded in six-well plates 24 h before transduction. Then adenoviruses (rAd-GFP, rAd-p53, rAd-TAp73β or rAd-Δ84p73β) were added to the culture medium and cells were incubated with the virus for 4 h.
Plasmids
A construct was generated containing 3.2 kb of the physiological sequence of the CD95 gene, that is, the 3′ part of the promoter, the complete exon 1 and the 5′ part of intron 1. This plasmid is denoted p1142CD95-luc and has been employed in all the transient transfection assays presented. Mutants of the intronic p53-binding site of p1142CD95-luc18 were established using the QuickChange Site-Directed Mutagenesis Kit (Stratagene, Heidelberg, Germany).
The Bax-luciferase reporter plasmid (Bax-Pr/luc) and TAp73α, β, γ, δ; ΔNp73β and Δ84p73β have been described previously.7, 32, 33
Transfections
Cells were transfected by the use of calcium-phosphate. After 18 h medium was changed. After 2 h, cells were infected with the adenovirus at a MOI of 10. After 24 h, cells were harvested and assayed for luciferase activity as described by the manufacturer (Promega, Heidelberg, Germany).
DAPI staining
DAPI (4′,6-diamidino-2-phenylindole, Sigma, Munich, Germany) staining of cellular DNA, was performed as described.17
Cytotoxicity MTT-assay
Cell viability was determined by a colorimetric MTT-assay as described.17
Detection of apoptosis
Apoptosis was assessed by fluorescence-activated cell sorting (FACS) analysis carried out in a FACScan® flow cytometer (Becton Dickinson, Heidelberg, Germany) using the CELLQuest® software system. Quantification of DNA fragmentation was performed by FACS analysis of propidium iodide-stained nuclei.18, 20
To block apoptosis, the broad spectrum caspase inhibitor ZVAD-FMK (z-Val-Ala-DL-Asp-fluoromethylketone, Bachem, Bubendorf, Germany), DEVD-FMK (Z-Asp(OCH3)-Glu(OCH3)-Val-Asp(OCH3)-FMK; inhibitor of caspase-3 as well as -6, -7, -8, and -10), Z-IETD-FMK (z-Ile-Glu(OMe)-Thr-Asp(OMe)-CH2F; inhibitor of caspase-8), Z-LEHD-FMK (z-Leu-Glu(OMe)-His-Asp(OMe)-CH2F; inhibitor of caspase-9, also -4 and -5) (all from Calbiochem, Schwalbach, Germany) and the antagonistic anti-human CD95L mAb (clone NOK-1, BD Biosciences, Heidelberg, Germany) were applied.
For caspase fluorometric assays, Hep3B cells were harvested 36 and 48 h following rAd-TAp73β or rAd-Δ84p73β or rAd-GFP transfer with or without concomitant bleomycin treatment (caspase-3, -8 and -9 fluorometric assay, R&D systems, Minneapolis, MN, USA).
Detection of CD95 ligand (CD95L) and CD95 receptor (CD95) mRNA
Total cellular RNA was prepared from 3 × 106 cells treated with bleomycin and/or 10 MOI rAd-TAp73β, using the Rneasy-kit® (Qiagen, Hilden, Germany). PCR conditions and primers have been described previously.17, 18
Detection of CD95 (APO-1/Fas)
Cell surface expression of the CD95 receptor was assessed by FACScan®. Hep3B or Saos2 cells were incubated for 30 min with anti- CD95 receptor antibody (anti-APO-1 IgG3, κ) and washed with PBS (+10% FCS). PE-labeled affinity purified F(ab)’2 fragment goat anti-mouse IgG Fc antibody (Dianova, Hamburg, Germany) was used as secondary detecting reagent. To examine staining specificity of the second antibody cells were incubated with isotype control (IgG3) alone.
Immunodetection of Bax, TAp73 and ΔNp73
For immunodetection of Bax, we applied the mouse anti-Bax monoclonal antibody sc-7480 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). For the analysis of TAp73 and ΔNp73 expression in hepatoma cells and in liver tissue from patients with HCC, we used highly specific antibodies raised in rabbits against Sam and ΔN domains, see below ‘Immunohistochemical analysis and Western blot of tissue samples’.30
Determination of mitochondrial membrane potential
Hep3B cells were incubated with 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1, 5 μg/ml; Molecular Probes, Eugene, OR, USA) for 20 min at room temperature in the dark, washed twice and analyzed by FACScan® as described.14
Microarray analysis
We have developed high-density cDNA arrays in cooperation with the Department of Molecular Genome Analysis, German Cancer Research Center, Heidelberg, Germany. Hep3B cells were infected with 10 MOI rAd-TAp73β or rAd-GFP respectively, and RNA was extracted after 12, 24, 36 and 48 h using the Quiagen RNAeasy-kit (Hilden, Germany). Two independent experiments were performed. Extracted RNA was checked for quality using Agilent 2100 bioanalyzer (Ambion, Waldbronn, Germany). Total RNA was labeled and rewritten to cDNA in a reverse transcriptase reaction, containing 100 ng/ml oligo-dT15 primer, 5 mmol DTT, 1 mmol of dATP, dDTP, dGTP and [33P]dCTP (Amersham, Freiburg, Germany) and 10 U superscript II reverse transcriptase (Gibco BRL, Eggenstein, Germany). cDNA was hybridized to nylon membranes containing PCR-products of 1066 specific cDNA clones. These clones represent genes that are involved in apoptosis and immunological signaling pathways. In total, 36 clones, of which three representative clones are shown in Table 1, served as positive controls. Array preparation and quality control was performed by the German Resource Center for Genome Research (RZPD, Berlin/Heidelberg, Germany). These arrays are now commercially available at www.rzpd.de (Immunofilter, RZPD). Hybridization was performed overnight at 68°C in Expresshyb® hybridization solution (Clontech, Heidelberg, Germany). Three hybridizations were performed for each experiment. After washing for 1 h in SDS-SSC buffer, arrays were exposed to phosphoimager screens and subsequently analyzed using a FujiBas® phosphoimager. We used Visual Grid Software (GPC Biotechnology, Martinsried, Germany) for matching the files. Expression levels were normalized over all positive spots using J-Express® software (J-Express V2.1, Molmine bioinformatics software solutions, Bergen, Norway) and subsequently evaluated by SAM (significance analysis of microarrays) software, which identifies genes whose expression has significantly changed.34
Patients and tissue samples
In total, 84 patients with HCC undergoing partial hepatectomy were included in a retrospective study. All patients underwent surgery with curative intent (R0 resections). Patients who received orthotopic liver transplantation were excluded from the study. No patient received preoperative or adjuvant chemotherapy or radiotherapy. Each tumor was evaluated with regard to typing, staging and Edmondson grading. Tumor typing and staging were performed by use of criteria of the World Health Organization35 and the International Union Against Cancer (UICC),36 respectively. The patients and their pathohistological data are summarized in Table 2. The slides were evaluated by two observers who were blinded to clinical and pathologic information.
Immunohistochemical analysis and Western blot of tissue samples
We have generated new antibodies with p73 isoform specificity, recognizing only the TA domain of p73 or only ΔN isoforms.30
The rabbit polyclonal antibody against ΔNp73 was used for immunohistochemical analysis of 84 HCCs and corresponding non-neoplastic liver tissue as described before.29
Western blot was performed using the rabbit polyclonal antibody against ΔNp73 and the monoclonal antibody against the TA domain of p73 as described previously.37
Statistical analysis
To examine whether synergy between rAd-TAp73β transfer and concurrent chemotherapeutic treatment is observed, a balanced two-way ANOVA (model with fixed effects) was performed. Furthermore, we applied MANOVA (multivariate analysis of variance) and Wilcoxon analysis to test for statistical significance. The log-rank test was used to detect differences between survival curves for stratified variables. Statistical analysis was carried out using the SAS software system (SAS Institute Inc., Cary, NC, USA).
Abbreviations
- ANOVA:
-
analysis of variance
- ΔNp73:
-
dominant-negative p73
- FACS:
-
fluorescence-activated cell sorting
- FADD:
-
Fas-associated death domain
- GFP:
-
green fluorescent protein
- HCC:
-
hepatocellular carcinoma
- MANOVA:
-
multivariate analysis of variance
- MEF:
-
mouse embryo fibroblast
- p53-IBS:
-
intronic p53-binding site
- SAM:
-
significance analysis of microarrays
References
Vousden KH and Lu X (2002) Live or let die: the cell's response to p53. Nat. Rev. Cancer 2: 594–604
Melino G, De Laurenzi V and Vousden KH (2002) p73: friend or foe in tumorigenesis. Nat. Rev. Cancer 2: 605–615
Moll UM and Slade N (2004) p63 and p73: roles in development and tumor formation. Mol. Cancer Res. 2: 371–386
Ramadan S, Terrinoni A, Catani MV, Sayan AE, Knight RA, Mueller M, Krammer PH, Melino G and Candi E (2005) p73 induces apoptosis by different mechanisms. Biochem. Biophys. Res. Commun. 331: 713–717
Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J, Vagner C, Bonnet H, Dikkes P, Sharpe A, McKeon F and Caput D (2000) p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 404: 99–103
Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, Minty A, Chalon P, Lelias JM, Dumont X, Ferrara P, McKeon F and Caput D (1997) Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 90: 809–819
De Laurenzi V, Costanzo A, Barcaroli D, Terrinoni A and Falco M (1998) Two new p73 splice variants, gamma and delta, with different transcriptional activities. J. Exp. Med. 188: 1768
Zaika AI, Slade N, Erster SH, Sansome C, Joseph TW, Pearl M, Chalas E and Moll UM (2002) DeltaNp73, a dominant-negative inhibitor of wild-type p53 and TAp73, is up-regulated in human tumors. J. Exp. Med. 196: 765–780
Casciano I, Mazzocco K, Boni L, Pagnan G, Banelli B, Allemanni G, Ponzoni M, Tonini GP and Romani M (2002) Expression of DeltaNp73 is a molecular marker for adverse outcome in neuroblastoma patients. Cell Death Differ. 9: 246–251
Petrenko O, Zaika A and Moll UM (2003) DNp73 facilitates cell immortalization and cooperates with oncogenic Ras in cellular transformation in vivo. Mol. Cell. Biol. 23: 5540–5555
Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D, Yang A, McKeon F and Jacks T (2005) Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 7: 363–373
Agami R, Blandino G, Oren M and Shaul Y (1999) Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature 399: 809
Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin Jr WG, Levrero M and Wang JY (1999) The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature 399: 806–809
Gressner O, Schilling T, Lorenz K, Schulze SE, Koch A, Schulze-Bergkamen H, Maria LA, Candi E, Terrinoni A, Valeria CM, Oren M, Melino G, Krammer PH, Stremmel W and Müller M (2005) TAp63alpha induces apoptosis by activating signaling via death receptors and mitochondria. EMBO J. 24: 2458–2471
Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F and Jacks T (2002) p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 416: 560–564
Senoo M, Manis JP, Alt FW and McKeon F (2004) p63 and p73 are not required for the development and p53-dependent apoptosis of T cells. Cancer Cell 6: 85–89
Müller M, Strand S, Hug H, Heinemann EM, Walczak H, Hofmann WJ, Stremmel W, Krammer PH and Galle PR (1997) Drug-induced apoptosis in hepatoma cells is mediated by the CD95 (APO-1/Fas) receptor/ligand system and involves activation of wild-type p53. J. Clin. Invest. 99: 403–413
Müller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M and Krammer PH (1998) p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J. Exp. Med. 188: 2033–2045
Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC and Kaelin WG (2003) Chemosensitivity linked to p73 function. Cancer Cell 3: 403–410
Nicoletti I, Migliorati G, Paggliacci MC, Grignani F and Riccardi C (1991) A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 139: 271–279
Eichhorst ST, Müller M, Li-Weber M, Schulze-Bergkamen H, Angel P and Krammer PH (2000) A novel AP-1 element in the CD95 ligand promoter is required for induction of apoptosis in hepatocellular carcinoma cells upon treatment with anticancer drugs. Mol. Cell. Biol. 20: 7826–7837
Wu GS, Burns TF, McDonald ER, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, Hamilton SR, Spinner NB, Markowitz S, Wu G and El-Deiry WS (1997) KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene [letter]. Nat. Genet. 17: 141–143
Liu X, Yue P, Khuri FR and Sun SY (2004) p53 upregulates death receptor 4 expression through an intronic p53 binding site. Cancer Res. 64: 5078–5083
Chau BN, Chen TT, Yisong Y, DeGregori J and Wang JYJ (2004) Tumor necrosis factor alpha-induced apoptosis requires p73 and c-ABL activation downstream of RB degradation. Mol. Cell. Biol. 24: 4438–4447
Micheau O, Solary E, Hammann A and Dimanche-Boitrel MT (1999) Fas ligand-independent, FADD-mediated activation of the Fas death pathway by anticancer drugs. J. Biol. Chem. 274: 7987–7992
Boldin MP, Mett IL, Varfolomeev EE, Chumakov I, Shemer-Avni Y, Camonis JH and Wallach D (1995) Self-association of the ‘death domains’ of the p55 tumor necrosis factor (TNF) receptor and Fas/APO1 prompts signaling for TNF and Fas/APO1 effects. J. Biol. Chem. 270: 387–391
Melino G, Bernassola F, Ranalli M, Yee K, Zong WX, Corazzari M, Knight RA, Green DR, Thompson C and Vousden KH (2003) p73 induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J. Biol. Chem. 279: 8076–8083
Terrasson J, Allart S, Martin H, Lule J, Haddada H, Caput D and Davrinche C (2005) p73-dependent apoptosis through death receptor: impairment by human cytomegalovirus infection. Cancer Res. 65: 2787–2794
Tannapfel A, Wasner M, Krause K, Geissler F, Katalinic A, Hauss J, Mossner J, Engeland K and Wittekind C (1999) Expression of p73 and its relation to histopathology and prognosis in hepatocellular carcinoma. J. Natl. Cancer Inst. 91: 1154–1158
Sayan AE, Paradisi A, Vojtesek B, Knight RA, Melino G and Candi E (2005) New antibodies recognizing p73: comparison with commercial antibodies. Biochem. Biophys. Res. Commun. 330: 186–193
He TC, Zhou S, da Costa LT, Yu J, Kinzler KW and Vogelstein B (1998) A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA 95: 2509–2514
Miyashita T and Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80: 293–299
De Laurenzi V, Raschella G, Barcaroli D, Annicchiarico-Petruzzelli M, Ranalli M, Catani MV, Tanno B, Costanzo A, Levrero M and Melino G (2000) Induction of neuronal differentiation by p73 in a neuroblastoma cell line. J. Biol. Chem. 275: 15226–15231
Tusher VG, Tibshirani R and Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 98: 5116–5121
Hamilton SR and Aaltonen LA (2000) WHO: Pathology and Genetics (Lyon: IARC Press)
Sobin LH and Wittekind CH (2002) UICC: TNM Classification of Malignant Tumours 6th edn (New York: Wiley-Liss)
Tannapfel A, Anhalt K, Hausermann P, Sommerer F, Benicke M, Uhlmann D, Witzigmann H, Hauss J and Wittekind CH (2003) Identification of novel proteins associated with hepatocellular carcinomas using protein microarrays. J. Pathol. 201: 238–249
Wada N, Matsumura M, Ohba Y, Kobayashi N, Takizawa T and Nakanishi Y (1995) Transcription stimulation of the Fas-encoding gene by nuclear factor for interleukin-6 expression upon influenza virus infection. J. Biol. Chem. 270: 18007–18012
El-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW and Vogelstein B (1992) Definition of a consensus binding site for p53. Nat. Genet. 1: 45–49
Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, Aach J, Ansorge W, Ball CA, Causton HC, Gaasterland T, Glenisson P, Holstege FC, Kim IF, Markowitz V, Matese JC, Parkinson H, Robinson A, Sarkans U, Schulze-Kremer S, Stewart J, Taylor R, Vilo J and Vingron M (2001) Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nat. Genet. 29: 365–371
Acknowledgements
We thank Petra Hill for expert technical assistance. This work was supported by grants of the Medizinische Forschungsförderung Heidelberg, of the Sonderforschungsbereich 601 and of the Tumorzentrum Heidelberg/Mannheim to MM and PHK. The work was in part performed thanks to grants from AIRC, EU (QLG1-1999-00739 and YLK-CT-2002-01956), MIUR, MinSan to GM, EU (QLK3-CT-2002-01956) to GM and MO and EU grant QLG1-1999-00739 to PHK.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by D Green
These authors contributed equally to this work
These authors share last authorship
Rights and permissions
About this article
Cite this article
Müller, M., Schilling, T., Sayan, A. et al. TAp73/ΔNp73 influences apoptotic response, chemosensitivity and prognosis in hepatocellular carcinoma. Cell Death Differ 12, 1564–1577 (2005). https://doi.org/10.1038/sj.cdd.4401774
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401774
Keywords
This article is cited by
-
TP73 Isoform-specific disruption reveals a critical role of TAp73beta in growth suppression and inflammatory response
Cell Death & Disease (2023)
-
RNA splicing: a dual-edged sword for hepatocellular carcinoma
Medical Oncology (2022)
-
Overexpression of splicing factor poly(rC)-binding protein 1 elicits cycle arrest, apoptosis induction, and p73 splicing in human cervical carcinoma cells
Journal of Cancer Research and Clinical Oncology (2022)
-
Neutralization of CD95 ligand protects the liver against ischemia-reperfusion injury and prevents acute liver failure
Cell Death & Disease (2018)
-
Improvement of gemcitabine sensitivity of p53-mutated pancreatic cancer MiaPaCa-2 cells by RUNX2 depletion-mediated augmentation of TAp73-dependent cell death
Oncogenesis (2016)
