
Abstract
Comprehensive characterization of cellular heterogeneity and the underlying regulatory landscapes of tissues and organs requires a highly robust and scalable method to acquire matched RNA and chromatin accessibility profiles on the same cells. Here, we describe a single-nucleus chromatin accessibility and mRNA expression sequencing 2 (SNARE-seq2) assay, implemented with cellular combinatorial indexing. This method involves tagmentation within permeabilized and fixed single-nucleus isolates to capture accessible chromatin (AC) regions, followed by the capture and reverse transcription of RNA transcripts. Through combinatorial split pool ligations, cDNA and AC within each single nucleus become appended with a common cell barcode combination. The captured cDNA and AC are then co-amplified before splitting and enrichment into single-nucleus RNA and single-nucleus AC sequencing libraries. This protocol is compatible with both nuclei and whole cells and can be completed in 3.5 d. SNARE-seq2 permits robust generation of high-quality, joint single-cell RNA and AC sequencing libraries from hundreds of thousands of single cells per experiment.
Similar content being viewed by others


Introduction
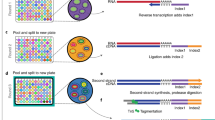
Significant progress in the development of next-generation sequencing and the decline in sequencing costs allow profiling of multiple-omics information from the genome1, transcriptome2,3,4,5,6,7,8,9 and epigenome (chromatin accessibility10,11,12,13,14 and DNA methylation15) within organisms at single-cell resolution. This progress also allows the capture of heterogeneity within the samples, especially in complex tissues such as brain, that could be missed by determining the average signals with traditional bulk analysis. RNA-seq at the single-cell level has been an effective approach for determining cellular functions from individual cell transcriptional states. With the development of these techniques, thousands to tens of thousands of transcriptomics data have been generated to define the molecular signatures of cellular populations and novel candidate cell subtypes in human postmortem brain4,16, mouse retina cells2, Caenorhabditis elegans5, developmental mouse brain and spinal cords9. Beyond transcriptomics profiling, it is also critical to define cell types by the upstream regulatory landscapes of gene expression associated with open or accessible chromatin (AC) regions. Single-cell chromatin accessibility assays can determine the chromatin status of DNA regulatory elements of individual cells. Studies in human brain16 and mouse kidney17 have relied on computational integration of transcriptome and chromatin accessibility assays to reveal the regulatory elements and transcription factors that underlie cell type–specific gene expression programs. Experimental joint profiling or ‘co-assay’ on the same cells or nuclei increases the sensitivity of rare-cell type detection over separate single-omics assays. We recently reported a single-cell dual-omics assay (single-nucleus chromatin accessibility and mRNA expression sequencing (SNARE-seq))18 and demonstrated the power of joint RNA/AC analysis at the single-cell level on identifying cell type–specific regulatory elements, especially on low-abundance cells. SNARE-seq was implemented with a micro-droplet platform, similar to Drop-seq2, and allowed us to generate informative data from several thousand cells per experiment. Because the community is moving toward the generation of multi-omics cell maps on various organs and tissues, a method with a higher scalability and comparable data quality is desirable. Here, we present an improved protocol, called ‘SNARE-seq2’ (Fig. 1; Extended Data Fig. 1 for SNARE-seq2 sequencing library generation details), that uses combinatorial indexing to increase the throughput by 10- to 100-fold and permits processing of multiple samples in the same batch to reduce technical variations. This scalable protocol enables the characterization of individual cells in a broad range of samples and has been implemented in a production pipeline for the construction of dual-omics single-cell atlases for frozen human brain, kidney and lung19.

Inside the permeabilized nuclei or cells, the AC is tagmented and ligated with AC Round 1 barcoded oligos while mRNA is primed with RT Round 1 barcoded reverse transcription primers. The Round 2 and Round 3 barcoded oligos are ligated to AC DNA and cDNA after two rounds of ligation reactions and split-pooling. The final split is to four to eight different pools. After cell/nucleus lysis, AC DNA and cDNA-mRNA hybrid molecules are captured, template switching is blocked on AC DNA and then template switching is performed on cDNA. Both types of molecules are co-amplified in the first-round PCR. The resulting PCR products are split into AC and RNA parts for AC DNA and RNA sequencing library generation. Sequencing libraries are sequenced on Illumina sequencers and analyzed. UMI, unique molecular identifier.
Development of the protocol
As a direct extension of our published SNARE-seq method, SNARE-seq2 uses a ligation-based combinatorial indexing scheme9 to jointly profile mRNA and chromatin accessibility in the same cells or nuclei. We chose ligation-based combinatorial indexing over droplet-based multiplexing because of the simplicity of experimental setup, scalability and flexibility in multi-sample processing. To accomplish this, we first optimized formaldehyde fixation and cross-linking on both whole cells and nuclei. We then identified the optimal Tn5 concentration from transposase titration on a large number of cells or nuclei in a single reaction (150,000 cells or nuclei/reaction) using the lymphoblastoid cell line GM12878. The results of tagmentation on formaldehyde-fixed samples and Tn5 titration were compared with the conventional bulk ATAC-seq20 on non-fixed nuclei (Supplementary Fig. 1). Next, we designed AC Round 1 barcoded oligos and AC Round 1 linker (Fig. 2a) that could hybridize with tagmented DNA at the Nextera adapter 2 that becomes appended to tagmented DNA during transposition. These were also designed to anneal with Round 2 barcode linker sequences at the 5′ end of AC Round 1 barcoded oligos the same way as reverse transcription (RT) Round 1 barcoded oligos (Fig. 2b). To capture mRNA, we used both oligo dT (dT) and random hexamer (N6) RT Round 1 barcoded oligos to capture polyadenylated and truncated mRNA (Fig. 2b), respectively. N6 RT oligos are expected to improve unique molecular identifier (UMI) counts per cell, especially for cases when the poly (A) tails of mRNA become degraded during sample storage or processing. We performed tagmentation using non-indexed Tn5 transposase to fragment AC of individual samples in bulk (pooled nuclei) to make the workflow simple and minimize the potential technical variability that could arise from multiple tagmentation reactions with separate Tn5 complexes. After AC Round 1 barcode ligation to tagmented DNA and RT of mRNA with RT Round 1 barcoded oligos, nuclei or cells were pooled and carried through two additional rounds of barcode ligation (Fig. 1). P5 ends of cDNA and AC DNA were designed differently (Fig. 2c). After co-amplification of the two types of libraries in the first PCR, we split the PCR products into two portions for enrichment and generation of AC and RNA sequencing libraries. By this approach, AC and RNA libraries sharing the same cell barcodes are generated, without requiring any subsampling of the original nucleus material, and at a cost of ~21 cents per cell or nucleus for library generation and sequencing (Fig. 1 and Supplementary Table 1). This protocol is further highly flexible on throughput, which can be scaled by increasing Round 1 unique cell barcodes (such as from 48 to 96 or 192 and then increasing the concentrations of enzymes and barcoded oligos in Round 2 and Round 3 barcoding plates to accommodate the higher nuclei concentration). Using SNARE-seq2, we have successfully generated high-quality data on >80,000 single nuclei from human motor cortex in a single batch while observing >80% shared cell barcodes between RNA and AC libraries (Table 1)21.

a, Ligation of AC Round 1 barcoded oligos and AC Round 1 linker oligos with tagmented DNA at Nextera adapter 2. b, Reverse transcription (RT) Round 1 barcoded oligos priming mRNA. c, Blocking of template-switching oligos on an AC DNA molecule and template-switching oligo on a cDNA molecule.
Application of the method
The effects of regulatory changes can be inferred only computationally when analyzing chromatin accessibility data and gene expression data that are generated from separate cells. Thus, an important application for dual-omics gene expression and chromatin accessibility profiling is the ability to directly measure regulatory changes that result in cell fate changes in response to a stimulus or during disease progression. Having a more robust and scalable approach can facilitate not only data generation for samples collected at different time points but also minimize the various technical variabilities that can cause further challenge in downstream analysis and integration.
In addition, the workflow of SNARE-seq2 can be extended to capture different types of molecules in cells or nuclei with some modifications and optimizations. A simple workflow could allow measuring proteins (via oligo-conjugated antibodies22) and mRNA or proteins and accessible DNA in parallel by using ligation-based combinatorial indexing instead of a droplet-based approach2 that has technical challenges limiting scalability (fixed barcode space with pre-produced beads, equipment setup and clogging issues with increased emulsion volume). In addition to capturing the whole transcriptome, this protocol can be adopted to capture specific mRNA targets using gene-specific RT primers.
Comparison with other methods
High-throughput assays that perform joint profiling of transcriptome and AC on the same cell rely on two main barcoding approaches, droplet-based sequencing and combinatorial indexing. The first high-throughput co-assay, sci-CAR17, uses a combinatorial indexing method to capture mRNA with well-specific barcodes together with chromatin accessibility using indexed Tn5 transposase on the same nucleus. One limitation of this strategy is sparsity of the chromatin accessibility data, because only half of the captured molecules are used to generate RNA or AC. SNARE-seq18 overcomes this by using Tn5 transposase to tag open chromatin in nuclei before generating nanoliter droplets. In these nanoliter reactions, mRNA and tagmented DNA that are ligated with splint oligonucleotides are both captured by uniquely barcoded primer beads. However, the lack of sample multiplexing and scalability are the major limitations of this protocol, with a dependency on equipment setup for droplet generation and the need to run samples one at a time. Paired-seq23 uses five levels of combinatorial indexing to provide up to 10 million theoretical cellular barcode combinations, which enables this protocol to generate a million datasets per batch. In comparison, SNARE-seq2 uses only four levels of combinatorial indexing (Supplementary Fig. 2a). Although SNARE-seq2 and Paired-seq both rely on similar molecular biology for cellular combinatorial indexing, they do differ in the molecular method connecting mRNA and AC information from the same cells. In Paired-seq, cells are first divided into aliquots in tubes for tagmentation with indexed transposomes, and then each tube of cells must be pelleted and resuspended into RT reactions with the corresponding barcoded primers for connecting mRNA and chromatin. SNARE-seq2 does not require indexed transposons to initially barcode the AC. Instead, following tagmentation, cells can be pooled and split to Round 1 barcoding reactions that contain both barcoded Round 1 adapters for chromatin and barcoded Round 1 primers for mRNA. SNARE-seq2 uses non-indexed Tn5 transposase to fragment AC of individual samples in a single reaction setup to ensure a simpler workflow and to minimize any potential differences in transposase activity that could arise from separately loading Tn5 with different indexed transposons. Cell and nuclei fixation and permeabilization were further optimized in SNARE-seq2 to best preserve the original chromatin conformation and maximize the capture of mRNA.
Overall, the SNARE-seq2 protocol was designed to be scalable and compatible with automation that allows measuring half a million whole cells or nuclei in a single batch. This method has demonstrated high-quality data sets from the brain21, including data on ~170,000 single nuclei from the primary motor cortices of humans and marmosets (http://biccn.org/data), and is contributing to ongoing efforts to profile the cellular composition of the adult human kidney and lung (https://hubmapconsortium.org/).
Experimental design
Nuclei isolation
If fresh tissue is not available, or dissociation of whole live cells is not feasible, then nuclei can be isolated from frozen archived tissues using a protocol that is compatible with both single-nucleus RNA-seq and single-nucleus ATAC-seq. For cell culture samples, single-cell dissociation can be done; thus, nuclei extraction is not required. Standard nuclei extraction described in the ATAC-seq protocol20 can also be used with the addition of RNase inhibitor to isolate nuclei from cell line samples if nuclei samples are required. Different tissues might require their individually optimized nuclei isolation protocols. Currently, we have optimized protocols available for a number of tissues, including brain (https://doi.org/10.17504/protocols.io.8tvhwn6), kidney (https://doi.org/10.17504/protocols.io.ufketkw) and lung (https://doi.org/10.17504/protocols.io.bh26j8he). During nuclei extraction, samples must be handled in cold buffer and kept on ice to protect RNA from degradation.
Formaldehyde fixation
For this protocol, nuclei or cells need to go through multiple rounds of pooling, splitting and incubating in reactions for ≥5 h. As such, formaldehyde fixation is required to keep all materials to be captured inside the nucleus or cell. Formaldehyde used for nucleus/cell fixation must be methanol free. We recommend using formaldehyde stock solution in 1× PBS. It must be freshly prepared just before fixation is performed; thus, we recommend using a formaldehyde stock that is stored in vacuum ampules and using a new ampule for every experiment. We have tested formaldehyde at different concentrations on the lymphoblastoid cell line GM12878 as either whole cells or nuclei and found that 0.5% (wt/vol) formaldehyde is an optimal concentration for nuclei fixation. This concentration must be high enough to keep nuclei intact but must also show the least negative effects on chromatin accessibility. For cell samples, 1% (wt/vol) formaldehyde was found to be the optimal concentration, because lower concentrations were not sufficient to keep the cell membrane, which is more fragile than the nuclear membrane, intact. We have applied these concentrations of formaldehyde for nucleus and cell fixation to frozen human and marmoset brain tissues.
ATAC-seq on fixed nuclei and cells
We initially determined the effects of different formaldehyde concentrations on chromatin accessibility by comparing bulk ATAC-seq on fixed nuclei or cells to a standard ATAC-seq without fixation on the same cell type (Box 1). The fraction of reads in peaks and transcription start site (TSS) enrichment scores were determined to compare the effect of different formaldehyde concentrations to non-fixed nuclei (Supplementary Fig. 1a). We found that increasing formaldehyde concentration reduces TSS enrichment score. We also validated the consistency of peak calling between technical and biological replicates of fixed nuclei against reference data of GM12878 from Omni-ATAC-seq24 (Supplementary Fig. 1b).
Ligation of AC Round 1 barcoded oligos to tagmented DNA
After in-nucleus tagmentation, DNA flanked with Nextera adapter 1 and Nextera adapter 2 is produced that can be used to generate sequencing libraries (Fig. 2a). We designed AC Round 1 barcoded oligos (Fig. 2a) that contain: (i) linker sequences on the 5′ end that are used to anneal with Round 2 linker sequences, (ii) AC Round 1 barcode sequences identical to the one used for dT RT Round 1 barcoded oligos and (iii) constant sequences of 24 bp at the 3′ end of AC Round 1 barcoded oligos that were designed to anneal with the 3′ end of AC Round 1 linker oligos (Fig. 2a). Next to 24 bp of constant sequences of AC Round 1 linker oligo, we designed reverse complementary sequences to Nextera adapter 2 (without mosaic end (ME)) sequences at the 5′ end. After annealing of Nextera adapter 2 of the tagmented DNA molecule and AC Round 1 linker oligo, the 5′ end of Nextera adapter 2 must be joined to the 3′ end of the AC Round 1 barcoded oligo; thus, Nextera adapter 2 requires a phosphate group on the 5′ end (Fig. 2a). To prevent polymerization at the 3′ end of the AC Round 1 linker molecule by reverse transcriptase during RT, the modified base 3′ inverted dT (3InvdT) was added to the 3′ end of the AC Round 1 linker molecule during oligo synthesis (Fig. 2a).
Single-cell AC library construction
After tagmentation, cell barcode ligation and cell/nuclei lysis, AC DNA and cDNA-mRNA hybrid molecules were captured on streptavidin beads. The cellular barcode region of the AC DNA and the cDNA at the 5′ end (assigned to the P7 end of the sequencing library structure) are the same (Supplementary Fig. 3). To selectively enrich the AC DNA or the cDNA in the second round of PCR, we designed the AC DNA and cDNA to have different P5 ends, allowing for the use of different primers. This was done by dividing the first-round PCR product for RNA or AC enrichment in the second round of PCR. The cDNA had the template switching oligo (TSO) on the P5 end9, while the AC DNA had Nextera adapter 1 (Fig. 2c). To prevent the addition of TSO to the P5 end of the AC DNA, we add a Nextera adapter 1 blocker oligo before the addition of RNase H minus reverse transcriptase, which is needed to complete RT of mRNA. This blocker oligo, which contains a 3InvdT at the 3′ end (Fig. 2c), works by preventing the addition of extra cytosines to the AC DNA. As a result, the 3′ end of the Nextera adapter 1 complementary strand (Nextera adapter 1 blocker) on AC DNA cannot be primed by rGrG+G of TSO (Fig. 2c). Blocked AC DNA can be selectively amplified by the Nextera adapter 1 sequence but not by the TSO_PCR primer specific for cDNA in first-round PCR. If template switching occurs on any unblocked AC DNA, the Nextera adapter 1 sequence can still be used to positively select for the AC DNA. Therefore, the presence of TSO sequences next to Nextera adapter 1 would not inhibit amplification of the AC DNA in the second round of PCR. However, the cDNA libraries will be contaminated with the AC DNA because AC DNA with TSO sequences will be amplified with cDNA by the TSO_PCR primer in the second-round PCR. After the Nextera tagmentation and barcoding of cDNA, both (RNA and AC products) will have the same structure at the P5 end (Supplementary Fig. 3b). Even if this occurs, the AC contaminants can be detected and filtered from the RNA sequencing reads by removing those reads containing AC Round 1 linker sequences next to the cellular barcode region. Although this reduces the fraction of RNA reads in RNA libraries, we found that blocking of TSOs on AC DNA is sufficient to limit the contamination of RNA libraries with AC DNA to <5% (Table 2). AC DNA is positively selected and barcoded by the PCR handle on the 3′ end (Extended Data Fig. 1) with AC-indexed PCR primer (Ad1_N5XX) that primes Nextera adapter 1 and contains a P5 sequence and P5 index (i5) (Extended Data Fig. 1) that serve as the fourth barcodes of each individual pool of the libraries. These AC libraries show nucleosome patterns that are larger than standard ATAC-seq (Supplementary Fig. 4a), because they contain extra sequences of cellular barcode, linker and constant sequences from AC Round 1 barcoded oligos of ~125 bp (Supplementary Fig. 4b).
Single-cell RNA sequencing library construction
RNA sequencing libraries are processed along with AC DNA libraries through the first-round PCR to co-amplify AC DNA and cDNA as described above. After splitting the first-round PCR products into two equal parts, cDNAs are selectively enriched over AC DNA in the second-round PCR using the TSO_PCR primer specific to cDNA but not template switch–blocked AC DNA. After that, RNA sequencing libraries are generated the same way as described9. Briefly, second-round PCR cDNA product is tagmented and amplified with the Nextera adapter 1 PCR handle to add a P5 sequence and with indexed P7 PCR primer (SPLiT_N7XX) that contains a P7 sequence and a P7 index (i7) (Extended Data Fig. 1) to target the barcoded 5′ ends of cDNA.
Spike-in controls
For this protocol, cell line samples show consistent results based on the success of sequencing library generation and data quality, but the mapping statistics such as UMI/gene counts per nucleus and the fraction of reads in peaks differ on the basis of cell type (Supplementary Fig. 5). When we perform the experiment on a specific tissue, either fresh or frozen, the sample quality could vary by RNA quality and chromatin integrity. These factors depend on how the tissues were collected and preserved before running the experiment. To determine the success of an experiment, we recommend including one or two wells of positive spike-in control cell lines (such as GM12878 or NIH/3T3 cells) that consistently show good-quality data. In addition, after tagmentation, an aliquot of tagmented cells/nuclei (5,000–20,000 cells/nuclei) of each sample can be taken as a bulk control for evaluation of the tagmentation step for the very first trial experiments (Box 1). For RNA, the RNA integrity number (RIN) for each sample should be determined as a bulk control of initial sample quality. For the entire procedure, we expect to see consistent results from both RNA and AC libraries from spike-in controls. If we observe poor data quality for the samples, but good data quality for the controls, this implies poor quality of the samples. On the other hand, if we observe poor data quality for both the samples and the spike-in control data, this implies technical issues during the experiment.
Expertise needed to implement the protocol
This protocol involves the generation of single-cell/nucleus RNA and AC libraries from various types of samples, fresh or frozen tissues and cell lines. Fresh and frozen tissue collection and preservation must be performed appropriately to minimize RNA degradation and maintain native chromatin structure. Single-nucleus or cell isolation techniques must be practiced to maintain cell/nuclei viability, to reduce clumping and to ensure a limited number of doublets present in the samples. The key steps of SNARE-seq2 involve mixing, pooling and distributing nuclei or cells with reaction mixture and unique cell barcodes in individual wells of the 96-well plate. Pipetting with a multichannel pipette (ideally, an electronic pipette) is required to make sure that the reactions are mixed homogeneously, to ensure that the reactions are performed under optimized conditions and to avoid sample and cell barcode cross-contamination.
Limitations
SNARE-seq2 allows processing of multiple samples within the same batch by seeding different samples into specific Round 1 barcode wells on the same 96-well plate, and then pooling and splitting into the next rounds of barcode ligation. As a result, it does not allow the selective sequencing of specific samples from the batch. This can become an issue when not all samples in the same batch have the same priorities or quality (e.g., tumor tissues versus normal tissues and samples with high versus low RIN). This limits the flexibility of subsampling of sequencing libraries for high-throughput sequencing. We therefore recommend co-processing samples of the same studies with the same priority or to separately process samples with different priorities on different plates.
When the quantity of RNA in a sample is known to be low (RIN value <8 or based on RNA integrity from gel electrophoresis or bioanalyzer trace), that sample may not be suitable for this assay for the following reasons: (i) it may be impossible to separate cDNA libraries from AC libraries or (ii) the resulting UMI/gene counts may become too limiting for answering most biological questions. For these reasons, samples from a CRISPR screen experiment that requires capture of rare guide RNAs to determine cellular genotypes would not be suitable.
Determination of experimental failures at an earlier time point during the 3.5-d SNARE-seq2 protocol is difficult. ATAC-seq on bulk samples could be useful for validation, but it takes ≥3 h to be able to observe nucleosome patterns. In addition, nucleosome patterns alone are not sufficient to represent the quality of bulk ATAC-seq data and sequencing, and data analysis is required to confirm a successful ATAC-seq experiment. We could, however, verify the success of tagmentation from nucleosome patterns in PAGE (Supplementary Fig. 4b) and the quality of RNA as a control for input samples by RIN. However, transcription and barcode ligation could not be verified until cDNA and AC DNA are amplified, enriched and validated by gel electrophoresis on day 2 of the experiment. The quality of the data could be determined after sequencing with some clues from gel electrophoresis and the yields of cDNA and AC DNA from the first and second rounds of PCR. In the cases when the assay was applied to multiple samples, only the overall success of the experiment could be validated by gel electrophoresis. It does not represent the success of each individual sample in terms of data quality. They must be sequenced, demultiplexed on the basis of Round 1 barcodes and assessed for quality of both RNA and AC libraries for each of the individual samples. Therefore, we routinely perform shallow sequencing of the RNA and AC libraries on an Illumina MiSeq instrument to determine the quality of these libraries before deeper sequencing on an Illumina NovaSeq instrument.
Materials
Biological materials
-
Human primary motor cortex nuclei were isolated according to the protocol described at https://www.protocols.io/view/nuclei-isolation-for-snare-seq2-8tvhwn6. Postmortem human brain tissue was collected under permission from decedent next of kin. Postmortem tissue collection was performed in accordance with the provision of the United States Uniform Anatomical Gift Act of 2006 described in the California Health and Safety Code section 7150 (effective 1/1/2008) and other applicable state and federal laws and regulations
-
GM12878 cell line (Coriell Institute, cat. no. GM12878; RRID: CVCL_7526) was grown in RPMI-1640 culture medium supplemented with 15% (vol/vol) FBS and 2 mM l-glutamine at 37 °C in a 5% CO2 humidified incubator
-
A549 cell line (American Type Culture Collection (ATCC), cat. no. CCL-185; RRID: CVCL_0023) was grown in F-12K medium supplemented with 10% (vol/vol) FBS at 37 °C in a 5% CO2 humidified incubator
-
NIH/3T3 (ATCC, cat. no. CRL-1658; RRID: CVCL_0594) was grown in DMEM supplemented with 10% (vol/vol) FBS at 37 °C in a 5% CO2 humidified incubator
Caution
The cell lines used in your research should be regularly checked to ensure that they are authentic and are not infected with mycoplasma.
Reagents
Critical
All reagents must be kept nuclease free.
-
EZ-Tn5 transposase (Lucigen, cat. no. TNP92110)
-
UltraPure DNase/RNase-free water (Thermo Fisher Scientific, cat. no. 10977015)
-
PEG 6000 (Sigma-Aldrich, cat. no. 81255-1KG)
-
Tris-HCl (1 M), pH 8.0 (Thermo Fisher Scientific, cat. no. 15568025)
-
Tris-HCl (2 M), pH 7.5 (Sigma-Aldrich, cat. no. T2319-100ML)
-
KCl (2 M) (Thermo Fisher Scientific, cat. no. AM9640G)
-
MgCl2 (1 M) (Sigma-Aldrich, cat. no. M1028-100ML)
-
IGEPAL CA-630 (used to replace NP-40) (Sigma-Aldrich, cat. no. I8896-100ML)
-
Pierce formaldehyde (16% (wt/vol)), methanol-free (Thermo Fisher Scientific, cat. no. 28906)
Caution
Formaldehyde is a carcinogenic, mutagenic and corrosive material. Contact with formaldehyde can cause severe irritatation and burn the skin, eyes, nose, mouth and throat. Inhaling formaldehyde can irritate the lungs. Formaldehyde must be handled with extreme caution.
Critical
We recommend using methanol-free Pierce formaldehyde (16% (wt/vol)) solution in 1× PBS in 1-ml ampules from Thermo Fisher Scientific (methanol can affect chromatin structure). Each 1-ml ampule of formaldehyde is for a single-use preparation of freshly diluted formaldehyde solution for nuclei or cell fixation.
-
PBS (1×), pH 7.4 (Thermo Fisher Scientific, cat. no. 10010023)
-
Bovine Albumin Fraction V, 7.5% (wt/vol) solution (BSA; Thermo Fisher Scientific, cat. no. 15260037)
-
RPMI 1640 medium (Thermo Fisher Scientific, cat. no. 11875093)
-
F-12K medium (ATCC, cat. no. ATCC 30-2004)
-
DMEM (Thermo Fisher Scientific, cat. no. 11995065)
-
FBS (Thermo Fisher Scientific, cat. no. 16141079)
-
Tango Buffer (10×) (Thermo Fisher Scientific, cat. no. BY5)
-
N,N-Dimethylformamide (DMF; Sigma-Aldrich, cat. no. 227056)
Caution
DMF is easily absorbed through the skin and can cause liver damage. Avoid skin contact.
-
Advantage UltraPure dNTP combination kit (100 mM each dNTP) (CloneTech, cat. no. 639132)
Critical
We recommend using a solution mixture of 25 mM of each dNTP (made by combining 100 mM of each dNTP) because of the limitation of reaction mixture volume.
-
SUPERase In RNase inhibitor (20 U/µl) (Thermo Fisher Scientific, cat. no. AM2696)
-
Enzymatics RNase inhibitor (40 U/µl) (Enzymatics, cat. no. Y9240L)
-
ATP solution (100 mM) (Thermo Fisher Scientific, cat. no. R0441)
Critical
100 mM ATP solution must be used instead of a 10 mM solution because the reaction volume is limiting.
-
DL-DTT (DTT; Sigma-Aldrich, cat. no. 10708984001)
Caution
DTT is harmful if swallowed. It causes skin and eye irritation and may cause respiratory irritation. Avoid breathing dust, fume, gas, vapors or spray.
-
SDS solution (10% (wt/vol)) (Thermo Fisher Scientific, cat. no. AM9822)
-
T7 DNA ligase (3,000 U/µl) (NEB, cat. no. M0318L)
-
Maxima H Minus reverse transcriptase (200 U/µl) (Thermo Fisher Scientific, cat. no. EP0753)
-
NEBuffer 3.1 (10×) (NEB, cat. no. B7203S)
-
BSA (20 mg/ml), molecular biology grade (NEB, cat. no. B9000S)
-
T4 DNA ligase (400 U/µl) (NEB, cat. no. M0202L)
-
T4 DNA ligase reaction buffer, 10× (NEB, cat. no. B0202S)
-
EDTA (0.5 M), pH 8.0 (Sigma-Aldrich, cat. no. 20-158)
-
Triton X-100 (Sigma-Aldrich, cat. no. X100-100ML)
-
Proteinase K solution (20 mg/ml), RNA grade (Thermo Fisher Scientific, cat. no. 25530049)
Critical
We recommended using RNA grade proteinase K from this supplier.
-
NaCl solution, 5 M (Sigma-Aldrich, cat. no. 71386-1L)
-
Tween 20 (Sigma-Aldrich, cat. no. P9416-100ML)
-
PMSF (Sigma-Aldrich, cat. no. P7626-5G)
Caution
PMSF is extremely corrosive and destructive to tissues, especially mucous membranes of the respiratory tract, eyes and skin. It may cause irreversible eye damage. Avoid inhaling, swallowing or absorbing on skin.
-
Dynabeads MyOne streptavidin C1 (Thermo Fisher Scientific, cat. no. 65001)
-
SSC Buffer (20× concentrate) (Sigma-Aldrich, cat. no. S6639)
-
Ficoll solution Type 400 (20% (wt/vol) in water) (Sigma-Aldrich, cat. no. F5415-50ML)
-
Hemo KlenTaq (NEB, cat. no. M0332L)
Critical
We recommend using Hemo KlenTaq from NEB for the gap-filling step on AC DNA because this polymerase does not contain strand displacement activity.
-
KAPA HiFi HotStart ReadyMix (2×) (KAPA Biosystems, cat. no. KK2602)
-
NEBNext high-fidelity PCR master mix (2×) (NEB, cat. no. M0541S)
-
KAPA pure beads (KAPA Biosystems, cat. no. KK8002)
-
Nextera XT DNA library prep kit (Illumina, cat. no. FC-131-1096)
-
Nextera DNA library prep kit (Illumina, cat. no. FC-121-1030)
-
Novex TBE gel 6%, 10 well (Thermo Fisher Scientific, cat. no. EC6265BOX)
-
SYBR Green I nucleic acid gel stain (10,000× concentrate in dimethyl sulfoxide) (Thermo Fisher Scientific, cat. no. S7563)
-
SYBR Gold nucleic acid gel stain, (10,000× concentrate in dimethyl sulfoxide) (Thermo Fisher Scientific, cat. no. S11494)
-
Sodium acetate (3 M), pH 5.5 (Thermo Fisher Scientific, cat. no. AM9740)
-
Tris-EDTA buffer (50×) (Fisher Scientific, cat. no. AAJ75834AP)
-
2-Propanol or isopropanol, for molecular biology (Sigma-Aldtrich, cat. no. I9516-500mL)
-
Ethanol, pure 200 proof, for molecular biology (Sigma-Aldtrich, cat. no. E7023-500mL)
-
Glycoblue coprecipitant (15 mg/ml) (Thermo Fisher Scientific, cat. no. AM9515)
-
Qubit double-stranded (ds) DNA HS assay kit (Thermo Fisher Scientific, cat. no. Q32854)
-
DNA Clean & Concentrator-5 (capped) (ZymoResearch, cat. no. D4014)
-
Low DNA mass ladder (Thermo Fisher Scientific, cat. no. 10068013)
-
TBE buffer (10×) (Thermo Fisher Scientific, cat. no. 15581044)
-
Oligonucleotides and primers (Integrated DNA Technologies)
-
MiSeq reagent kit v2 (300 cycles) (Illumina, cat. no. MS-102-2002)
-
MiSeq reagent kit v3 (150 cycles) (Illumina, cat. no. MS-102-3001)
Equipment
-
1.5-, 2- and 5-ml DNA LoBind tubes (Eppendorf, cat. nos. 22431021, 30108078 and 30108310)
-
0.2-ml eight-strip PCR tube, without cap (VWR, cat. no. 20170-002)
-
0.2-ml eight-strip PCR tube, with individually attached bubble (VWR, cat. no. 53509-304)
-
15-ml Corning polypropylene tube (Fisher Scientific, cat. no. 0553859B)
-
50-ml Corning polypropylene tube (Fisher Scientific, cat. no. 0553868)
-
Twin.tec 96-well LoBind PCR plate, semi-skirted (Eppendorf, cat. no. 30129504)
-
25-ml reservoir for eight-channel pipettes (individually wrapped) (Olympus Plastics, cat. no. 28-132)
-
Microseal ‘B’ PCR plate-sealing film, adhesive, optical (Bio-Rad, cat. no. MSB1001)
-
Qubit assay tubes (Thermo Fisher Scientific, cat. no. Q32856)
-
Eppendorf ThermoMixer C with Thermo top (Eppendorf, cat. no. 2231000574)
-
IKA MS3 digital orbital shaker, with MS 1.32 tube insert (Coleparmer, cat. no. UX-04304-04)
-
Tube revolver/rotator (Thermo Fisher Scientific, cat. no. 88881001)
-
DynaMag-2 magnet for 1.5-ml microtube (Thermo Fisher Scientific, cat. no. 12321D)
-
0.2-ml PCR strip/1.5-ml Microfuge magnetic separator (Permagen Labware, SKU: MSR1224B)
-
Millex-GP syringe filter unit, 0.22 µm (Merck Millipore, cat. no. SLGP033RS)
-
Pall Nanosep 0.2-µm column (Pall Corporation, cat. no. ODM02C35)
-
MiSeq (Illumina)
-
Qubit 3.0 fluorometer (Thermo Fisher Scientific, cat. no. Q33216)
-
E1-ClipTip multichannel pipette, 12 channel, 0.5–12.5, 1–30 and 2–125 µl (Fisher Scientific, cat. nos. 14-387-972TI, 14-387-973TI and 14-387-974TI)
-
ClipTip 384 12.5-, 30- and 125-µl multichannel pipette tip, racked, filter, sterile (Thermo Fisher Scientific, cat. nos. 94420053, 94420103 and 94420153)
-
Refrigerated centrifuge (Eppendorf)
-
Bench top centrifuge (Eppendorf)
-
mySPIN minicentrifuge (Thermo Fisher Scientific, cat. no. 75004061)
-
CFX96 Touch deep-well real-time PCR detection system (Bio-Rad)
-
T100 Thermo cycler (Bio-Rad, cat. no. 1861096)
-
TC20 automated cell counter (Bio-Rad, cat. no. 1450102)
-
Dual-chambered counting slide (Bio-Rad, cat. no. 145-0011)
-
Eppendorf PCR-cooler (Eppendorf, cat. no. 022510525)
-
XCell SureLock mini-cell electrophoresis system (Thermo Fisher Scientific, cat. no. EI0001)
-
UV transilluminator
-
Scalpel (Integra, cat. no. 4-410)
-
CellTrics 30-µm strainer (Fisher Scientific, cat. no. NC9682496)
Software
-
Linux/Unix operating system that is a Debian-based distribution such as Ubuntu with Python 2.7 installed
-
Samtools25 (https://github.com/samtools/samtools/releases/tag/1.10)
-
Cutadapt26 (https://github.com/marcelm/cutadapt)
-
deindexer (https://github.com/ws6/deindexer)
-
dropEst27 (https://github.com/hms-dbmi/dropEst)
-
Minimap228 (https://github.com/lh3/minimap2)
-
snapTools30 (https://github.com/r3fang/SnapTools)
Reagent setup
40% (wt/vol) PEG 6000
Weigh 16.0 g of PEG 6000 in a 50-ml tube. Add nuclease-free water and bring the total volume to 40 ml. Rotate the tube at room temperature until PEG 6000 is completely dissolved. Spin down the tube at 200g for 2 min at room temperature to remove tiny bubbles. Continue to prepare 4× GLR buffer.
Critical
40% (wt/vol) PEG 6000 is very viscous and difficult to filter through a 0.22-µm filter. We suggest preparing 40% PEG 6000 freshly before making 4× GLR buffer. When PEG is diluted in 4× GLR buffer, it is easier to filter.
4× GLR buffer
To prepare 40 ml of 4× GLR buffer, add 2.64 ml of nuclease-free water, 10.56 ml of 1 M Tris-HCl (pH 8.0), 0.8 ml of 1 M MgCl2 and 4 ml of 2 M KCl into a 50-ml tube. Gently mix well by vortexing. Add 22 ml of 40% (wt/vol) PEG 6000 and gently mix well by vortexing. Filter through a 0.22-µm filter into a new 50-ml tube and briefly spin the tube at room temperature for 30 s. Place 1.8-ml aliquots into 2-ml tubes to minimize contamination from each use and store at 4 °C for ≤1 year.
10% (vol/vol) Triton X-100
Slowly aspirate 2.0 ml of Triton X-100 liquid with a low-retention pipette tip and slowly dispense into 18.0 ml of nuclease-free water in a 50-ml tube. Dissolve Triton X-100 by slowly rotating the tube until the solution is clear. Filter 10% (vol/vol) Triton X-100 solution through a 0.22-µm syringe filter into a new 50-ml tube and store at 4 °C for ≤1 year.
Critical
If it is difficult to pipette Triton X-100 accurately because it is a viscous liquid, you may warm it at 37 °C before pipetting.
10% (vol/vol) Tween 20
Tween 20 is a very viscous liquid and difficult to pipette accurately. We convert the volume in cubic centimeters into grams using the density of Tween 20 at 25 °C, which is 1.1 g/cm3. To prepare 20 ml of 10% (vol/vol) Tween 20, weigh 2.2 g of Tween 20 in a 50-ml tube. Add 18.0 ml of nuclease-free water and invert or rotate the tube slowly at room temperature until Tween 20 is completely dissolved in water. Filter the 10% (vol/vol) Tween 20 solution through a 0.22-µm syringe filter into a new 50-ml tube and store at room temperature for 1 year.
Transposon preparation
Resuspend Nextera adapter 1, 5P-Nextera adapter 2 and ME oligos with nuclease-free water to 100 µM. Mix 500 µl of 100 µM Nextera adapter 1 and 500 µl of 100 µM ME in a 1.5-ml microtube, place a 30-µl aliquot of non-annealed transposons into each 0.2-ml PCR tube and store at −20 °C for ≤1–2 years. Prepare 5P-Nextera adapter 2 and ME oligos the same way as Nextera adapter 1. All sequences of oligos can be found in Table 3.
Loading Tn5 transposase (Tn5) with transposons
-
1
Convert Tn5 transposase (expressed/purified using the Sandberg protocol26) or EZ-Tn5 transposase (Lucigen #TNP92110) concentration from milligrams per milliliter to µM units. For example, if the concentration of unloaded Tn5 is 0.40 mg/ml, the monomer concentration in µM is 7.55 µM based on the molar mass of monomer Tn5 (~53,000 g/mol; 0.4 mg/ml × (1 mol/53,000 g)).
-
2
Load transposons at 1.5× molar ratio to Tn5 transposase. The volume of each transposon is calculated by ([7.55 µM monomer Tn5 × 75 µl of monomer Tn5 × (1.5/50 µM transposons)]/2 = ~8.5 µl).
-
3
Thaw non-annealed transposons (Nextera adapter 1/ME and 5P-Nextera adapter 2/ME) on ice. Briefly vortex and quick-spin the tubes. Anneal transposons on a thermocycler using the following program: 95 °C for 5 min, slowly cool down to 65 °C (0.1 °C/s), 65 °C for 5 min, slowly cool down to 4 °C (0.1 °C/s) and hold at 4 °C. The concentration of each transposon after annealing is 50 µM.
-
4
To load Tn5 with transposons, add 8.5 µl of 50 µM annealed Nextera adapter 1/ME and 8.5 µl of 50 µM annealed 5P-Nextera adapter 2/ME into the bottom of a 1.5-ml DNA LoBind microtube, mix well by pipetting 10 times or gently vortexing and briefly spin the tube in a minicentrifuge for 3 s. Add 75 µl of 7.55 µM unloaded Tn5 and mix by gently pipetting 20 times (set the volume of p200 pipette to 80 µl). Quickly spin the tube and incubate at 25 °C for 1 h at 350 rpm in a thermomixer. The final concentration of loaded Tn5 is 6.15 µM (monomer Tn5 concentration).
Critical
Anneal transposons and load Tn5 freshly on the day of the experiment (1–2 h before the start of the experiment). Add transposons and mix well before adding Tn5 to make sure that both are homogeneous to mix with Tn5. Avoid generating bubbles when mixing Tn5 with transposons by pipetting slowly, and do not fill the pipette tip with air. If the experiment is not ready, store loaded Tn5 at −20 °C for ≤1 month.
25 mM dNTP mixture
Mix 250 µl each of 100 mM dATP, dCTP, dGTP and dTTP in a 1.5-ml tube. Mix well by vortexing and briefly spin the tube at room temperature for 5 s. Place a 250-µl aliquot into each 1.5-ml tube and store at −20 °C for 2 years.
2× lysis buffer
To prepare 25 ml of 2× lysis buffer, add 6.5 ml of nuclease-free water into a 50-ml tube. Add 0.5 ml of 1 M Tris-HCl (pH 8.0), 2 ml of 5 M NaCl, 5 ml of 0.5 M EDTA and 11 ml of 10% (wt/vol) SDS. Gently mix and place 1.8-ml aliquots into 2-ml tubes and store at room temperature for 1 year.
1× B&W-T buffer
To prepare 40 ml of 1× B&W-T buffer, add 31.56 ml of nuclease-free water into a 50-ml tube. Add 200 µl of 1 M Tris-HCl (pH 8.0), 8 ml of 5 M NaCl, 40 µl of 0.5 M EDTA and 200 µl of 10% (vol/vol) Tween 20. Gently mix by vortexing and store at room temperature for 1 year.
2× B&W buffer
To prepare 25 ml of 2× B&W buffer, add 14.7 ml of nuclease-free water into a 50-ml tube. Add 250 µl of 1 M Tris-HCl (pH 8.0), 10 ml of 5 M NaCl and 50 µl of 0.5 M EDTA. Mix well by vortexing and store at room temperature for 1 year.
0.1 M PMSF
Weigh 34.8 mg of PMSF and transfer into a 2-ml microtube. Add 100% isopropanol to 2 ml and vortex vigorously to dissolve PMSF. Quickly spin the tube down and transfer all the solution into a 3-ml syringe. Filter through a 0.22-µm syringe filter into a new 2-ml tube. Place a 50-µl aliquot into each 0.2-ml PCR tube and store at −20 °C for ≤4 months.
Critical
To maintain the activity of PMSF in solution, store PMSF in single-use aliquots.
Barcoded oligo plates
Prepare barcoded oligos in working plates as described in Box 2.
Procedure
Nuclei isolation and fixation
Timing 3 h
-
1
Isolate nuclei from tissue following the tissue-specific nuclei extraction protocol (https://doi.org/10.17504/protocols.io.8tvhwn6, https://doi.org/10.17504/protocols.io.ufketkw or https://doi.org/10.17504/protocols.io.bh26j8he) with 0.1 U/µl SUPERase In RNase inhibitor and 0.2 U/µl Enzymatics RNase inhibitor in the isolation buffers. For cell lines, extract nuclei with ATAC lysis buffer with 0.1% (vol/vol) NP-40 as previously described20 with the addition of RNase inhibitors; increase lysis volume proportionally to the number of input cells such that 1 ml of lysis buffer is used for 1 million cells. For whole-cell samples, see Box 3.
-
2
Prepare 1 ml of 1× PBS + RI per sample (Box 3) and 1% (wt/vol) formaldehyde in 1× PBS and keep on ice.
Component
Volume per sample (µl)
PBS, pH 7.4 (1×)
937.5
Formaldehyde (16% (wt/vol))
62.5
Total volume (µl)
1,000
Critical step
We recommend using methanol-free formaldehyde solution in 1× PBS.
-
3
Resuspend nuclei (1–2 million) with 1 ml of 1× PBS + RI and keep on ice. Add 1 ml of 1% (wt/vol) formaldehyde to the nuclei suspension and pipette gently eight times. Leave the tube on ice for 10 min. For a small number of nuclei (100,000 to 0.5 million), resuspend with 0.5 ml of 1× PBS + RI and add 0.5 ml of 1% (wt/vol) formaldehyde.
Critical step
This protocol assumes one sample per plate in Round 1 barcoding (48 wells of Round 1 barcodes; an expected number of cells or data sets of ~60,000–100,000 per plate). Nuclei recovery decreases exponentially through multiple rounds of washing and barcoding when the starting number of nuclei is low.
-
4
Pellet nuclei at 900g for 8 min at 4 °C.
-
5
During centrifugation, prepare 1× PBS + 0.1% (wt/vol) BSA + RI as outlined below and leave on ice.
Component
Volume per plate (µl)
PBS, pH 7.4 (1×)
986.7
BSA, 7.5% (wt/vol)
13.4
SUPERase In RNase inhibitor (20 U/µl)
1.50
Enzymatics RNase inhibitor (40 U/µl)
0.75
Total volume (µl)
1,002.4
In addition, prepare 1× Tango buffer + RI and leave on ice:
Component
Volume per plate (µl)
Tango buffer (10×)
100
DMF
160
SUPERase In RNase inhibitor (20 U/µl)
5
Enzymatics RNase inhibitor (40 U/µl)
2.5
Nuclease-free water
732.5
Total volume (µl)
1,000
-
6
Remove the supernatant and resuspend nuclei with 1 ml of 1× PBS + 0.1% (wt/vol) BSA + RI to wash residual formaldehyde.
Caution
Formaldehyde is toxic. The waste must be collected separately.
-
7
Pellet nuclei at 900g for 8 min at 4°C, aspirate the supernatant and resuspend with 200 µl of 1× Tango buffer + RI.
Critical step
Resuspend nuclei in a small volume to have a minimum concentration of 3,400 nuclei/µl. If different numbers of starting nuclei are used, adjust the suspension volume proportionally.
-
8
Count nuclei concentration with a cell counter and adjust nuclei to a concentration of 3,400 nuclei/µl with 1× Tango buffer + RI. Nuclei are ready for tagmentation.
Tagmentation
Timing 1 h
-
9
In this part of the procedure, perform tagmentation on 150,000 nuclei in a total volume of 75 µl/tube. A total of four tubes are required per full run. In each tagmentation reaction, there are 150,000 nuclei resuspended in 45 µl of 1× Tango buffer and 30 µl of tagmentation mixture. The final concentration of Tn5 transposase and DMF in the tagmentation reaction is 0.8 µM and 16% (vol/vol), respectively.
-
10
Set up a tagmentation mixture following the table below:
Component
Volume per tagmentation reaction (µl)
Tango buffer (10×)
3.00
DMF (100% (vol/vol))
4.80
Loaded Tn5 (6.15 µM)
9.77
SUPERase In RNase inhibitor (20 U/µl)
1.50
Enzymatics RNase inhibitor (40 U/µl)
0.75
Nuclease-free water
10.18
Total volume (µl)
30.00
-
11
If one sample is to be processed in one plate (48 wells), set up a total of four tagmentation reactions in one tube before splitting into four tubes in Step 12. Add 180 µl of 3,400 nuclei/µl nuclei into a 1.5-ml DNA LoBind tube (total number of nuclei is 150,000 nuclei/tube × four tubes = 600,000 nuclei) and then add 120 µl of tagmentation mixture and mix gently by pipetting 10 times (pipetting volume: 280 µl). If multiple samples are being processed, the ratio of nuclei suspension and tagmentation mixture is 3:2 or 45 µl:30 µl per individual tube.
-
12
Briefly spin the tube in a minicentrifuge at room temperature for 3 s and distribute 75-µl aliquots of tagmentation reaction into four 1.5-ml DNA LoBind tubes.
Critical step
Set up four tubes of tagmentation reaction (four tubes × 150,000 nuclei/tube = 600,000 nuclei) to have enough nuclei for 48 wells of Round 1 barcoding in Step 22 (8,000 nuclei/well × a total of 48 wells = 384,000 nuclei). The required minimum number of wells for Round 1 barcoding is 8 (for up to 50,000 fixed nuclei total input to Round 1). Set up reactions on ice. The reaction volume is kept maximally at 75 µl so that nuclei can stay in suspension with gentle shaking.
-
13
Place the tubes on a thermomixer and incubate at 37 °C for 30 min at 300 rpm.
-
14
In the last 5 min of incubation, prepare 1 ml of 1× PBS + 0.1% (wt/vol) BSA + RI (see Step 5) and keep on ice.
-
15
Remove the tubes from the thermomixer and place on ice. Then, pool tagmented nuclei of the same sample into the same tube (4 × 75 µl = 300 µl) to pellet together.
-
16
Add 2.5 volumes of 1× PBS + 0.1% (wt/vol) BSA + RI (e.g., 750 µl of buffer to 300 µl of tagmented nuclei) to pooled tagmented nuclei and mix by pipetting gently five times and centrifuge at 900g for 8 min at 4 °C.
-
17
During centrifugation, prepare 0.5× PBS + RI:
Component
Volume per plate (µl)
PBS, pH 7.4 (1×)
500
SUPERase In RNase inhibitor (20 U/µl)
2.5
Enzymatics RNase inhibitor (40 U/µl)
1.25
Nuclease-free water
500
Total volume (µl)
1,003.75
-
18
Aspirate the supernatant and resuspend nuclei with 300 µl of 0.5× PBS + RI by pipetting gently eight times.
Critical step
Resuspend nuclei with 300 µl of 0.5× PBS + RI to have a minimum concentration of nuclei not lower than 1,000 nuclei/µl. If a different number of nuclei is used for tagmentation, adjust the suspension volume while ensuring that a minimum of eight Round 1 barcoding wells are seeded.
-
19
Count the nuclei concentration with a cell counter and dilute nuclei to 1,000 nuclei/µl with 0.5× PBS + RI. Nuclei are ready for Round 1 DNA barcoding.
Round 1 DNA barcoding: AC oligo ligation
Timing 45 min
-
20
Thaw the AC/RT Round 1 barcoded oligo working plate (Box 2, ‘Round 1 barcoded oligos’) on ice and spin the plate on a swinging bucket rotor at 200g for 1 min at 4 °C and then leave the plate on ice.
-
21
Prepare GLR-A mixture following the table below:
Component
Volume per reaction (µl)
Final concentration in the reaction
Volume per plate (µl)
GLR buffer (4×)
5.00
1×
260.00
ATP (100 mM)
0.40
2 mM
20.80
dNTPs (25 mM each)
1.00
1.25 mM
52.00
SUPERase In RNase inhibitor (20 U/µl)
0.25
0.25 U/µl
13.00
Enzymatics RNase inhibitor (40 U/µl)
0.13
0.26 U/µl
6.76
DTT (1 M)
0.20
10 mM
10.40
T7 DNA ligase (3,000 U/µl)
1.50
225 U/µl
78.00
Nuclease-free water
0.02
–
1.04
Total volume (µl)
8.50
–
442
Critical step
Prepare GLR-A mixture during the washing step (Step 16) but add ATP, RNase inhibitor and T7 DNA ligase just before you are ready to load it into the Round 1 barcoding plate.
-
22
Add 8 µl of nuclei (1,000 nuclei/µl) to each well (rows A–D) of the AC/RT Round 1 barcoded oligo working plate with a multichannel pipette.
Critical step
To avoid cross-contamination of barcoded oligos, make sure that all barcoded oligos are spun to the bottom of the well. Do not touch the tips that dispense nuclei too close to the oligos in the well. Apply this technique when adding nuclei to barcoded oligo plates in other steps.
-
23
Add 8.5 µl of GLR-A mixture to each well with a multichannel pipette.
Critical step
If different samples are loaded on the same plate, avoid sample cross-contamination by touching the tip of the pipette tip that contains GLR-A mixture to the opposite side of the well to the side where nuclei were loaded. Apply this technique when adding reaction mixture to a barcoded oligo plate in other steps.
-
24
Seal and quick-spin the plate at 160g for 15 s at 4 °C. Gently mix the reaction mixture, Round 1 barcoded oligos and nuclei by pipetting five times (mixing volume: 18 µl).
-
25
Seal and quick-spin the plate at 160g for 10 s at 4 °C and incubate the plate on a thermomixer at 25 °C for 30 min at 300 rpm.
Round 1 DNA barcoding: RT
Timing 45 min
-
26
Distribute aliquots of Maxima H Minus reverse transcriptase into PCR strip tubes (i.e., 20 µl to each tube × six tubes for a full plate).
-
27
Remove the Round 1 barcoding plate from the thermomixer, put it on a PCR cooling rack and add 2.2 µl of Maxima H Minus reverse transcriptase to each well with a multichannel pipette.
Critical step
If using an electronic pipette, set the multiple dispensing function and add enzyme to the same side of the well that was used to add the GLR-A mixture to prevent sample cross-contamination. To get an accurate volume, set the speed of the multichannel pipette to be very slow for aspirating and dispensing, because the enzyme is very viscous.
-
28
Seal and quick-spin the plate at 160g for 15 s at 4 °C to bring the enzyme to the bottom of the well and mix by gently pipetting five times (mixing volume: 18 µl). Seal and quick-spin the plate at 160g for 10 s at 4 °C.
-
29
Incubate the plate on a thermocycler using the following program:
Segment
Temperature (°C)
Duration
No. of cycles
1
50
10 min
1
2
8
12 s
3
15
45 s
–
20
45 s
–
30
30 s
–
42
2 min
–
50
3 min
–
3
50
5 min
1
4
4
Forever
1
Round 2 DNA barcoding
Timing 1.5 h
-
30
Five minutes before RT finishes, prepare 3 ml of 1× PBS + 0.1% BSA + RI (see Step 5, scale volumes 3×).
-
31
Remove the plate from the thermocycler and put on ice or a PCR cooling rack; then, pool all reactions into a chilled 25-ml reservoir and transfer the pooled reaction into a chilled 5-ml DNA LoBind tube.
Critical step
Before pooling, pipette each well in the Round 1 barcoding plate two times to disperse nuclei from the bottom of the wells, and make sure to transfer the entire reaction from the well by aspirating slowly and setting the pipette volume to 26 µl. This technique should be done for all pooling steps. In addition, keep reactions on ice all the time when handling samples or reaction mixture to prevent RNA degradation and preserve enzyme activity and all temperature-sensitive reagents like ATP.
-
32
Add 2.8 ml of 1× PBS + 0.1% (wt/vol) BSA + RI (2.5× volume) to rinse the basin and transfer buffer to the tube containing the pooled reactions. Adjust the volume for a different number of Round 1 reactions.
-
33
Add 19.5 µl of 10% (vol/vol) Triton X-100 (final concentration of Triton X-100 is 0.05% (vol/vol); adjust the volume for a different number of Round 1 reactions), mix by inverting the tube five times and centrifuge at 900g for 8 min at 4 °C.
-
34
Thaw the Round 2 barcoded oligo working plate (Box 2, ‘Ligation Round 2 barcoded oligos’) on ice and spin the plate at 200g for 1 min at 4 °C and leave the plate on ice.
-
35
During centrifugation, prepare 1× NEBuffer 3.1 following the tables below:
Component
Volume per plate (µl)
NEBuffer 3.1 (10×)
210
Enzymatics RNase inhibitor (40 U/µl)
21
Nuclease-free water
1,890
Total volume (µl)
2,121
Also prepare the ligation mixture as follows:
Component
Volume per plate (µl)
Final concentration in the reaction
T4 DNA ligase buffer (10×)
510
1×
Enzymatics RNase inhibitor (40 U/µl)
40.8
0.32 U/µl
SUPERase In RNase inhibitor (20 U/µl)
12.75
0.05 U/µl
BSA (20 mg/ml)
51
0.2 mg/ml
T4 DNA ligase (400 U/µl)
127.5
12.5 U/µl
Nuclease-free water
1,338.75
–
Total volume (µl)
2,080.8
–
-
36
Remove the supernatant (from Step 33) as much as possible (~20 µl should be left) and for each Round 2 plate, resuspend the nuclei pellet with 0.12 ml of 1× NEBuffer 3.1; then, add 1.90 ml of 1× NEBuffer 3.1 and 2.04 ml of ligation mixture and mix by pipetting 10 times.
Critical step
All wells of the Round 2 barcoding plate must be used to maximize the number of barcode combinations from three rounds of cellular barcoding. For fewer wells seeded in the Round 1 plate (up to one half of the plate), resuspend nuclei in one half of the volumes above (1.02 ml of 1× NEBuffer 3.1 and 1.02 ml of ligation mixture). Use 5 µl of Round 2 barcoded oligos and seed with only 20 µl of nuclei suspension in the next step. The volume of blocking oligo is also reduced by half for Steps 40 and 41.
-
37
Transfer nuclei in the ligation mixture into a 25-ml reservoir and add 40 µl of nuclei suspension into each well of the Round 2 barcoded oligo working plate and mix gently by pipetting five times (mixing volume: 45 µl).
-
38
Seal the plate with sealing film and quick spin the plate at 160g for 10 s at 4 °C.
-
39
Incubate the plate on a thermomixer at 37 °C for 30 min at 300 rpm.
-
40
Prepare Round 2 blocking solution containing Round 2 blocking oligos (BC_0216) (Table 3) following the table below.
Component
Volume per plate (µl)
Final concentration
BC_0216 (1,000 µM)
47.52
39.6 µM
T4 DNA ligase Buffer (10×)
300
2.5×
Nuclease-free water
852.48
–
Total volume (µl)
1,200
–
Critical step
Round 2 blocking oligos are used to hybridize with the free Round 2 linker sequences to prevent further hybridization of a free Round 2 linker to Round 1 barcoded oligos that can cause cross-contamination (Supplementary Fig. 2b).
-
41
Remove the Round 2 DNA barcoding plate from the thermomixer and quick-spin at 160g for 10 s at 4 °C. Add 10 µl of Round 2 blocking solution to each well with a multichannel pipette and mix by pipetting gently five times (mixing volume: 55 µl).
-
42
Seal the plate with sealing film and quick-spin at 160g for 10 s at 4 °C and then incubate the plate on a thermomixer at 37 °C for 30 min at 300 rpm.
Round 3 DNA barcoding
Timing 1 h
-
43
Thaw the Round 3 barcoded oligo working plate (Box 2, ‘Ligation Round 3 barcoded oligos’) on ice, spin the plate on a swinging bucket rotor at 200g for 1 min at 4 °C and leave the plate on ice.
-
44
Place the Round 2 DNA barcoding plate from Step 43 on a PCR cooling rack and pool nuclei from the Round 2 ligation reaction into a 25-ml reservoir. Add 100 µl of T4 DNA ligase (400 U/µl) into the basin with nuclei from the Round 2 barcoding plate and mix well by gently pipetting 10 times and rock the basin from side to side 10 times.
-
45
Add 50 µl of nuclei suspension to each well of the Round 3 DNA barcoded oligo working plate and mix gently by pipetting five times (mixing volume: 55 µl).
Critical step
For a smaller number of wells seeded to Round 1 (up to one half of the plate), remove 5 µl from each Round 3 barcoding well and seed with only 25 µl of nuclei suspension in this step. The volume of blocking oligo is also reduced by one half for Steps 48 and 49.
-
46
Seal the plate with sealing film and briefly spin the plate at 160g for 10 s at 4 °C. Incubate the plate on a thermomixer at 37 °C for 30 min at 300 rpm.
-
47
Prepare Round 3 blocking solution containing Round 3 blocking oligos (BC_0066) (Table 3) following the table below.
Component
Volume per plate (µl)
Final concentration
BC_0066 (1,000 µM)
41.4
17.25 µM
EDTA (500 mM)
600
125 mM
Nuclease-free water
1,758.6
–
Total volume (µl)
2,400
–
-
48
Remove the Round 3 DNA barcoding plate from the thermomixer, add 20 µl of Round 3 blocking solution to each well and gently mix by pipetting three times (mixing volume: 75 µl).
Critical step
Round 3 blocking oligos are used to hybridize with the free Round 3 linker sequences to prevent cross-hybridization of the Round 3 linker to Round 2 barcoded oligos during nuclei pooling (Supplementary Fig. 2b).
-
49
Without incubation, pool the reaction into a 25-ml reservoir placed on ice before transferring into a 15-ml tube and centrifuging at 1,000g for 8 min at 4 °C.
-
50
In parallel, prepare chilled wash buffer:
Component
Volume per plate (µl)
Final concentration
PBS, pH 7.4 (1×)
4,000
1×
Triton X-100 (10% (vol/vol))
40
0.1% (vol/vol)
SUPERase In RNase inhibitor (20 U/µl)
10
0.05 U/µl
Total volume (µl)
4,050
–
-
51
Remove the supernatant and add 4 ml of wash buffer. Pipette gently five times with a p1000 pipette and then centrifuge at 1,000g for 8 min at 4 °C.
-
52
Carefully remove the supernatant as much as possible and resuspend nuclei with 300 µl of 1× PBS + RI (0.5 ml of PBS (1×), 5 µl of SUPERase In RNase inhibitor (20 U/µl) and 2.5 µl of Enzymatics RNase inhibitor (40 U/µl)).
Critical step
For a smaller number of wells seeded to Round 1 (up to one half of the plate), resuspend nuclei in 200 µl of 1× PBS + RI so that a minimum of four pools are generated.
-
53
Count the nuclei concentration with a cell counter and distribute nuclei aliquots at 1,000–20,000 nuclei in each pool in 1.5-ml DNA LoBind tubes and adjust the total volume of nuclei to 50 µl with 1× PBS + RI.
Critical step
If required to sequence all nuclei/cells, we recommend using aliquots of equal numbers of nuclei by splitting 50 µl of nuclei suspension into six tubes (pools) for a full plate and four tubes for less than one half plate of Round 1 barcoding.
Nuclei lysis and reversal of formaldehyde cross-linking
Timing 2 h
-
54
Add 50 µl of 2× lysis buffer and 10 µl of 20 mg/ml proteinase K to each pool, mix well by gently vortexing, briefly spin the tubes down at room temperature for 10 s to collect all nuclei suspension to the bottom of the tube and incubate on a thermomixer at 55 °C for 2 h at 350 rpm to lyse nuclei and reverse cross-link formaldehyde fixation.
-
55
Put nuclei lysate at −80 °C to inactivate proteinase K before continuing to day 2 of the experiment.
Critical step
The lysis buffer’s components precipitate at room temperature or lower. Re-dissolve by incubating the tube at 37 °C until components are completely dissolved before adding to the nuclei suspension.
Pause point
The nuclear or cell lysate can be stored at −80 °C for ≤6 months before continuing to day 2 of the experiment.
cDNA/AC DNA capture
Timing 1.5 h
Critical
cDNA and AC DNA with biotin (5Biosg) tagged on Round 3 barcoded oligos are conjugated to streptavidin beads in the following steps.
-
56
Prepare Dynabeads MyOne streptavidin C1 as described in Box 4.
-
57
During bead preparation, take the tubes of lysate (from Step 55) out of −80 °C and place onto a thermomixer set at 55 °C for 2 min until the lysate is completely thawed.
-
58
Add 5 µl of 0.1 M PMSF (from −20 °C) to each tube, pulse-vortex for 10 s, pulse-spin on a minicentrifuge at room temperature for 5 s and incubate at room temperature for 10 min with no shaking.
-
59
Add 100 µl of streptavidin beads in 2× B&W + RI (from Box 4, step 8) to each tube of lysate (no pipetting to mix required) and then agitate the tubes on a mixer at room temperature for 1 h at 1,200 rpm.
Critical step
We recommend using an orbital shaker for microtubes (see Equipment). The speed of the mixer can be adjusted to keep beads suspended in the solution.
-
60
Pulse-spin the tubes in a minicentrifuge at room temperature for 5 s, place the tubes onto the magnetic rack until the solution is clear and remove the supernatant with a p200 pipette.
Critical step
Each time before placing the tube back on the magnet, quickly spin the tubes in a minicentrifuge for 3–5 s to collect all supernatant/lysate/buffer and beads to the bottom of the tube. Use a p200 pipette (rather than a p1000 pipette) to remove the supernatant to avoid disturbing or losing the beads.
-
61
Add 250 µl of 1× B&W-T + RI and agitate the beads at room temperature for 5 min at 1,500 rpm to wash the beads.
-
62
Place the tubes onto the magnetic rack until the solution is clear and remove the supernatant. Continue to template-switching oligo blocking on AC DNA immediately.
Blocking template-switching oligos on AC DNA
Timing 30 min
-
63
While working on Steps 61 and 62, prepare Nextera adapter 1 blocking solution containing Nextera adapter 1 blocker (Table 3) following the table below.
Component
Volume per pool (µl)
SSC (6×)
250
Nextera adapter 1 blocker (100 µM)
2.5
SUPERase In RNase inhibitor (20 U/µl)
1
Total volume (µl)
253.5
-
64
Add 250 µl of 6× SSC to each tube without resuspending the beads, wait until the supernatant is clear and then remove the supernatant with a p200 pipette.
-
65
Add 250 µl of Nextera adapter 1 blocking solution to each tube and agitate the tubes on a mixer at room temperature for 1 min at 1,500 rpm and then reduce the speed to 500 rpm for 14 min.
Critical step
Start agitation of the tubes at a high speed of 1,500 rpm for 1 min to make sure the beads are resuspended well, and then shake gently at 500 rpm for 14 min to allow hybridization of Nextera adapter 1 blocker and Nextera adapter 1 on AC DNA and ensure that the beads do not settle.
Critical step
Nextera adapter 1 blocker with 3InvdT at the 3′ end blocks the template-switching oligo on AC DNA to prevent AC DNA amplification in the RNA second PCR, as described in Experimental design (Fig. 2c).
-
66
Place the tubes on the magnetic rack until the solution is clear and remove the supernatant with a p200 pipette.
-
67
Wash the beads twice with 1× B&W-T + RI as described in Steps 61 and 62 at room temperature for 5 min each round. In parallel, prepare Tris-T + RI during the second bead wash.
Component
Volume per pool (µl)
Tris-HCl, pH 8.0 (10 mM)
250
Tween 20 (10% (vol/vol))
2.5
SUPERase In RNase inhibitor (20 U/µl)
0.63
Total volume (µl)
253.13
-
68
Wash the beads with 250 µl of Tris-T + RI the same way as the 1× B&W-T + RI wash (at room temperature for 5 min at 1,500 rpm). In parallel, prepare the GLR-B mixture (described in Step 69).
Critical step
If the GLR-B mixture is not ready, leave the beads in the tube with Tris-T + RI on ice until the GLR-B mixture is ready.
Gap filling, ligation and complete RT
Timing 2 h 15 min
-
69
During washing in Step 68, prepare the GLR-B mixture.
Component
Volume per reaction (µl)
Final concentration in the reaction
GLR buffer (4×)
50
1×
ATP (100 mM)
4
2 mM
Ficoll PM 400 (20% (wt/vol))
20
2% (wt/vol)
dNTPs (25 mM each)
10
1.25 mM
SUPERase In RNase inhibitor (20 U/µl)
5
0.5 U/µl
DTT (1 M)
2
10 mM
Hemo KlenTaq
12.5
–
T7 DNA ligase (3,000 U/µl)
2.5
37.5 U/µl
Nuclease-free water
89
–
Total volume (µl)
195
–
-
70
Place the tubes onto the magnetic rack until the solution is clear, remove the supernatant with a p200 pipette and add 250 µl of nuclease-free water to each tube without resuspending the beads.
-
71
Remove the water, resuspend the beads with 195 µl of the GLR-B mixture by gently vortexing and quick-spin the tubes on a minicentrifuge for 3 s.
-
72
Rotate the tubes in the incubator at 37 °C at a slow speed (10–20 rpm) for 15 min to allow for gap filling on AC DNA and ligation of Nextera adapter 1 blocker to AC DNA (Fig. 2c).
Critical step
For any step that requires a rotator, a thermomixer can be alternatively used as long as the tube is shaken gently and the beads do not settle.
-
73
Remove the tubes from the incubator and add to each tube 5 µl of 100 µM TSO (Table 3) and 5 µl of Maxima H Minus reverse transcriptase and mix well by gently vortexing. Continue to incubate at room temperature for 30 min followed by 42 °C for 90 min with slow rotation.
Pause point
The beads can be stored in Tris-T buffer at 4 °C overnight before continuing to the first PCR in the next step. However, we recommend continuing to the first PCR immediately, if possible.
First PCR, cDNA/AC DNA amplification
Timing 1.5 h
Critical
These steps describe how to co-amplify cDNA and AC DNA from the same cell/nucleus before splitting amplified materials into ‘AC’ and ‘RNA’ in the next sections.
-
74
In the last 5 min of incubation at 42 °C, set up a PCR mixture to amplify both cDNA and AC DNA and prepare PCR strip tubes with individual attached caps (four tubes per pool).
Component
Volume per reaction (µl)
Final concentration in the reaction
KAPA HiFi HotStart ReadyMix (2×)
110
1×
BC_0108 (TSO_PCR) (10 µM)
8.8
0.4 µM
BC_0062 (10 µM)
17.6
0.8 µM
BC_0082 (10 µM)
8.8
0.4 µM
Nuclease-free water
74.8
–
Total volume (µl)
220
–
-
75
After the incubation at 42 °C for 90 min is complete (Step 73), place the tubes on the magnetic rack until the liquid is clear.
-
76
Remove the supernatant with a p200 pipette and add 250 µl of nuclease-free water to each tube without resuspending the beads. Once the liquid is clear, remove the supernatant.
-
77
Resuspend the beads with 220 µl of the first PCR mixture, quick-spin and distribute an aliquot of 55 µl of bead suspension in PCR mixture to each of four PCR strip tubes. Transfer all the leftover beads to the four PCR tubes equally.
Critical step
Transfer the beads in PCR mixture directly to the bottom of the tube, so that there is no need to spin the tube before placing on a thermocycler. Try to transfer the PCR mixture with the beads into PCR strip tubes as quickly as possible to minimize the beads settling before the reaction starts. We recommend increasing the PCR cycle number to a total of 10–11 cycles if working with a very low number of cells/nuclei as input (fewer than 100,000 nuclei in Round 1 barcoding) or cells/nuclei that have a very low quantity of RNA.
-
78
Place the tubes on a thermocycler and run the following program:
Segment
Temperature (°C)
Duration
No. of cycles
1
95
3 min
1
2
98
20 s
9–11
58
45 s
–
72
3 min
–
3
72
5 min
1
4
4
Forever
1
Pause point
The PCR reaction can be stored at −20 °C for 1 month or 4 °C for 1 week.
-
79
Place the strip tubes on a 0.2-ml magnetic rack until the supernatant is clear and pool the first-round PCR products from four strip tubes of the same pool into a 1.5-ml DNA LoBind tube.
-
80
Vortex the tube and quick-spin in a minicentrifuge for 3 s before splitting PCR products into two equal parts (100 µl each), ‘AC’ for chromatin accessibility library preparation and ‘RNA’ for RNA library preparation.
Critical step
AC and RNA in this step are the same PCR products but will be bead size–selected at different KAPA Pure bead volume ratios in Steps 81–94 for AC library preparation and Steps 95–104 for RNA library preparation.
Single-nucleus AC library preparation
AC libraries: first PCR purification and validation
Timing 1 h
-
81
Perform one round of 1.2× KAPA Pure bead purification following the manufacturer’s instructions by using 120 µl of KAPA Pure beads with 100 µl of PCR products from Step 80 and elute with 40 µl of nuclease-free water.
-
82
Transfer the eluent to new PCR strip tubes. The resultant product is called ‘AC-A’.
-
83
Use 2 µl of AC-A product to determine DNA concentration with a Qubit dsDNA HS assay kit following the manufacturer’s instructions.
-
84
Check the quality of ~5–10 ng of AC-A on a 6% (wt/vol) TBE PAGE run in 1× TBE buffer at 250 volts for 23 min with 0.5 µl of Low DNA Mass Ladder as a reference. An example of AC-A on a 6% TBE polyacrylamide gel is shown in Fig. 3a.
Fig. 3: Validation of AC DNA and RNA library preparation. 
a, Gel image showing first-round PCR products from co-amplification of AC DNA and cDNA purified with 1.2× KAPA Pure beads (~5 ng each lane). b, Gel image showing SNARE-seq2 AC libraries of individual pools after second-round PCR for AC-DNA enrichment and column purification (pools 1–6, ~10 ng each lane). c, PAGE size selection of pooled AC libraries (pools 1–6) in 6% TBE PAGE to cut the band at the size range of ~300–1,000 bp. d, AC library fragment size distribution measured by TapeStation. e, Gel image showing the first-round PCR products from co-amplification of AC DNA and cDNA purified with 0.8× KAPA Pure beads (~5 ng each lane). f, Gel image showing amplified cDNA in a second-round PCR and purified with 0.8× KAPA Pure beads (~12 ng each lane). g, Gel image showing SNARE-seq2 RNA libraries of individual pools after tagmentation on 1 ng of amplified cDNA, PCR amplification and two rounds of 0.7× KAPA Pure bead purification (pools 1–6, ~4 ng each lane). h, RNA library size distribution measured by TapeStation.
Pause point
The AC-A products can be stored at 4 °C for 1 month or −20 °C for long-term storage.

AC libraries: second AC PCR and AC library preparation
Timing 6.5 h
Critical
These steps describe how to enrich AC DNA over cDNA and prepare AC libraries.
-
85
In PCR strip tubes, use 5 ng of AC-A as a template for enrichment of AC DNA over cDNA. Adjust the volume of template to 10 µl with nuclease-free water and quick-spin the tubes.
Critical step
If the concentration of PCR products is >5 ng/µl, pipetting a volume smaller than 1 µl tends to have a higher error. We recommend preparing aliquots of the AC-A, diluting them to 0.5 ng/µl in a total of 30–50 µl and using 10 µl as a template for the second AC PCR.
-
86
Prepare the second AC PCR mixture following the table below.
Component
Volume per reaction (µl)
Final concentration in the reaction
KAPA HiFi HotStart ReadyMix (2×)
25
1×
SPLiT_N701 (10 µM)
2.5
0.5 µM
SYBR Green (50×)
0.8
0.8×
Nuclease-free water
9.2
–
Total volume (µl)
37.5
–
-
87
Add 37.5 µl of the second AC PCR mixture and 2.5 µl of 10 µM Ad1_N5XX (AC-indexed PCR primer; Supplementary Table 7) into the tube containing 10 µl of AC-A template. Mix by gently vortexing and quick-spin PCR tubes in a minicentrifuge at room temperature for 3 s.
-
88
Run the reactions on a real-time PCR machine with the following program:
Segment
Temperature (°C)
Duration
No. of cycles
1
95
3 min
1
2
98
20 s
9
58
45 s
–
72
1 min
–
3
72
5 min
1
4
4
Forever
1
-
89
Purify the PCR product with a DNA Clean & Concentrator kit following the manufacturer’s instructions and elute with 40 µl of DNA elution buffer. The resultant product is called ‘AC-B’.
-
90
Use 2 µl of AC-B to determine the DNA concentration with a Qubit dsDNA HS assay kit following the manufacturer’s instructions.
-
91
Check the quality of ~10 ng of AC-B on a 6% TBE PAGE run in 1× TBE buffer at 250 volts for 23 min with 0.5 µl of Low DNA Mass Ladder as a reference. The pattern of nucleosomes by SNARE-seq2 is shown in Fig. 3b.
Critical step
We expect to see a nucleosome pattern with a larger size (~125 bp larger) than that of standard ATAC-seq (Supplementary Fig. 4a), because of the presence of cell barcodes and linker sequences.
-
92
Pool equimolar amounts of AC-B (~200–250 ng/pool) and perform PAGE size selection for fragments between 300 and 1,000 bp (Box 5 and Fig. 3c).
-
93
Use 2 µl of PAGE size-selected AC sequencing libraries to determine the DNA concentration with a Qubit dsDNA HS assay kit.
-
94
Check the quality of ~5–10 ng of AC sequencing libraries on a 6% TBE PAGE run in 1× TBE buffer at 250 volts for 23 min or by TapeStation. An example of AC sequencing libraries by TapeStation can be found in Fig. 3d.
Pause point
The AC-B products can be stored at 4 °C for 1 month or −20 °C for long-term storage.
Single-cell RNA library preparation
RNA libraries: first PCR purification and validation
Timing 1 h
-
95
Perform one round of 0.8× KAPA Pure bead purification following the manufacturer’s instructions by mixing 100 µl of first-round PCR product (from Step 80) with 80 µl of KAPA Pure beads and elute with 40 µl of nuclease-free water. The resultant product is called ‘RNA-A’.
-
96
Use 2 µl of RNA-A to determine the DNA concentration with a Qubit dsDNA HS assay kit following the manufacturer’s instructions.
-
97
Check the quality of 5–10 ng of RNA-A on a 6% TBE PAGE run in 1× TBE buffer at 250 volts for 23 min with 0.5 µl of Low DNA Mass Ladder as a reference. An image of RNA-A on a 6% TBE polyacrylamide gel is shown in Fig. 3e.
Pause point
The RNA-A products can be stored at 4 °C for 1 month or −20 °C for long-term storage.
RNA libraries: second RNA PCR, purification and validation
Timing 2 h
-
98
In PCR strip tubes, add 5 ng of RNA-A as a template for enrichment of cDNA over AC DNA, adjust the volume of the template to 10 µl with nuclease-free water and quick-spin the tubes.
-
99
Prepare PCR mixture for second-round amplification of cDNA following the table below.
Component
Volume per reaction (µl)
Final concentration in the reaction
KAPA HiFi HotStart ReadyMix (2×)
25
1×
BC_0108 (TSO_PCR) (10 µM)
2
0.4 µM
BC_0062 (10 µM)
2
0.4 µM
SYBR Green (50×)
0.8
0.8×
Nuclease-free water
10.2
–
Total volume (µl)
40
–
-
100
Add 40 µl of RNA second PCR mixture to each tube. Gently vortex and quick-spin PCR tubes in a minicentrifuge at room temperature for 3 s.
-
101
Run the reactions on real-time PCR using the following program:
Segment
Temperature (°C)
Duration
No. of cycles
1
95
3 min
1
2
98
20 s
9–11
67
45 s
–
72
3 min
–
3
72
5 min
1
4
4
Forever
1
-
102
Purify PCR products with one round of 0.8× KAPA Pure beads and elute with 40 µl of nuclease-free water. The resultant purified DNA is called ‘RNA-B’.
-
103
Determine DNA concentration with a Qubit dsDNA HS assay kit using 2 µl of RNA-B.
-
104
Verify 10 ng of RNA-B on a 6% TBE PAGE by running at 250 volts for 23 min with 0.5 µl of Low DNA Mass Ladder as a reference. The pattern of RNA-B on a 6% TBE polyacrylamide gel is shown in Fig. 3f.
Critical step
If the smear of RNA-B is 375 bp or larger, continue to cDNA tagmentation. If there is a strong smear smaller than 375 bp, perform another round of 0.8× KAPA Pure Bead purification (Step 102) and elute with 40 µl of nuclease-free water.
Pause point
The RNA-B products can be stored at 4 °C for 1 month or −20 °C for long-term storage.
RNA libraries: cDNA tagmentation
Timing 3 h
Critical
These steps describe how to generate RNA sequencing libraries by Nextera tagmentation of cDNA (using a Nextera XT DNA libray preparation kit) and PCR following the manufacturer’s instructions using 1 ng of RNA-B as a template.
-
105
Dilute RNA-B to 0.2 ng/µl in a PCR strip tube in a total volume of 50 µl. Use 5 µl (1 ng) of diluted RNA-B for tagmentation.
-
106
Prepare the tagmentation mixture in a 1.5-ml tube and distribute 15-µl aliquots into 0.2-ml PCR strip tubes with hinged caps.
Component
Volume per reaction (µl)
Tagment DNA Buffer (2×)
10
Amplicon Tagment Mix
5
Total volume (µl)
15
-
107
Transfer 5 µl of diluted cDNA (RNA-B, total of 1 ng) with a multichannel pipette into the tagmentation mixture, mix by pipetting 10 times, gently vortex and quick-spin in a minicentrifuge at room temperature for 5 s.
-
108
Place the tubes on a thermocycler set at 55 °C for 5 min with a heated lid.
-
109
Remove the tube from the thermocycler and stop the reaction by adding 5 µl of Neutralize Tagment Buffer (equivalent to 0.2% (wt/vol) SDS), mix by pipetting five times, gently spin down the tubes in a minicentrifuge for 5 s and incubate at room temperature for 5 min.
-
110
Set up the tagmentation PCR mixture during the incubation in Step 108.
Component
Volume per reaction (µl)
Final concentration in the reaction
Nextera PCR Master Mix
15
–
BC_0118 (N501, 10 µM)
1
0.2 µM
Nuclease-free water
8
–
Total volume (µl)
24
–
-
111
Add 24 µl of the tagmentation PCR mixture and 1 µl of 10 µM SPLiT_N7XX (RNA-indexed PCR primers; Supplementary Table 8) to each tube of tagmented cDNA, respectively, mix well and gently spin down the tubes in a minicentrifuge for 3 s.
-
112
Run PCR on a thermocycler using the following program:
Segment
Temperature (°C)
Duration
No. of cycles
1
95
30 s
1
2
95
10 s
12
55
30 s
–
72
30 s
–
3
72
5 min
1
4
4
Forever
1
-
113
Purify PCR products with at least two rounds using 0.7× KAPA Pure Beads to make sure that no adapter dimers are left over in the sequencing libraries. Elute the last round of bead-purified RNA libraries with 30 µl of nuclease-free water. The resultant purified PCR product is called ‘RNA-C’.
Critical step
Adapter dimers contaminating a sequencing library can cluster more efficiently on Illumina flow cells (this is especially true for the patterned flow cells such as NovaSeq S4.) Significant adapter dimers can lead to a reduction of sequencing output and quality.
-
114
Use 1 µl of RNA sequencing libraries (RNA-C) to determine DNA concentration with a Qubit dsDNA HS assay kit.
-
115
Check the quality of ~5 ng of RNA libraries on a 6% TBE PAGE run at 250 volts for 23 min with 0.5 µl of Low DNA Mass Ladder as a reference. The pattern of RNA-C on a 6% TBE polyacrylamide gel is shown in Fig. 3g.
-
116
Pool equimolar ratios of RNA-C (~30–70 ng/pool) and determine the pooled RNA sequencing library concentration using a Qubit dsDNA HS assay kit. Determine the size distribution of RNA libraries by TapeStation (Fig. 3h).
Pause point
The RNA-C products can be stored at 4 °C for 1 month or −20 °C for long-term storage.
Sequencing library quality validation by Illumina MiSeq sequencing
AC libraries: quality validation by Illumina MiSeq sequencing
Timing 1 d
-
117
Validate AC sequencing libraries from Step 94 with Illumina MiSeq sequencing (Box 6) using a v2 reagent kit with loading at 20 pM based on Qubit dsDNA HS quantification with a ≥5% PhiX spike-in in accordance with the Illumina loading guide. The sequencing scheme of AC libraries can be found in Supplementary Fig. 3a.
Critical step
For validation sequencing, quantification of the sequencing library concentration by a Qubit dsDNA HS assay kit and determination of average library size by 6% TBE PAGE are sufficient. For high-throughput sequencing, we recommend running sequencing libraries on a TapeStation (or Bioanalyzer or an equivalent instrument) to determine an accurate average library size and assess contamination of adapter dimer peaks at ~125–175 bp. We recommend running qPCR to quantify sequencing library concentration using the average library size derived from TapeStation. For a two-channel sequencing system such as Illumina NovaSeq, we recommend spiking in with a minimum of 5–10% PhiX or consulting your sequencing core facility.
RNA libraries: quality validation by Illumina MiSeq sequencing
Timing 1 d
-
118
Validate RNA sequencing libraries from Step 116 with Illumina MiSeq sequencing (Box 6) using a v3 reagent kit with loading at 22 pM based on Qubit dsDNA HS quantification with a ≥5% PhiX spike-in in accordance with the Illumina loading guide. Follow the instructions for accurate sequencing library concentration determination described in Step 116. The sequencing scheme of RNA libraries can be found in Supplementary Fig. 3b.
Critical step
For Read 2, we suggest sequencing a total of 102 cycles to capture the region encompassing the UMI and cellular barcodes at positions 1–94 bp and the AC Round 1 linker sequences that would occur at positions 95–102 (ACGTACTG). This will permit identification and removal of any contaminating AC reads in RNA libraries (Supplementary Fig. 3b).
Bioinformatic analysis
Critical
One wrapper script (each for RNA and ATAC libraries) is used for processing from raw fastq files to count matrix files. The workflow of the wrapper script for RNA libraries is as follows: after fastq files for each sample have been generated by demultiplexing with Round 1 barcodes, Cutadapt26 is used to filter AC sequences for RNA libraries, and then dropEst27 is used to process RNA reads and cell barcode reads using the same configuration as SPLiT-seq. The workflow of the wrapper script for ATAC libraries is as follows: after fastq files for each sample have been generated by demultiplexing with Round 1 barcodes, we use a custom script to extract cell barcodes from cell barcode reads and append cell barcodes to the paired-end chromatin reads; then, chromatin reads are given as inputs to snapTools30. The chosen alignment software for RNA reads with the dropEst pipeline is STAR29, and the alignment software for chromatin reads with snapTools is minimap228. The final output from dropEst is a cell-by-genes matrix in the form of an R data file, and the final output from snapTools is a bed-formatted fragment file and a ‘snap’ object that contains data to generate a cell-by-bins matrix when imported into R using snapATAC30.
Software installation
Timing 1 h
-
119
Install samtools by typing the following commands into a console window (this software is required for processing alignment files):
$ cd && wget https://github.com/samtools/samtools/releases/download/1.10/samtools-1.10.tar.bz2 && tar -vxjf samtools-1.10.tar.bz2 && cd samtools-1.10 && make
-
120
Install git by typing the following command into a console window (git is used for downloading software packages; it is not required if users directly download packages from their github links):
$ sudo apt-get install git-all
Critical step
Admin privilege is required to install git this way.
-
121
Install Cython and then pysam by typing the following commands into a console window (Cython is required for pysam, and pysam is necessary for some steps in processing sam and fastq files):
$ pip install Cython && pip install pysam
-
122
Install Cutadapt by typing the following command into a console window (Cutadapt is required to remove contaminating AC reads):
$ python3 -m pip install --user --upgrade cutadapt
-
123
Install deindexer in the home directory by typing the following commands into a console window (deindexer is required for demultiplexing fastqs by sample barcodes):
$ cd && git clone https://github.com/ws6/deindexer && cd deindexer && make
-
124
Install R in the home directory by typing the following commands into a console window (R is required for obtaining and working with count matrices):
$ wget https://cran.revolutionanalytics.com/src/base/R-3/R-3.6.2.tar.gz && tar -zxf R-3.6.2.tar.gz && cd R-3.6.2 && ./configure --prefix=$HOME/R && make && make check
-
125
Add software and libraries to the environment’s path by running these export commands (note: add export lines to a user bashrc or profile file to make the changes permanent):
$ export PATH="$HOME/samtools-1.10:$PATH" && export PATH="$HOME/deindexer:$PATH" && export PATH="$HOME/R/bin:$PATH" && export LD_LIBRARY_PATH="$HOME/R/lib/R/lib:$LD_LIBRARY_PATH"
-
126
Install R packages by typing the following commands into a console window (R packages are required for analysis of count matrices):
$ R -e ‘install.packages(c(“httr”, “broom”, “igraph”, “png”, “rlang”, “crayon”, “digest”, “assertthat”, “glue”, “purr”, “backports”, “dplyr”, “tidytext”), repos="http://cran.us.r-project.org")’
$ R -e ‘devtools::install_github(“hms-dbmi/dropEst/dropestr”, dependencies = T)’
$ R -e ‘source("https://bioconductor.org/biocLite.R"); biocLite("DropletUtils")’
$ R -e ‘install.packages("Seurat", repos="http://cran.us.r-project.org", dependencies = T)’
-
127
Ensure that system requirements for dropEst are met using the setup guide here (dropEst is required for processing alignment files to cell-by-genes count matrices): https://dropest.readthedocs.io/en/latest/setup.html.
-
128
Install dropest in the home directory by typing the following commands into a console window:
$ cd && R -e ‘chooseCRANmirror(ind=52); install.packages(c("devtools", "Rcpp","RcppEigen", "RInside", "Matrix", "optparse", "rmarkdown", "withr"))’ && git clone https://github.com/hms-dbmi/dropEst.git && mkdir dropEst/build && cd dropEst/build && cmake .. && make
-
129
Install minimap2 in the home directory by typing the following commands into a console window (minimap2 is required for alignment of ATAC data):
$ cd && git clone https://github.com/lh3/minimap2 && cd minimap2 && make
-
130
Install STAR in the home directory by typing the following commands into a console window (STAR is required for alignment of RNA data):
$ cd && git clone https://github.com/alexdobin/STAR.git && cd STAR/source && make STAR
-
131
Install snapTools in the home directory by typing the following commands into a console window (snapTools is required for processing ATAC data):
$ cd && git clone https://github.com/r3fang/snaptools.git && cd snaptools && pip install -e.
-
132
Download the SNARE-seq2 code and data tar file and then copy or move it to the user’s home directory. Extract the files using the tar commands:
$ cd && tar -xzf snare-seq2_code_data.tar.gz
Download of reference genomes for alignment
Timing 30 min
-
133
Go to https://support.10xgenomics.com/single-cell-atac/software/downloads/latest and follow the instructions to download GRCh38 (or mm10) chromatin accessibility reference files to the home directory.
-
134
Go to https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest and follow the instructions to download GRCh38 (or mm10) RNA reference files to the home directory.
Gene expression data (single-nuclei RNA-seq data)
Timing 8 h
-
135
The timing of 8 h was estimated by assuming that we process ~10,000 cells or data sets with an average read depth of 50,000 reads/data set.
-
136
Generate the <sample>.list text files to provide the list of sample-specific RT primers used per sample. The dT and N6 pairs are listed separately in case no N6 primers were used in the experiment. For example, if sample 1 was added to wells A1–A6 of the Round 1 barcode plate with N6 primers, we list ‘A1 A2 A3 A4 A5 A6 E1 E2 E3 E4 E5 E6’ because the original wells for N6 primers added to wells A1–A6 were E1–E6. Make sure that each well identifier listed is separated by a space or new line.
-
137
Make a directory named ‘plate1_lists’ and move all the sample list files into that folder by typing the following commands into the console:
$ mkdir plate1_lists && mv *.list plate1_lists/
-
138
Map reads using SeqToRDS.sh by typing the following command into the console and replacing <raw.fastq.dir> with the path to the folder containing the raw fastq files. Replace <fastq.id> with the prefix of the fastq file names for the specific library that you are analyzing (i.e., ‘N713_S1’). The main output will be an alignment bam file, a filtered bam file and a filtered count matrix in both ‘.mtx’ and ‘.rds’ format.
$ $HOME/snare-seq2_code_data/bin/SeqToRDS.sh -d <raw.fastq.dir> -f <fastq.id> -s plate1_lists -p $HOME/dropEst -a $HOME/STAR-2.5.1b/bin/Linux_x86_64/ -c ~/.local/bin/ -i $HOME/refdata-cellranger-GRCh38-3.0.0/star/ -g $HOME/refdata-cellranger-GRCh38-3.0.0/genes/genes.gtf
-
139
Create a Seurat object with dT and N6 RNA counts combined using create_seurat_kneeplot_2.r script by typing the following command into the console. Note that the file containing dTN6 barcode pairs must be available at ‘$HOME/snare-seq2_code_data/config/R1_dTN6_pairs.txt’. To perform this step, ensure that the required R packages are installed.
$ Rscript $HOME/snare-seq2_code_data/bin/create_seurat_kneeplot_2.r 03_dropEst
Chromatin accessibility data (single-nuclei ATAC-seq data)
Timing 8 h
The timing of 8 h was estimated by assuming that we process ~10,000 data sets with an average read depth of 50,000 reads/data set.
-
140
Sample list files for ATAC-seq data to provide the list of sample-specific ligation adapters used per sample also come from Round 1 barcode plate wells. Use the same list files generated from Steps 136–137 because ATAC shares the same barcodes as N6 primers.
-
141
Map reads using SeqToFrag.sh by typing the following command into the console and replacing <raw.fastq.dir> with the path to the folder containing the raw fastq files. Replace <fastq.id> with the prefix of the fastq file names for the specific library that you are analyzing (i.e., ‘N531_S3’). The main output will be an alignment bam file, a bed-formatted fragment file and a snap file.
$ $HOME/snare-seq2_code_data/bin/SeqToFrag.sh -d <raw.fastq.dir> -f <fastq_id> -s plate1_lists -a $HOME/minimap2/ -g $HOME/refdata-cellranger-atac-GRCh38-1.2.0/fasta/genome.fa -c $HOME/refdata-cellranger-atac-GRCh38-1.2.0/fasta/genome.fa.fai -n hg38
Troubleshooting
Troubleshooting advice can be found in Table 4.
Timing
-
Steps 1–8, nuclei isolation and fixation: 3 h
-
Steps 9–19, tagmentation: 1 h
-
Steps 20–25, Round 1 DNA barcoding, AC oligo ligation: 45 min
-
Steps 26–29, Round 1 DNA barcoding, RT: 45 min
-
Steps 30–42, Round 2 DNA barcoding: 1.5 h
-
Steps 43–53, Round 3 DNA barcoding: 1 h
-
Steps 54–55, nuclei lysis and formaldehyde reverse cross-linking: 2 h
-
Steps 56–62, cDNA/AC DNA capture: 1.5 h
-
Steps 63–68, blocking template-switching oligos on AC DNA: 30 min
-
Steps 69–73, gap filling, ligation and complete RT: 2 h 15 min
-
Steps 74–80, first PCR, cDNA/AC DNA amplification: 1.5 h
-
Steps 81–84, AC libraries, first PCR purification and validation: 1 h
-
Steps 85–94, AC libraries, second AC PCR and AC library preparation: 6.5 h
-
Steps 95–97, RNA libraries, first PCR purification and validation: 1 h
-
Steps 98–104, RNA libraries: second RNA PCR, purification and validation: 2 h
-
Steps 105–116, RNA libraries, cDNA tagmentation: 3 h
-
Step 117, AC libraries, quality validation by Illumina MiSeq sequencing: 1 d
-
Step 118, RNA libraries, quality validation by Illumina MiSeq sequencing: 1 d
-
Steps 119–141, bioinformatic analysis: 17.5 h
Anticipated results
The success of the SNARE-seq2 assay can be assessed during sequencing library generation. For the sequencing library generation, we expect to see a smear of cDNA and AC DNA mixture on a 6% TBE polyacrylamide gel above 375 bp after co-amplification in the first-round PCR and 0.8× bead purification (Step 97) for RNA (Fig. 3e). The background from adapter dimers should not be present after 0.8× bead purification (Step 97) or could be very faint after 1.2× bead purification (Step 84) for AC DNA (Fig. 3a). The yields of cDNA and AC mixture from nine cycles of amplification in the first PCR in Step 83 or 93 by Qubit quantification should be between 150 and 300 ng. The yields could be slightly lower or higher on the basis of input and cell types. We do not expect to see a strong nucleosome pattern in the first round of PCR. If strong discrete bands of nucleosomes are observed over the smear of cDNA, this implies poor quality of RNA because much of the product derives from chromatin. In Step 91, we expect to see a clear pattern of mono-, di- and tri-nucleosomes at ~300, ~525 and ~750 bp after AC DNA enrichment in the second PCR of AC DNA (Fig. 3b) to generate AC libraries. For RNA sequencing libraries, we should observe a similar result as from the SPLiT-seq protocol9 (Fig. 3g).
For sequencing library quality validation, uniquely mapped reads should be >80% and >70% for RNA (70-bp single-end reads by STAR mapping) and AC (75-bp paired-end reads by minimap2 mapping) libraries, respectively. RNA library quality can also be validated by the fraction of AC library contamination in the final RNA libraries as explained in Experimental design. For samples with good RNA quality, the percentage of contaminated AC libraries in the final RNA libraries would be negligible (<1%) as shown in human brain samples (Table 2). For some samples, the numbers could vary between 1% and 5% on the basis of the expression level of mRNA or RNA quality variation. If AC library contamination in the RNA library is >5%, it indicates poor RNA quality in the starting sample.
The median ratio of UMI count from the dT (y axis) and N6 (x axis) pairs should be equal or >1. This ratio suggests that most mRNA was intact, permitting capture by dT barcodes more so than N6 oligos.
For AC libraries, we expect to see barcodes that passed QC by considering two parameters: fragment in promoter ratio and number of unique fragments. However, these two parameters could vary on the basis of tissue or cell type. For our analyses, we set the cutoff of the fraction of reads in promoters and unique reads at 0.1–0.8 and ≥1,000, respectively. Table 1 shows statistics of two human brain samples processed on the same plate and from the two independent experiments. When high-throughput sequencing is performed, assuming 25,000–30,000 reads/cell barcode, we expect to get ~60,000–80,000 usable data sets for either RNA or AC libraries from one plate of seeding. The shared number of passed-filter cell barcodes between the two assays should be >80% of data sets that pass filtering (Table 1).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Raw and processed SNARE-seq2 data of human brain samples shown in Tables 1 and 2 are available for download from the Brain Cell Data Center (https://biccn.org/data) under U01 ZhangKun grant ID (U01MH114828). Raw data were deposited to the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/) with accession number GSE157660. Processed SNARE-seq2 data of GM12878, 3T3 and A549 cells shown in Supplementary Fig. 5 are available at https://github.com/dinhdiep/snare_seq2.
Code availability
All codes used in this protocol and example test data are available at https://github.com/dinhdiep/snare_seq2.
References
Fu, Y. et al. High-throughput single-cell whole-genome amplification through centrifugal emulsification and eMDA. Commun. Biol. 2, 1–10 (2019).
Macosko, E. Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Klein, A. M. et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015).
Lake, B. B. et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352, 1586–1590 (2016).
Cao, J. et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667 (2017).
Zilionis, R. et al. Single-cell barcoding and sequencing using droplet microfluidics. Nat. Protoc. 12, 44–73 (2017).
Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Gierahn, T. M. et al. Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nat. Methods 14, 395–398 (2017).
Rosenberg, A. B. et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182 (2018).
Buenrostro, J. D. et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015).
Jin, W. et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 528, 142–146 (2015).
Sos, B. C. et al. Characterization of chromatin accessibility with a transposome hypersensitive sites sequencing (THS-seq) assay. Genome Biol. 17, 20 (2016).
Preissl, S. et al. Single-nucleus analysis of accessible chromatin in developing mouse forebrain reveals cell-type-specific transcriptional regulation. Nat. Neurosci. 21, 432–439 (2018).
Cusanovich, D. A. et al. The cis-regulatory dynamics of embryonic development at single-cell resolution. Nature 555, 538–542 (2018).
Mulqueen, R. M. et al. Highly scalable generation of DNA methylation profiles in single cells. Nat. Biotechnol. 36, 428–431 (2018).
Lake, B. B. et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 36, 70–80 (2018).
Cao, J. et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385 (2018).
Chen, S., Lake, B. B. & Zhang, K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol. 37, 1452–1457 (2019).
Snyder, M. P. et al. The human body at cellular resolution: the NIH Human Biomolecular Atlas Program. Nature 574, 187–192 (2019).
Buenrostro, J. D., Wu, B., Chang, H. Y. & Greenleaf, W. J. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109, 21.29.1–21.29.9 (2015).
Bakken, T. E. et al. Evolution of cellular diversity in primary motor cortex of human, marmoset monkey, and mouse. Nature https://doi.org/10.1038/s41586-021-03465-8 (2021).
Stoeckius, M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017).
Zhu, C. et al. An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome. Nat. Struct. Mol. Biol. 26, 1063–1070 (2019).
Corces, M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Petukhov, V. et al. dropEst: pipeline for accurate estimation of molecular counts in droplet-based single-cell RNA-seq experiments. Genome Biol. 19, 78 (2018).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Fang, R. et al. Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat. Commun. 12, 1337 (2021).
Verckist, L. et al. Selective gene expression analysis of the neuroepithelial body microenvironment in postnatal lungs with special interest for potential stem cell characteristics. Respir. Res. 18, 87 (2017).
Acknowledgements
We thank the QB3 MacroLab for purifying the Tn5 transposase enzyme. We thank Andrew Shiau’s laboratory for sharing the optimized protocol for purifying Tn5 transposase enzyme. This project was supported by National Institutes of Health grants U01MH098977, U01MH114828, R01HL123755 and U54HL145608 to K.Z. This publication includes data generated at the UC San Diego IGM Genomics Center and La Jolla Institute for Immunology using Illumina NovaSeq 6000 that were purchased with funding from National Institutes of Health SIG grant #S10 OD026929 and National Institutes of Health grant U19 AI142742, respectively.
Author information
Authors and Affiliations
Contributions
N.P. and K.Z. conceived the study. N.P. and S.C. designed the experiment. N.P. and B.B.L conducted the experiments. D.D. developed the pipeline for bioinformatic analysis. N.P., D.D. and K.Z. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Protocols thanks Linas Mazutis and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this protocol
Bakken, T. E. et al. Preprint at: https://www.biorxiv.org/content/10.1101/2020.03.31.016972v2 (2020)
SNARE-seq2 analysis tools: https://github.com/dinhdiep/snare_seq2
Extended data
Extended Data Fig. 1 Schematics of SNARE-seq2 sequencing library generation.
a, The sequences of barcoded oligos and linkers in SNARE-seq2 protocol. b, The sequences of tagmented accessible DNA and mRNA molecules that are available for capture. c, The sequence of tagmented accessible DNA after being ligated to Round 1 barcodes. d, Round 1 barcoded oligos prime polyadenylated and truncated mRNA, respectively, followed by reverse transcription. e and f, The sequences of AC DNA and mRNA after Round 2 and Round 3 barcoding ligation, respectively. g, AC DNA and cDNA are captured with streptavidin beads. h, The sequence of AC DNA after binding to Nextera adapter 1 blocker. i, The sequence of cDNA after reverse transcription is complete and a PCR handle is added to the 3′ end of the cDNA by template-switching oligos. j, The sequence of AC DNA after gap-filling and priming sites for the first PCR. k, The sequence of AC DNA and priming sites for the second (indexing) PCR. l and m, The sequence of AC DNA sequencing library, with priming sites for sequencing primers. n, The sequence of RNA and priming sites for the first PCR. o, The sequence of RNA and priming sites for the second PCR. p, The sequence of RNA after end tagmentation and priming sites for indexing PCR. q and r, The sequence of the RNA sequencing library, with priming sites for sequencing primers. ‘/Phos/’ denotes phosphorylated, ‘/Biosg/’ denotes a 5′ biotin, a lower case ‘x’ denotes a genomic or transcriptomic sequence, ‘//’ denotes genomic or transcriptomic sequences of variable lengths, a dashed arrow denotes the direction of reverse transcription, ‘#’ denotes either DNA or cDNA sequences, ‘…’ denotes only partial sequences shown and ‘/3InvdT/’ denotes a 3′ inverted thymine base. The bolded numbers in brackets denote the order in which Illumina sequencing data are generated. BC, barcode; UMI, unique molecular identifier.
Supplementary information
Supplementary Information
Supplementary Figs. 1–5 and Supplementary Tables 1–9.
Rights and permissions
About this article

Cite this article
Plongthongkum, N., Diep, D., Chen, S. et al. Scalable dual-omics profiling with single-nucleus chromatin accessibility and mRNA expression sequencing 2 (SNARE-seq2). Nat Protoc 16, 4992–5029 (2021). https://doi.org/10.1038/s41596-021-00507-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-021-00507-3
