
Abstract
The gastric microbiome has been proposed as an etiological factor in gastric carcinogenesis. We compared the gastric microbiota in subjects presenting with gastric cancer (GC, n = 12) and controls (functional dyspepsia (FD), n = 20) from a high GC risk population in Singapore and Malaysia. cDNA from 16S rRNA transcripts were amplified (515F-806R) and sequenced using Illumina MiSeq 2 × 250 bp chemistry. Increased richness and phylogenetic diversity but not Shannon’s diversity was found in GC as compared to controls. nMDS clustered GC and FD subjects separately, with PERMANOVA confirming a significant difference between the groups. H. pylori serological status had a significant impact on gastric microbiome α-diversity and composition. Several bacterial taxa were enriched in GC, including Lactococcus, Veilonella, and Fusobacteriaceae (Fusobacterium and Leptotrichia). Prediction of bacterial metabolic contribution indicated that serological status had a significant impact on metabolic function, while carbohydrate digestion and pathways were enriched in GC. Our findings highlight three mechanisms of interest in GC, including enrichment of pro-inflammatory oral bacterial species, increased abundance of lactic acid producing bacteria, and enrichment of short chain fatty acid production pathways.
Similar content being viewed by others


Introduction
Gastric cancer (GC) is the fifth most commonly diagnosed cancer and third leading cause of cancer-related deaths worldwide1. It accounted for an estimated 723,000 deaths (8.8% of total deaths from cancer) in 20121. Almost two-thirds of GC cases occur in East Asia, Eastern Europe, and Central and South America, with Chinese ethnicity being identified as an independent risk factor for the development of GC in multiracial studies2,3. In countries such as Malaysia and Singapore, individuals with Chinese ethnicity have the highest age-standardized rate (ASR) (25.7 per 100,000 men) as compared with individuals of Malayan (6.6 per 100,000) and Indian (8.4 per 100,000) backgrounds1.
While Helicobacter pylori infection is the most important risk factor for GC, the etiology of GC is clearly multifactorial as evidenced by the fact that only 1–3% of H. pylori-infected patients develop GC, and that progression to GC in some subjects occurs even after eradication of the bacterium4. In addition to host and environmental factors, the gastric microbiome is now believed to contribute to the progression of disease, particularly when H. pylori disappears in patients who progress to the metaplastic and dysplastic stages. For example, Lertpiriyapong et al.5, using a transgenic H. pylori INS-GAS mouse model, reported that colonization with microbiota limited to three species of Altered Schaedler’s Flora, including ASF356 (Clostridium sp.), ASF361 (Lactobacillus murinus) and ASF519 (Bacteroides sp.), were sufficient to induce neoplasia. Further, a range of independent studies assessing gastric microbiota in relation to Correa’s Cascade have demonstrated that significant differences exist in the microbiota of those with gastritis, intestinal metaplasia (IM) and GC6,7,8,9,10, suggesting that dysbiosis in the stomach is dynamic and correlates with progression to gastric carcinogenesis.
In contrast, a study by Yang et al.11 which included subjects presenting with non-atrophic gastritis, AG and IM, from a high GC risk area (Túquerres) and a low GC risk area (Tumaco) in Colombia, failed to identify any correlation between gastric microbiota composition and histological diagnosis. However, the authors did identify that Leptotrichia spp. and Veillonella spp., were significantly more abundant in Túquerres while 16 OTUs, including a Staphylococcus spp., were significantly more abundant in Tumaco.
To our knowledge, no study to date has assessed the active microbiome (rRNA transcripts) in the context of gastric carcinogenesis. Here, we compared the active gastric microbiome of GC patients and functional dyspepsia (FD) controls using 16S rRNA transcript amplicon sequencing on an Illumina MiSeq platform, and identified a number of microbial changes and predicted functional changes associated with carcinogenesis.
Methods
Ethics approval and consent to participate
Ethics approval was obtained from the Human Ethics Committee (HREC) of the University of New South Wales (UNSW) (HREC 08115 and HREC 02144). All subjects recruited to the study signed the written informed consent. All experiments were performed in accordance with relevant guidelines and regulations.
Subjects
This study comprised 36 predominantly ethnic Chinese subjects who underwent upper gastrointestinal endoscopy at the University Hospital of Malaysia (Kuala Lumpur) and the Changi General Hospital (Singapore) in the period between January 2004 and April 2007. Subjects who had been prescribed antibiotics, non-steroidal anti-inflammatory drugs (NSAIDs) or acid suppressants in the two-month period prior to recruitment as well as subjects known to be infected with the Human Immunodeficiency Virus (HIV) were excluded.
The case group comprised 12 patients newly diagnosed with primary non-cardia GC (International Classification of Diseases, 9th revision, code 151). Subjects diagnosed with FD (n = 20) — defined as persistent or recurrent symptoms (pain or discomfort centred in the upper abdomen) in the absence of organic disease, in accordance with Rome II classification system, were invited to participate as controls. An additional four subjects presenting with gastric ulcers were recruited; however, their results were not reported on in the main text due to low numbers. Information pertaining to gender and age was obtained from each subject through a brief socio-demographic questionnaire.
Sample collection
Antral gastric biopsies were obtained from symptomatic subjects undergoing routine endoscopic examination of gastrointestinal symptoms. These biological specimens were frozen in liquid nitrogen immediately after collection and transported to Australia on dry ice, where they were stored at −80 °C until further processing. In addition, peripheral whole blood was collected from all study subjects to determine H. pylori status.
Microbiota sequencing and analysis
RNA was extracted from biopsies using the Isolate II RNA mini kit (Bioline; Sydney, NSW, Australia) and cDNA synthesized using the Tetro cDNA synthesis kit (Bioline) according to the manufacturer’s instructions.
The 16S rRNA gene was amplified using the KAPA HiFi HotStart ReadyMix (95 °C for 3 min, 25 cycles of 95 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s, followed by a final step of 72 °C for 5 min) and the earth microbiome primers (515F-806R). Indices and Illumina sequencing adapters were attached using the Nextera XT Index Kit according the manufacturer’s instructions. Amplicon sequencing was performed with Illumina MiSeq 2 × 250 bp chemistry (n = 16192 ± 2237 total clean reads/sample derived from a total of 774,558 raw sequences; 24.7% sequences filtered out) at the Ramaciotti Centre for Genomics. All relevant positive (bacterial DNA) and negative (blank) PCR controls as well as extraction controls (extraction kit buffers) were included in the experimental procedures. Raw reads were analyzed using the MiSeq standard operating procedures within Mothur v1.37.312,13. Differences across α-diversity and phylogenetic diversity (according to Faith) measures were examined using GraphPad Prism 6 employing both subsampled (n = 2000 reads) and non-subsampled read counts, and reported patterns were consistent across both datasets. To determine global differences in microbial composition, multivariate analyses such as non-metric multidimensional scaling (nMDS), permutational MANOVA (PERMANOVA), and PERMDISP were performed on relative abundances using Primer-E14. To identify specific microbial taxa that differed significantly across conditions, Linear Discriminant Analysis Effect Size (LEfSe)15 was performed using the Galaxy web application16. PICRUSt17 was used to predict bacterial metabolic contributions and both global and specific changes were examined using Primer-E and LEfSe, respectively. SparCC18 correlations were calculated in Mothur v1.39.113 and plotted using Cytoscape v3.5.119 in an attempt to identify bacterial networks within the gastric microbiota.
Helicobacter pylori detection in serum
Detection of human IgG antibodies to H. pylori was performed using the MPD Helico Blot 2.1 kit (MP Biomedicals, Australia) according to the manufacturer’s instructions. In addition, this kit includes recombinant antigens with high predictive value for the indication of current H. pylori infection. This kit has been shown to exhibit higher sensitivity and specificity when compared to commercial ELISAs20.
Data Availability
Raw data (fastq files) have been submitted to the European Nucleotide Archive (Accession number: PRJEB21497). Patient metadata and relative abundance are provided in Supplementary File S1.
Results
Impact of Helicobacter pylori infection and disease on α- and phylogenetic diversity of gastric microbiota
Species richness and phylogenetic diversity were found to be significantly increased in GC patients as compared to FD controls (Fig. 1). Evenness and Shannon’s diversity did not differ across subgroups (Fig. 1). H. pylori seropositive patients also had increased species richness and phylogenetic diversity but not evenness and Shannon’s diversity (Supplementary File 1: Figure S1A). Evidence that this was not due to an artefact arising from higher seropositivity levels in GC patients comes from the finding that FD controls positive for H. pylori had higher species richness and phylogenetic diversity as compared to FD controls that were seronegative (Supplementary File 1: Figure S1B). Further, these analyses indicated that the increased richness and phylogenetic diversity in GC patients was independent of H. pylori seropositivity (Supplementary File 1: Figure S1B).

Alpha-diversity and phylogenetic diversity of the gastric microbiota. Phylogenetic diversity was calculated according to Faith. P-values were calculated using one-way ANOVA with a Tukey’s multiple comparisons test.
Associations between Helicobacter pylori infection and disease and the composition of gastric microbiota
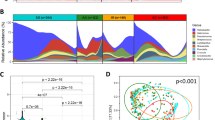
The global profile of the gastric microbiome was significantly altered during GC as compared to FD subjects (Fig. 2A–C), with differences observed up to the phylum level (Fig. 2B). H. pylori serological status of the subject also appeared to have a significant impact, with the gastric microbiota composition in subjects testing positive for the bacterium being different to those testing negative (Fig. 2B). Despite this, the differences observed between FD controls and GC patients, were independent of serological status, given that these comparisons showed significant differences when tested only in subjects that were H. pylori positive by serology (Supplementary File 1: Figure S2A). Further, the differences across disease and serological status did not arise from the relative abundance of H. pylori in the samples, as removal of the contribution of Helicobacter to the overall composition showed no differences in the results (Supplementary File 1: Figures S2B and S3).

Composition of the gastric microbiota. (A) Non-metric multidimensional scaling plot and (B) PERMANOVA following square-root transformation and Bray-Curtis similarity resemblance. (C) Relative abundance of bacterial genera across FD and GC subjects following the removal of the contribution of Helicobacter. Colour legend is provided in Supplementary File 1. (D) Cladogram of LEfSe analysis between FD controls and GC patients nested for H. pylori serological status. Higher resolution colour legend is provided in Supplementary File 1. FD: Functional dyspepsia; GC: Gastric cancer; HP-NEG: negative for H. pylori; HP-POS: positive for H. pylori.
We observed no significant differences in dispersion of the data (PERMDISP) across disease, serological status, indicating that the significant differences arising from the PERMANOVA analyses were locational and not dispersion effects. We also confirmed that the differences observed in our cohort did not arise due to age, gender or country of origin. PERMANOVA at both OTU and genus taxonomic levels comparing the gastric microbiome of FD patients from Singapore and Malaysia found no significant differences (OTU: t = 0.97, P = 0.48, Permutations = 992, Df = 18; Genus: t = 0.98, P = 0.45, Permutations = 993, Df = 18). No significant differences were found between male and female FD patients using PERMANOVA (OTU: t = 0.88, P = 0.76, Permutations = 995, Df = 18; Genus: t = 0.88, P = 0.72, Permutations = 995, Df = 18) and no correlation with age was observed using distance-based linear modelling (OTU: Pseudo-F = 1.32, P = 0.14, Df = 18; Genus: Pseudo-F = 1.28, P = 0.21, Df = 18).
We then identified the bacterial taxa contributing to these differences using LEfSE (Fig. 2D, Supplementary File S1). 91 taxa were different when comparing FD controls and GC patients (Supplementary File S1: LEfSe2). H. pylori serological status was also found to enrich for a range of bacterial taxa, with 36 taxa enriched in negative subjects and 28 in positive subjects (Supplementary File S1: Figure S4, LEfSe6). Given this, we conducted a further comparison between FD controls and GC patients with serological status nested as a subclass. Thirty-eight bacterial taxa were identified to be significantly different between GC patients and FD controls (Fig. 2D, Supplementary File S1: LEfSe5), 23 of which were enriched in GC. These included Lactococcus OTU0002 (100% similarity to Lactococcus lactis), Fusobacterium OTU0087 (100% similarity to Fusobacterium mortiferum), Pasteurellaceae_unclassified OTU0022 (100% similarity to Haemophilus parahaemolyticus and H. sputorum), Staphylococcus OTU0045 (100% similarity to up to seven Staphylococcus species), and Methylobacterium OTU0018 (99% similarity to Methylobacterium adhaesivum). Other bacterial taxa of interest identified to be enriched in GC in the non-nested analysis (Supplementary File S1: LEfSe2) include Veillonella OTU0005 (100% similarity to Veillonella atypica and V. dispar), Dialister OTU0132 (100% similarity to Dialister pneumosintes), and Leptotrichia OTU0042 (100% similarity to Leptotrichia buccalis). The relative abundances of bacterial genera showing large changes across disease were plotted in Fig. 3. Most OTUs corresponding to Methylobacterium were found to be decreased in GC except for OTU0018 (Fig. 3).

Relative abundance of bacterial genera of interest in FD controls and GC patients. Methylobacterium relative abundance was broken down to OTU contribution.
Associations between Helicobacter pylori infection and disease and the predicted bacterial metabolic contribution
Bacterial metabolic contributions were then predicted using PICRUSt and nearest sequence Taxon index (NSTI) calculated for these predictions (Supplementary File S1: PICRUSt counts, PICRUSt NSTI scores). The average NSTI score for all samples was 0.039 ± 0.004 which is well below the accepted threshold of 0.1, suggesting that the predictions were reliable.
Serological status of the subject but not disease status was associated with a significantly shift in the predicted global microbial metabolic output. Subjects that were negative or positive were divided into two separate clusters (Fig. 4A,B). Despite some inconsistencies in the clusters, PERMANOVA identified significant differences between negative and positive subjects (t = 3.05, P(Perm) = 0.002, Permutations: 998). In contrast, no significant difference was found across disease (FD vs. GC: t = 1.48, P(Perm) = 0.12, Permutations: 999).

Prediction of the metabolic contribution of the gastric microbiota using PICRUSt. (A) Non-metric multidimensional scaling plot and (B) Cluster analysis with SIMPROF testing following Log(X + 1) transformation and Bray-Curtis similarity resemblance. (C) KEGG pathways (Level 3) enriched in GC using LEfSe analysis. P-values are presented at the end of the bars.
Predicted KEGG pathways enriched across disease and H. pylori serological status were then identified using LEfSe (Supplementary File S1: PICRUSt LEfSe1-5). Both simple and nested designs were performed to account for confounding factors. We observed 20 predicted pathways (KEGG Level 3) to be enriched in GC patients when compared to FD controls (serological status nested as subclass, Fig. 4C). A complete list of enriched pathways is provided in Supplementary File S1 (PICRUSt LEfSe3). Of interest, a number of predicted pathways related to bacterial carbohydrate metabolism were found to be enriched in GC (Fig. 4C). Moreover, carbohydrate digestion and absorption, which is partly responsible for the production of short chain fatty acids (SCFAs) such as butyrate, propionate and acetate, was also enriched in GC (Fig. 4C).
Identification of bacterial interactions in gastric carcinogenesis
To determine the interactions across different bacterial OTUs, we then utilised SparCC and Cytoscape to calculate and visualize correlations among the first 100 OTUs. Interaction network across bacterial OTUs was found to be notably denser in GC patients as compared to FD controls (Fig. 5A, Supplementary File S1: SparCC FD, SparCC GC). This could be related to an increase in abundance of other organisms following the decreased dominance of H. pylori in GC patients (Fig. 3). Comparison of the FD and GC networks to identify shared interactions (independent of correlation direction) between OTUs found six interactions across 15 OTUs to be common across the networks (Fig. 5B). The relationships between OTU0011 (Lactobacillus) and OTU0031 (Clostridium sensu stricto), OTU0021 (Prevotella), OTU0041 (Faecalibacterium), and OTU0068 (Megamonas), OTU0021, OTU0041, and OTU0068, as well as OTU0043, OTU0056 and OTU0063 were all consistent across FD and GC networks. In contrast, the correlations between OTU0020 and OTU0050, and OTU0045 and OTU0074 were in opposite directions in the FD and GC networks.

Network analysis of bacterial OTUs within the gastric microbiota. (A) Network plots for FD controls and GC patients. Correlations for OTUs 1–100 were calculated using SparCC and correlations greater than 0.6 or lower than −0.5 were visualized using Cytoscape. Co-occurrence and co-exclusion interactions were drawn in green and red, respectively. Thickness of the line indicates strength of the correlation. Full lists of correlations are provided in Supplementary File 1. (B) Similarities between the FD and GC networks, and the shared interactions between OTUs. Numbers represent OTUs that interact with each other (i.e. 10 = OTU0010). (C) Network analysis of OTUs that were identified to be enriched in GC using LEfSe. OTUs in dark purple were identified as enriched in GC in the nested analysis; OTUs in pink were identified as enriched in GC in the non-nested analysis; OTUs in yellow were enriched in FD in either the nested or non-nested analysis; OTUs in light blue were not found to be enriched using LEfSe. A full list of correlations is provided in Supplementary File 1.
To define the network most relevant to disease, we then built a network comprising the five OTUs (OTU0002, OTU0018, OTU0022, OTU0045, and OTU0087) that were found to be enriched in GC patients in the nested analysis (Supplementary File S1: LEfSe5), included their interaction partners, and if their interaction partners had been associated with disease in the non-nested analysis (Supplementary File S1: LEfSe2). We visualized the network in Cytoscape (Fig. 5C) and the list of correlations is provided in the supplementary material (Supplementary File S1: SparCC GC LEfSe). One notable interaction node in this GC network is that among OTU0002 (Lactococcus), OTU0038 (Aneurinibacillus) and OTU0046 (Bacillus). Further, the co-exclusion interaction between OTU0001 (Helicobacter) and OTU0027 (100% similarity to Campylobacter concisus) is likely to be relevant in the context of gastrointestinal disease.
Discussion
It is widely accepted that GC is a multifactorial disease involving the interplay of host, microbial and environmental factors. Despite H. pylori being recognised as the main etiological factor in the development of GC, and the ability to eradicate this organism using triple or quadruple regimens, GC remains the third leading cause of cancer-related deaths worldwide. With this in mind, studies have focussed on host genetics, and more recently, the gastric microbiome’s contributions to disease etiology. Here, the viable gastric microbiome was profiled in FD and GC subjects to identify microbial changes across disease that may be relevant to gastric carcinogenesis.
Previous studies have reported conflicting results on the α-diversity of the gastric microbiome across the GC cascade6,7,9. To shed light on these discrepancies, we examined all the different α-diversity measures in detail and found species richness but not evenness or Shannon’s diversity to increase in GC. Phylogenetic diversity was found to follow similar patterns as those observed for species richness. It is likely that large fluctuations in H. pylori relative abundances across subjects contribute to highly variable evenness measures but do not impact richness measures. In support of this conclusion, a strong inverse association between the relative abundance of H. pylori and α-diversity has been previously reported8. We did identify serological status as an important contributor, with H. pylori-positive subjects exhibiting higher species richness and phylogenetic diversity. These results are of significance as they suggest that the ability of the infection to illicit an immune response in the host plays an important role in shaping the composition of the gastric microbiome.
The global composition of the gastric microbiome in our cohort was also significantly different in GC as has been previously observed6,8,9,10; however, we were able to confirm that these differences were independent of H. pylori contribution to the overall relative abundance. However, serological status was associated with a significant shift in the gastric microbiome composition. Importantly, the differences across gastric carcinogenesis and H. pylori serological status were independent of each other, highlighting the importance of host- H. pylori interactions, but also suggesting a role for the gastric microbiome in disease progression. Notably, there was an increase in co-occurrence interactions across the gastric microbiome in GC as compared to FD. While this may arise from the depletion of H. pylori in GC allowing other microbial species to flourish, it may also lead to other consequences in the host, with one recent study reporting an increased bacterial load in GC patients as compared to chronic gastritis patients9.
A marked increase in the relative abundance of lactic acid producing bacteria (Lactococcus and Lactobacillus) was observed in GC patients. Further, Lactococcus OTU0002 was found to have strong co-occurrence interactions with two other OTUs associated with GC (Aneurinibacillus OTU0038 and Bacillus OTU0046). Indeed, previous studies have observed a similar increase in abundance of Lactobacillus species in GC6,7,9. While Lactobacillus species are often utilized as probiotics and assumed to be beneficial to the host, in the context of cancer, elevated levels of lactic acid can be highly detrimental. For example, lactate can serve as an energy source for tumor cells, inducing glycolytic enzymes which leads to an increase in ATP supply; this metabolite can also promote inflammation and stimulate tumor angiogenesis21,22,23,24. Of particular significance, Lertpiriyapong et al.5, using a transgenic H. pylori INS-GAS mouse model, showed that colonisation with microbiota limited to three species of Altered Schaedler’s Flora, including ASF356 (Clostridium species), ASF361 (Lactobacillus murinus) and ASF519 (Bacteroides species), were sufficient to induce neoplasia. Most studies have focussed on the role of host lactic acid production in cancer cell metabolism, but these findings suggest that at least in relation to GC, the role of exogenous bacterial lactic acid should be investigated further.
An increased abundance of Escherichia/Shigella was observed in our GC patients, consistent with previous findings9. It is difficult to conclude what role this may play in GC given the diversity in metabolic and pathogenic potential within this group of organisms. However, specific genotoxin strains or invasive pathotypes of E. coli have been linked with inflammatory bowel diseases (IBD)25 and colorectal cancer26. Thus, the increased abundance of Escherichia/Shigella has the potential to be harmful to the host.
Bacterial species commonly found in the oral cavity including Fusobacterium, Veillonella, Leptotrichia, Haemophilus, and Campylobacter were found to have higher relative abundances in GC patients. This enrichment of oral organisms is increasingly being reported across several types of cancer such as esophageal adenocarcinomas, colorectal adenocarcinomas, and breast cancers27,28,29. Of these, Fusobacterium species have received a lot of attention due to their pro-inflammatory nature, with TLR4 and autophagy playing a very important role in the inflammation they induce30,31,32. Notably, polymorphisms in TLR4 and autophagy are well known to increase the risk of developing GC in Chinese individuals33,34,35,36, thus, it would be interesting to determine the impact of these polymorphisms in the context of increased levels of Fusobacterium. Interestingly, a recent analysis of the gastric microbiome in two populations from Colombia (Túquerres and Tumaco), found Leptotrichia and Veillonella to be significantly more abundant in inhabitants of Túquerres, these individuals exhibiting a 25-fold higher risk of GC than Tumaco inhabitants despite both populations having similar H. pylori prevalence11. Fusobacterium species have been shown to co-aggregate with other bacteria to form biofilms that play a key role in colorectal adenocarcinoma initiation and progression29. In this context, Veillonella species could be a plausible pro-oncogenic partner as these bacteria have been found to be increased in oral, lung and colorectal cancer patients37,38,39. Given this, the enrichment of these oral bacterial species should not only be further investigated in GC, but also in other types of cancer.
An additional interaction of interest was the co-exclusion relationship between Helicobacter OTU0001 and Campylobacter OTU0027 (putatively identified as C. concisus). Emerging Campylobacter species such as C. concisus are members of the oral microbiota that have been associated, through epidemiological studies, with a range of gastrointestinal diseases28,40,41. A recent meta-analysis on the relationship between H. pylori and IBD hypothesized that a co-exclusion relationship between this gastric bacterium and related members of the Helicobacter and Campylobacter genera in the intestinal tract may be one mechanism by which H. pylori exerts a protective effect in IBD42.
Another important finding was the effect of H. pylori serological status on the predicted global metabolic contribution of the gastric microbiome, an effect that was not observed in disease. This highlighted the potential capacity for the immune response against H. pylori to regulate the overall composition and predicted metabolic output of the gastric microbiome. We did observe a number of bacterial metabolic pathways that were predicted to be significantly enriched in GC. In addition to an array of carbohydrate metabolism pathways likely related to the enrichment of Lactococcus and Lactobacillus species in GC, we identified enrichment of carbohydrate digestion and absorption, which is involved in the production of SCFAs such as butyrate, propionate and acetate. Importantly, increased levels of bacterial SCFAs have been shown to induce hyperproliferation of colonic cells43, and have been proposed to induce oesophageal trans-differentiation in Barrett’s esophagus44. However, the results derived from PICRUSt are predictive in nature and should be interpreted with caution.
This study is not without limitations. Despite the significant differences identified, this is a pilot study with a low number of subjects. Additional studies with a larger cohort of subjects are warranted. Further, it remains to be seen whether the changes in the gastric microbiome are involved in the progression of disease or are a consequence of disease.
In conclusion, the viable gastric microbiome appears to be significantly affected by H. pylori serological status and altered in gastric carcinogenesis. Our study identifies a number of potential mechanisms of interest such as enrichment of pro-inflammatory oral bacterial species, increased abundance of lactic acid producing bacteria, and enrichment of SCFA production pathways. Further investigations should focus on whether these changes are a cause or consequence of GC.
References
Ferlay, J. et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide. IARC CancerBase No. 11 [Internet]. Lyon, France: International Agency for Research on Cancer (2013).
Brenner, H., Rothenbacher, D. & Arndt, V. Epidemiology of stomach cancer. Methods Mol. Biol. 472, 467–477 (2009).
Goh, K. L., Cheah, P. L., Md, N., Quek, K. F. & Parasakthi, N. Ethnicity and H. pylori as risk factors for gastric cancer in Malaysia: A prospective case control study. Am. J. Gastroenterol. 102, 40–45 (2007).
Wroblewski, L. E., Peek, R. M. Jr. & Wilson, K. T. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin. Microbiol. Rev. 23, 713–739 (2010).
Lertpiriyapong, K. et al. Gastric colonisation with a restricted commensal microbiota replicates the promotion of neoplastic lesions by diverse intestinal microbiota in the Helicobacter pylori INS-GAS mouse model of gastric carcinogenesis. Gut 63, 54–63 (2014).
Aviles-Jimenez, F., Vazquez-Jimenez, F., Medrano-Guzman, R., Mantilla, A. & Torres, J. Stomach microbiota composition varies between patients with non-atrophic gastritis and patients with intestinal type of gastric cancer. Sci. Rep. 4, 4202 (2014).
Eun, C. S. et al. Differences in gastric mucosal microbiota profiling in patients with chronic gastritis, intestinal metaplasia, and gastric cancer using pyrosequencing methods. Helicobacter 19, 407–416 (2014).
Li, T. H. et al. Alterations in Gastric Microbiota After H. Pylori Eradication and in Different Histological Stages of Gastric Carcinogenesis. Sci. Rep. 7, 44935 (2017).
Wang, L. et al. Bacterial overgrowth and diversification of microbiota in gastric cancer. Eur. J. Gastroenterol. Hepatol. 28, 261–266 (2016).
Yu, G. et al. Gastric microbiota features associated with cancer risk factors and clinical outcomes: A pilot study in gastric cardia cancer patients from Shanxi, China. Int. J. Cancer 141, 45–51 (2017).
Yang, I. et al. Different gastric microbiota compositions in two human populations with high and low gastric cancer risk in Colombia. Sci. Rep. 6, 18594 (2016).
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K. & Schloss, P. D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120 (2013).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
Clarke, K. R. Non-parametric multivariate analyses of changes in community structure. Austral. J. Ecol. 18, 117–143 (1993).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60 (2011).
Afgan, E. et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 44, W3–W10 (2016).
Langille, M. G. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821 (2013).
Friedman, J. & Alm, E. J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 8, e1002687 (2012).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Mitchell, H. et al. Immunoblotting using multiple antigens is essential to demonstrate the true risk of Helicobacter pylori infection for gastric cancer. Aliment. Pharmacol. Ther. 28, 903–910 (2008).
Doherty, J. R. & Cleveland, J. L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Invest. 123, 3685–3692 (2013).
Kennedy, K. M. et al. Catabolism of exogenous lactate reveals it as a legitimate metabolic substrate in breast cancer. PLoS One 8, e75154 (2013).
Sonveaux, P. et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS One 7, e33418 (2012).
Sonveaux, P. et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Invest. 118, 3930–3942 (2008).
Rolhion, N. & Darfeuille-Michaud, A. Adherent-invasive Escherichia coli in inflammatory bowel disease. Inflamm. Bowel Dis. 13, 1277–1283 (2007).
Arthur, J. C. et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120–123 (2012).
Hieken, T. J. et al. The Microbiome of Aseptically Collected Human Breast Tissue in Benign and Malignant Disease. Sci. Rep. 6, 30751 (2016).
Kaakoush, N. O., Castano-Rodriguez, N., Man, S. M. & Mitchell, H. M. Is Campylobacter to esophageal adenocarcinoma as Helicobacter is to gastric adenocarcinoma? Trends Microbiol. 23, 455–462 (2015).
Li, S., Konstantinov, S. R., Smits, R. & Peppelenbosch, M. P. Bacterial Biofilms in Colorectal Cancer Initiation and Progression. Trends Mol. Med. 23, 18–30 (2017).
Chen, Y. et al. Invasive Fusobacterium nucleatum activates beta-catenin signaling in colorectal cancer via a TLR4/P-PAK1 cascade. Oncotarget 8, 31802–31814 (2017).
Tang, B. et al. Fusobacterium nucleatum-Induced Impairment of Autophagic Flux Enhances the Expression of Proinflammatory Cytokines via ROS in Caco-2 Cells. PLoS One 11, e0165701 (2016).
Yang, Y. et al. Fusobacterium nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-LikeReceptor 4 Signaling to Nuclear Factor-kappaB, and Up-regulating Expression of MicroRNA-21. Gastroenterology 152, 851–866 e824 (2017).
Castano-Rodriguez, N., Kaakoush, N. O., Goh, K. L., Fock, K. M. & Mitchell, H. M. The role of TLR2, TLR4 and CD14 genetic polymorphisms in gastric carcinogenesis: a case-control study and meta-analysis. PLoS One 8, e60327 (2013).
Castano-Rodriguez, N., Kaakoush, N. O., Goh, K. L., Fock, K. M. & Mitchell, H. M. Autophagy in Helicobacter pylori Infection and Related Gastric Cancer. Helicobacter 20, 353–369 (2015).
Castano-Rodriguez, N., Kaakoush, N. O. & Mitchell, H. M. Pattern-recognition receptors and gastric cancer. Front. Immunol. 5, 336 (2014).
Castano-Rodriguez, N. et al. Genetic polymorphisms in the Toll-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer. Human Immunol. 75, 808–815 (2014).
Geng, J. et al. Co-occurrence of driver and passenger bacteria in human colorectal cancer. Gut Pathog. 6, 26 (2014).
Guerrero-Preston, R. et al. 16S rRNA amplicon sequencing identifies microbiota associated with oral cancer, human papilloma virus infection and surgical treatment. Oncotarget 7, 51320–51334 (2016).
Yan, X. et al. Discovery and validation of potential bacterial biomarkers for lung cancer. Am J Cancer Res 5, 3111–3122 (2015).
Kaakoush, N. O., Castano-Rodriguez, N., Mitchell, H. M. & Man, S. M. Global Epidemiology of Campylobacter Infection. Clin. Microbiol. Rev. 28, 687–720 (2015).
Kaakoush, N. O., Mitchell, H. M. & Man, S. M. Role of emerging Campylobacter species in inflammatory bowel diseases. Inflamm. Bowel Dis. 20, 2189–2197 (2014).
Castano-Rodriguez, N., Kaakoush, N. O., Lee, W. S. & Mitchell, H. M. Dual role of Helicobacter and Campylobacter species in IBD: a systematic review and meta-analysis. Gut 66, 235–249 (2017).
Belcheva, A. et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell 158, 288–299 (2014).
Kaakoush, N. O. & Morris, M. J. The oesophageal microbiome: an unexplored link in obesity-associated oesophageal adenocarcinoma. FEMS Microbiol. Ecol. 92 (2016).
Acknowledgements
NCR is supported by a National Health and Medical Research Council, Australia Early Career fellowship (APP1111461) and Cancer Australia priority-driven collaborative cancer research grant (1129488). NOK is supported by a Cancer Institute NSW Career Development Fellowship (15/CDF/1-11). HMM acknowledges funding from the Ministry of Higher Education, Malaysia (UM.C/625/HIR/MOHE/CHAN/13/1) and from the University of New South Wales.
Author information
Authors and Affiliations
Contributions
K.L.G., K.M.F. and H.M.M. were involved in study subject recruitment and sample collection. H.M.M., N.C.R. and N.O.K. contributed reagents and funds. N.C.R. and N.O.K. performed the experimental work and microbiota analyses. N.O.K. and N.C.R. drafted the manuscript. N.C.R., K.L.G., K.M.F., H.M.M. and N.O.K. read, corrected and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Castaño-Rodríguez, N., Goh, KL., Fock, K.M. et al. Dysbiosis of the microbiome in gastric carcinogenesis. Sci Rep 7, 15957 (2017). https://doi.org/10.1038/s41598-017-16289-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16289-2
