
Abstract
In coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, the relationship between disease severity and the host immune response is not fully understood. Here we performed single-cell RNA sequencing in peripheral blood samples of 5 healthy donors and 13 patients with COVID-19, including moderate, severe and convalescent cases. Through determining the transcriptional profiles of immune cells, coupled with assembled T cell receptor and B cell receptor sequences, we analyzed the functional properties of immune cells. Most cell types in patients with COVID-19 showed a strong interferon-α response and an overall acute inflammatory response. Moreover, intensive expansion of highly cytotoxic effector T cell subsets, such as CD4+ effector-GNLY (granulysin), CD8+ effector-GNLY and NKT CD160, was associated with convalescence in moderate patients. In severe patients, the immune landscape featured a deranged interferon response, profound immune exhaustion with skewed T cell receptor repertoire and broad T cell expansion. These findings illustrate the dynamic nature of immune responses during disease progression.
Similar content being viewed by others


Main
COVID-19 caused by SARS-CoV-2 infection causes a spectrum of illness from mild to severe disease and death1. Though SARS-CoV-2 like SARS-CoV uses angiotensin-converting enzyme 2 (ACE2) as its receptor for entry in target cells2,3, the viral shedding pattern is different between the two viruses. The SARS-CoV-2 viral load is detectable during the presymptomatic stage4,5,6 and peaks soon after disease onset7,8,9, which is significantly earlier than that of SARS-CoV10. These factors contribute to the high contagious nature of SARS-CoV-2 and its rapid spread, leading to the global pandemic of COVID-19. Both SARS-CoV-2 infection and viral infection-mediated immune responses can directly and/or indirectly damage cells in the respiratory tract of patients with COVID-19 (refs. 11,12). The majority of patients exhibit mild to moderate symptoms, up to 15% progress to severe pneumonia and approximately 5% eventually develop acute respiratory distress syndrome and/or multiple organ failure11. Higher fatality rates have been observed in elderly individuals with comorbidities and those who are immunocompromised13,14,15,16.
As there are no effective drugs or vaccines available at this time against SARS-CoV-2, there is an urgent need to better understand the host immune response during disease to better devise prognostic and diagnostic markers and to design appropriate therapeutic interventions for patients with severe disease presentation.
Viral infection and the antiviral host immune response interact in vivo and shape disease severity as well as clinical outcomes, especially during acute viral infection. Therefore, the immunopathology of COVID-19 has received much attention. Immune responses in a patient with COVID-19 with moderate disease presentation17, showed that a robust cellular and humoral immune response can be elicited upon acute SARS-CoV-2 infection. However, it remained unknown how the uncontrolled innate and impaired adaptive immune responses were associated with pulmonary tissue damage. Patients with COVID-19 with severe disease presentation showed pronounced lymphopenia and elevation of serum proinflammatory cytokines18,19. We recently reported a fatal case where significant interstitial lymphocytic infiltrates in both lung tissues and overactivation of T cells in peripheral blood were observed20. More recently, inflammatory FCN1+ macrophages were found to replace FABP4+ macrophages in the bronchoalveolar lavage fluid from severe SARS-CoV-2-infected patients, whereas highly expanded and functional competent tissue-resident clonal CD8+ T cells were observed in patients with moderate SARS-CoV-2 infection21. These observations have revealed possible immunopathogenic mechanisms underlying COVID-19 progression at a first glance. However, a global characterization of the antiviral or pathogenic immune responses in different clinical settings is still lacking.
Here, we implemented single-cell RNA sequencing (scRNA-seq) to obtain an unbiased and comprehensive visualization of the immunological responses in peripheral blood mononuclear cells (PBMCs) from patients with COVID-19 with moderate to severe symptoms. Our study depicts a high-resolution transcriptomic landscape of blood immune cells during disease progression of COVID-19, which will facilitate a better understanding of the protective and pathogenic immune responses of the disease.
Results
Single-cell transcriptional profiling of peripheral immune cells
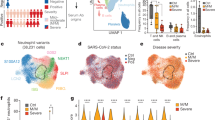
To characterize the immunological features of patients with COVID-19, we performed droplet-based scRNA-seq (10X Genomics) to study the transcriptomic profiles of PBMCs from 13 patients and 5 healthy donors (HDs) as controls (Fig. 1a). The 13 patients with COVID-19 were classified into three clinical conditions: moderate (n = 7), severe (n = 4) and convalescent (conv; n = 6, of whom 4 were paired with moderate cases) (Fig. 1a,b, Supplementary Table 1 and Supplementary Fig. 1). The clinical characteristics and laboratory findings of enrolled patients are detailed in Supplementary Table 1. Single-cell T cell receptor (TCR) and B cell receptor (BCR) sequencing was also performed for each subject. After the unified single-cell analysis pipeline (see Methods), ~0.6 billion unique transcripts were obtained from 122,542 cells from PBMCs of all samples. Among these cells, 22,711 cells (18.5%) were from the HD condition, 37,901 cells (30.9%) were from the moderate condition, 24,640 cells (20.1%) were from the severe condition and 37,290 cells (30.4%) were from the conv condition. All high-quality cells were integrated into an unbatched and comparable dataset and subjected to principal component analysis after correction for read depth and mitochondrial read counts (Supplementary Fig. 2a,b).

a, A schematic showing the overall study design. The scRNA-seq was applied to PBMCs across four conditions and the output data were used for TCR and BCR profiling and expression analyses. b, Timeline of the course of disease for 13 patients infected with SARS-CoV-2 and enrolled in our study. RT–qPCR indicates PCR test for SARS-CoV-2 nucleic acids. RT–qPCR positive indicates nasopharyngeal or sputum samples that were positive for SARS-CoV-2 nucleic acids. The color bars on the left represent conditions with the same color as in a. c, Cellular populations identified. The UMAP projection of 122,542 single cells from HD (n = 5), moderate (n = 7), severe (n = 4) and conv (n = 6) samples, showing the formation of 14 clusters with the respective labels. Each dot corresponds to a single cell, colored according to cell type. d, Canonical cell markers were used to label clusters by cell identity as represented in the UMAP plot. Data are colored according to expression levels and the legend is labeled in log scale. e, Violin plots showing the expression distribution of selected canonical cell markers in the 14 clusters. The rows represent selected marker genes and the columns represent clusters with the same color as in c.
Using graph-based clustering of uniform manifold approximation and projection (UMAP), we captured the transcriptomes of 14 major cell types or subtypes according to the expression of canonical gene markers (Fig. 1c–e and Supplementary Fig. 2a,b). These cells included naive-state T (naive T) cells (CD3+CCR7+), activated-state T (activated T) cells (CD3+PRF1+), mucosal-associated invariant T (MAIT) cells (SLC4A10+TRAV1-2+), γδ T cells (TRGV9+TRDV2+), proliferative T (pro T) cells (CD3+MKI67+), natural killer (NK) cells (KLRF1+), B cells (MS4A1+), plasma B cells (MZB1+), CD14+ monocytes (CD14+ mono; LYZ+CD14+), CD16+ monocytes (CD16+ mono; LYZ+FCGR3A+), monocyte-derived dendritic cells (mono DCs; CD1C+), plasmacytoid dendritic cells (pDCs; LILRA4+), platelets (PPBP+) and hemopoietic stem cells (HSCs; CYTL1+GATA2+). As such, we clearly defined the composition of cell subpopulations in peripheral blood.
Differences in cell compositions across disease conditions
To reveal the differences in cell composition across three conditions (moderate, severe and conv) and to compare with that in HDs, we calculated the relative percentage of the 14 major cell types in the PBMCs of each individual on the basis of scRNA-seq data (Fig. 2a–d). The relative percentage of the activated T cell cluster peaked in moderate patients and did not return to normal levels, even at convalescence. Of note, the relative abundance of naive T cells, MAIT cells and mono DCs decreased with disease severity and these populations were later restored in conv patients (Fig. 2d). In contrast, the relative percentage of pro T cells, plasma B cells, CD14+ mono and platelets increased with disease severity and later declined in conv patients (Fig. 2d). The massive increase of CD14+ mono in patients with severe disease was in accordance with a recent study demonstrating that inflammatory monocytes, induced by pathogenic T cells, incite the inflammatory storm in COVID-19 (ref. 22).

a, UMAP projection of the HD, moderate, severe and conv conditions. Each dot corresponds to a single cell, colored according to cell type. b, Average proportion of each cell type derived from HDs (n = 5), moderate (n = 7), severe (n = 4) and conv (n = 6) samples. c, The top dot plot shows the sum of the absolute counts of lymphocytes and monocytes in the PBMCs of each sample. The bottom bar plot shows cell compositions at a single sample level. d, Condition preference of each cluster. y axis, average percentage of samples across four conditions. Conditions are shown in different colors. Each bar plot represents one cell cluster. Error bars represent ± s.e.m. for 5 HDs and 13 patients. All differences with P < 0.05 are indicated; two-sided unpaired Mann–Whitney U-test was used for analysis. e, Box plots of the expression levels of two GO biological process terms across clusters derived from HD (n = 5), moderate (n = 7), severe (n = 4) and conv (n = 6) samples. Conditions are shown in different colors. Horizontal lines represent median values, with whiskers extending to the farthest data point within a maximum of 1.5 × interquartile range. All differences with P < 0.01 are indicated. **P < 0.001; ***P < 0.0001; using two-sided unpaired Dunn’s (Bonferroni) test.
Next, to investigate the antiviral and pathogenic immune responses during SARS-CoV-2 infection, we evaluated expression levels of two important pathways (Gene Ontology (GO) biological process terms: response to interferon (IFN)-α and acute inflammatory response) in major cell types across four conditions. We found that the response to IFN-α was uniformly and significantly upregulated in all major cell types from the PBMCs of patients with COVID-19 and showed the highest value in almost every major cell type in severe patients, with the exception of plasma B cells, in which the IFN-α response was greatest in moderate patients (Fig. 2e). In addition, with the exception of pro T cells, the acute inflammatory response showed consistent and significant differences across conditions in the selected cell types. Several cell types showed trends in the acute inflammatory response that roughly corresponded with disease severity, including activated T cell, γδ T cell, NK cell and CD16+ mono (Fig. 2e). Moreover, the plasma levels of type I IFN, IFN-γ and other inflammatory cytokines displayed the highest levels in severe patients (Supplementary Fig. 2c). These results suggest a strong overall proinflammatory response in patients with COVID-19 (Fig. 2e).
Strong interferon responses were observed in innate immune cells
To further investigate the transcriptomic changes of innate immune cells (Fig. 3a,b) after SARS-CoV-2 infection, we compared the expression patterns of the moderate or severe condition with that of the HD condition in CD14+ and CD16+ monocytes. We found that significantly differentially expressed genes (DEGs) were involved in IFN responses, myeloid leukocyte activation, cytokine production and nuclear factor (NF)-κB signaling pathway in patients with COVID-19 (Fig. 3c,d). In addition, more DEGs in monocytes from the severe condition were enriched in molecule metabolic and catabolic processes, as well as cytokine secretion (Supplementary Fig. 3a). For NK cells, similarly to monocytes, DEGs associated with IFN responses, cytokine production, NF-κB signaling pathway and leukocyte cytotoxicity were significantly enriched in patients with COVID-19 (Fig. 3e,f), suggesting a consistent response by innate immune cells to SARS-CoV-2 infection. Furthermore, in comparison with moderate patients, the DEGs of NK cells of the severe condition, such as ITGB2, CCL5 and CXCR2, were more closely related to migration-associated processes (Supplementary Fig. 3b,c).

a, UMAP projection of monocytes (39,276) and NK cells (7,479). Each dot corresponds to a single cell, colored according to cell type. b, UMAP projection of the HD, moderate, severe and conv conditions. c, Scatter-plot showing DEGs in the monocytes of moderate (n = 7) or severe patients (n = 4) in comparison with those of HDs (n = 5). Each red dot denotes an individual gene with Benjamini–Hochberg adjusted P value (two-sided unpaired Mann–Whitney U-test) ≤0.01 and average log2 fold change ≥0.5 for the moderate/HD and severe/HD comparisons. Example genes are labeled with gene name. d, Gene enrichment analyses of the DEGs colored in red in c. GO terms are labeled with name and ID, and sorted by −log10 (P) value. A darker color indicates a smaller P value. The top 20 enriched GO terms are shown. Interesting terms are labeled in red. e, Scatter-plot similar to c, but for NK cells. f, Gene enrichment analysis similar to d, but for NK cells. g, Box plots of expression levels of two GO biological process terms in monocytes and NK cells across clusters derived from HD (n = 5), moderate (n = 7), severe (n = 4) and conv (n = 6) samples. Conditions are shown in different colors. Horizontal lines represent median values, with whiskers extending to the furthest data point within a maximum of 1.5 × interquartile range. All differences with P < 0.01 are indicated. **P < 0.001; ***P < 0.0001; using two-sided unpaired Dunn’s (Bonferroni) test.
In line with the DEG enrichment results, we found that monocytes and NK cells both showed significantly upregulated IFN and acute inflammatory responses after SARS-CoV-2 infection, especially in severe patients (Fig. 2e). Levels of cellular apoptosis and migration were also upregulated in both monocytes and NK cells compared to the HD condition (Fig. 3g). Unlike the comparable apoptosis levels in monocytes and NK cells, innate immune cells in severe patients were more prone to migration than those in moderate patients (Fig. 3g and Supplementary Fig. 3c). These results suggest that most innate immune cell types in patients with COVID-19 show strong IFN responses.
Features of T cell subsets in patients with COVID-19
To characterize changes in individual T cell subsets among individuals across four conditions, we subclustered T cells from PBMCs and obtained 12 subsets according to the expression and distribution of canonical T cell markers (Fig. 4a,b): 6 subtypes of CD4+ T cells (CD3E+CD4+), 3 subtypes of CD8+ T cells (CD3E+CD8A+) and 3 subtypes of NKT cells (CD3E+CD4–CD8A–TYROBP+).

a, UMAP projection of 55,655 T cells. Each dot corresponds to a single cell, colored according to cell type. b, Violin plots showing expression distribution of canonical cell markers in 12 T cell subsets. c, Condition preference of each cluster. Error bars represent ± s.e.m. for 5 HDs and 13 patients. All differences with P < 0.05 are indicated; two-sided unpaired Mann–Whitney U-test was used for analysis. d, Box plots of the cytotoxicity and exhaustion scores across different clusters and conditions. The squares along the x axis indicate subsets corresponding to subsets in a. Horizontal lines represent median values, with whiskers extending to the farthest data point within a maximum of 1.5 × interquartile range. All differences with P < 0.01 are indicated. **P < 0.001; ***P < 0.0001; using two-sided unpaired Dunn’s (Bonferroni) test. e, Dot plot showing the expression levels of well-defined cytotoxicity and exhaustion-related genes in NKT CD160 cells across four conditions. f, DEGs of moderate (n = 7) or severe (n = 4) patients in comparison with HDs (n = 5). Each red dot denotes an individual gene with Benjamini–Hochberg adjusted P value (two-sided unpaired Mann–Whitney U-test) ≤0.01 and average log2 fold change ≥0.5 in the moderate/HD and severe/HD comparisons. Example genes are labeled with the gene name. g, Gene enrichment analyses of DEGs are colored in red as in f. Interesting GO terms are labeled in red. h, Box plots of the median cell scores for each cluster for two GO biological process terms across HD (n = 5), moderate (n = 7), severe (n = 4) and conv (n = 6) samples. Horizontal lines represent median values, with whiskers extending to the furthest data point within a maximum of 1.5 × interquartile range. All differences with P < 0.05 are indicated and were analyzed by a two-sided paired Mann–Whitney U-test. i, The expression levels of two GO biological process terms across clusters derived from HD (n = 5), moderate (n = 7), severe (n = 4) and conv (n = 6) samples. Horizontal lines represent median values, with whiskers extending to the furthest data point within a maximum of 1.5 × interquartile range. All differences with P < 0.01 are indicated. **P < 0.001; ***P < 0.0001; using two-sided unpaired Dunn’s (Bonferroni) test.
Of the six subtypes of CD4+ T cell clusters, in addition to naive CD4+ (CD4+ naive) T cell (CCR7+SELL+), memory CD4+ (CD4+ memory) T cell (S100A4+GPR183+), effector memory CD4+ (CD4+ effector memory) T cell (S100A4+GPR183+GZMA+) and regulatory T (Treg) cell (FOXP3+IL2RA+) subtypes, we defined two effector CD4+ T subtypes, CD4+ effector-GZMK and CD4+ effector-GNLY. The CD4+ effector-GNLY cluster was characterized by high expression of genes associated with cytotoxicity, including NKG7, GZMA, GZMB, GZMH and GNLY, whereas the CD4+ effector-GZMK cluster showed relatively high expression of the GZMK gene, but low expression of other cytotoxic genes (Fig. 4b and Supplementary Fig. 4a,b). Furthermore, CD4+ effector-GNLY cells showed high expression of TBX21, implying that they were type 1 helper T (TH1)-like cells (Supplementary Fig. 4c). In contrast, CD4+ effector-GZMK and CD4+ effector memory harbored type 2 helper T (TH2) cell-like features with high expression of GATA3 (Supplementary Fig. 4c). The three subtypes of CD8+ T cell clusters included a naive CD8+ (CD8+ naive) T cell subset (CCR7+SELL+) and two effector CD8+ T cell subsets (CD8+ effector-GZMK and CD8+ effector-GNLY), which both had high expression levels of GZMA and NKG7. In detail, CD8+ effector-GZMK uniquely expressed GZMK, whereas CD8+ effector-GNLY showed relatively high expression levels of GZMB/H and GNLY (Fig. 4b and Supplementary Fig. 4a,b). The three subsets of NKT cell clusters were defined as naive NKT (NKT naive) cells (CCR7+SELL+), CD56+ NKT (NKT CD56) cells and CD160+ NKT (NKT CD160) cells (Fig. 4a,b).
To gain insights into features in T cell subsets, we evaluated the distribution of each cluster across four conditions (Fig. 4c and Supplementary Fig. 4d,e). Notably, the proportions of naive-state T cell subsets, including CD4+ naive, CD4+ memory, CD4+ effector memory, Treg, CD8+ naive and NKT naive subsets, decreased in patients with COVID-19 in comparison with HDs (Fig. 4c and Supplementary Fig. 4d). Even in the conv condition, the proportions of CD4+ naive, CD8+ naive and Treg clusters did not restore to the levels of HDs (Fig. 4c and Supplementary Fig. 4d). In contrast, the proportions of active state T cell subsets, including CD4+ effector-GNLY, CD8+ effector-GNLY, NKT CD56 and NKT CD160 subsets, increased in patients with COVID-19, and these cytotoxic subsets were present in high proportions even in conv patients (Fig. 4c). Of particular note, the CD4+ effector-GNLY subset was almost absent in HDs but highly enriched in moderate, severe and conv patients. In addition, the abundance of the NKT CD160 subset was significantly reduced in severe patients compared to moderate patients.
We then evaluated the cytotoxicity and exhaustion scores of different effector state T cell subsets across four conditions (Fig. 4d and Supplementary Fig. 4f). The CD4+ effector-GNLY, CD8+ effector-GNLY, NKT CD56 and NKT CD160 subsets showed higher cytotoxicity scores than those of the other subsets. Within these highly cytotoxic clusters, HDs all had the lowest cytotoxicity scores, whereas the moderate condition showed the highest cytotoxic state, with the exception of the CD4+ effector-GNLY subset (Fig. 4d,e and Supplementary Fig. 4f). Meanwhile, the CD4+ effector-GZMK, CD8+ effector-GZMK and NKT CD160 clusters showed higher exhaustion scores than those of the other subsets. Within these highly exhausted subsets, HDs all had the lowest exhaustion scores, whereas severe patients showed the most exhausted state (Fig. 4d,e and Supplementary Fig. 4f), in agreement with previous functional studies which examined CD8+ T cells from severe patients and found highly exhausted status and functional impairment23.
To further investigate differential transcriptomic changes in T cells after SARS-CoV-2 infection, we compared the expression profiles of effector T cells (excluding CD4+ naive, CD4+ memory, CD8+ naive and NKT naive clusters) between the moderate or severe and HD conditions. We observed that DEGs upregulated in patients with COVID-19 were involved in processes including IFN responses, cytokine production, cell killing, leukocyte cell–cell adhesion and cytoskeleton organization (Fig. 4f,g and Supplementary Fig. 4i). In addition, using an apoptosis and migration scoring system, we observed that T cells in severe patients likely underwent migration and apoptosis (Fig. 4h,i and Supplementary Fig. 4g,h). Significant activation of cell death and migration pathways in the PBMCs of severe patients suggests that cell death and lymphocyte migration may be associated with lymphopenia, a common phenomenon observed in patients with severe COVID-19 (refs. 18,19,24).
Clonal expansion in T cells and preferred usage of V(D)J genes in patients with COVID-19
Next, to gain insight into the clonal relationship among individual T cells and usage of V(D)J genes across four conditions, we reconstructed TCR sequences (Supplementary Table 2) from the TCR sequencing. Briefly, there were more than 70% of cells in all subsets with matched TCR information, except for the three NKT subsets (Fig. 5a,b). First, compared to the HDs, clonal expansion was obvious in patients with COVID-19 and patients in convalescence (Fig. 5c–e). The extent of clonal expansion in the moderate and conv conditions was higher than that of the severe condition. Meanwhile, large clonal expansions (clonal size >100) were absent in the severe condition (Fig. 5e), indicating that severe patients might lack efficient clonal expansion of effector T cells. We observed different degrees of clonal expansion among T cell subsets (Fig. 5c,d). Effector T cell subsets CD4+ effector-GNLY, CD8+ effector-GZMK and CD8+ effector-GNLY showed high proportions of clonal cells (Fig. 5a,d and Supplementary Fig. 5a) and contained high proportions of inter-cluster clonal cells (Fig. 5f), suggesting that effector T cells underwent dynamic state transitions (Fig. 5a,f).

a, UMAP of T cells derived from PBMCs. Clusters are denoted by colors labeled with inferred cell types (top left), TCR detection (top right), selected TCR clones belonging to the same clusters (bottom left) and different clusters (bottom right). b, Bar plots showing the percentage of TCR detection in each T cell cluster. c, The association between the number of T cell clones and the number of cells per clonotype. The dashed line separates nonclonal and clonal cells. LOESS fitting is labeled as the solid line showing negative correlation between the two axes. d, The distribution of the clone state of T cells in each cluster. e, The clonal status percentage of T cells (left) and percentage of different levels of clonal T cells (right) across four conditions. f, Comparison between the fraction of clonal cells in each subset (x axis) and percentage of cells with TCRs shared across clusters (y axis). g, Usage of some TRAV (top left), TRAJ (bottom left), TRBV (top right) and TRBJ (bottom right) genes across four conditions. Error bars represent ± s.e.m. for 5 HDs and 13 patients. All differences with P < 0.05 are indicated and were analyzed using two-sided unpaired Mann–Whitney U-test. h, The top ten CDR3 usages are shown. Each bar is colored by condition identity. Shared CDR3 sequences are in a red font. i, TRA/B rearrangement differences across four conditions. The colors indicate the usage percentage of specific V-J gene pairs.
To study the dynamics and gene preference of TCRs in patients with COVID-19 and HDs, we compared the usage of V(D)J genes across four conditions (Fig. 5g–i and Supplementary Fig. 5b). The top 10 complementarity determining region 3 (CDR3) sequences were different across four conditions (Fig. 5h). The moderate and conv conditions shared some CDR3 sequences because four samples from these conditions were paired. The usage percentage of the top 10 CDR3 sequences in the HD condition was lower and more balanced compared to those of the other three conditions. Of note, we discovered a different usage of V(D)J genes with decreased diversity in patients with COVID-19, which was more pronounced in TRA genes (Fig. 5i). We also identified over-representation of TRAJ39 and TRAJ43 in severe patients compared to moderate and conv patients (Fig. 5g). The preferred TRBJ gene in severe patients was TRBJ1-1, whereas TRBJ2-1 was preferred in moderate and conv patients (Fig. 5i). The selective usage of V(D)J genes indicates that different immunodominant epitopes may drive the molecular composition of T cell responses and may be associated with SARS-CoV-2-specific infection.
Features of B cell subsets in patients with COVID-19
To trace the dynamic changes of different B subtypes, we subclustered B cells into six subsets according to the expression and distribution of canonical B cell markers (Fig. 6a,b and Supplementary Fig. 6a). We identified one naive B subset (MS4A1+IGHD+), one memory B subset (MS4A1+CD27+), one intermediate transition memory B subset (intermediate memory B; IGHD+CD27+), one germinal center B subset (MS4A1+NEIL1+) and two plasma subsets, plasma B (MZB1+CD38+) and dividing plasma B (MZB1+CD38+MKI67+).

a, UMAP projection of 11,377 B cells. Each dot corresponds to a single cell, colored according to cell type. b, Canonical cell markers were used to label clusters by cell identity as represented in the UMAP plot. Data are colored according to expression level and the legend is labeled in log scale. c, Average proportion of each B cell subtype derived from HD (n = 5), moderate (n = 7), severe (n = 4) and conv (n = 6) samples. d, Bar plot showing B cell compositions at the single sample level. e, Condition preference of each cluster. The y axis shows the average percentage of samples across four conditions. Conditions are shown in different colors. Each bar plot represents one cell cluster. Error bars represent ± s.e.m. for 5 HDs and 13 patients. All differences with P < 0.05 are indicated; two-sided unpaired Mann–Whitney U-test was used for analysis. f, Scatter-plot showing DEGs in moderate (n = 7) or severe (n = 4) patients in comparison with those of HDs (n = 5). Each red dot denotes an individual gene with Benjamini–Hochberg adjusted P value (two-sided unpaired Mann–Whitney U -test) ≤0.01 and average log2 fold change ≥0.5 in the moderate/HD and severe/HD comparisons. Example genes are labeled with the gene name. g, Gene enrichment analyses of DEGs are colored in red as in f. GO terms are labeled with name and ID and are sorted by −log10 (P) values. A darker color indicates a smaller P value. The top 20 enriched GO terms are shown. Notable terms are labeled in red.
Notably, the proportions of active state B subsets, including germinal center B, plasma B and dividing plasma B subsets, increased in patients with COVID-19 in comparison with those of HDs. In contrast, the proportion of memory B cells decreased in patients with COVID-19 compared to that of HDs (Fig. 6c–e).
To further investigate differential transcriptomic changes in B cells after SARS-CoV-2 infection, we compared the expression profiles of B/plasma cells of the moderate or severe condition to those of the HD condition. DEGs that were most significantly enriched in patients with COVID-19 were involved in genes associated with the IFN response (Fig. 6f,g and Supplementary Fig. 6c). Moreover, DEGs in severe patients were associated with protein synthesis, maturation and transport-related biological processes (Supplementary Fig. 6b). These results reveal the transcriptomic features of B cell subsets in patients with COVID-19.
Expanded B cells and specific rearrangements of V(D)J genes in severe patients
We also reconstructed BCR sequences (Supplementary Table 3) from BCR sequencing and analyzed the state of BCR clonal expansion. Briefly, the detection percentage of BCRs was more than 75% in each cluster (Fig. 7a,b). We found that B cells from severe patients showed obvious clonal expansions (Fig. 7c and Supplementary Fig. 6d) than the other three conditions, indicating that B cell activity and humoral immune responses were strongly activated in severe patients, reminiscent of a previous observation that higher antibody titers are associated with worse clinical outcomes25,26,27. This raises the concern that, pathogen-directed antibodies can promote disease pathology, resulting in antibody-dependent enhancement similar to that observed in SARS28.

a, UMAP of B cells derived from PBMCs. Clusters are denoted by color and labeled with inferred cell types (left). The UMAP of B cells is also colored based on BCR detection (right). b, Bar plot showing the percentage of BCR detection in each B cell cluster. c, Bar plot showing the distribution of the clone state of B cells in each cluster. d, Bar plot showing the percentages of IGHA, IGHD, IGHG and IGHM in each condition, with error bars representing ± s.e.m. for 5 HDs and 13 patients. All differences with P < 0.05 are indicated and were analyzed using two-sided unpaired Mann–Whitney U-test. e, Bar plots showing the percentages of IGHA, IGHD, IGHG and IGHM at the single sample level. f, Usage of some IGHV (top left), IGHJ (top right), IGKV (middle left), IGKJ (middle right), IGLV (bottom left) and IGLJ (bottom right) genes across the HD, moderate, severe and conv conditions. Conditions are shown in different colors. Error bars represent ± s.e.m. for 5 HDs and 13 patients. All differences with P < 0.05 are indicated and were analyzed using two-sided unpaired Mann–Whitney U-test. g, Heat maps showing IGH/K/L rearrangement differences across the HD, moderate, severe and conv conditions. Colors indicate the usage percentages of specific V-J gene pairs.
Next, we evaluated the distribution of IgA, IgD, IgG and IgM (IgE not detected) in each patient at the moderate, severe and conv conditions, respectively. In most patients, IgM was the predominant immunoglobulin (Fig. 7d,e). Compared to HDs, the abundance of IgG increased in patients with COVID-19, whereas IgM decreased. In convalescent patients, levels of IgG and IgM returned to levels similar to those of HDs.
To study biased V(D)J rearrangements of the BCR, we compared the usage of V(D)J genes across four conditions (Fig. 7f,g and Supplementary Fig. 6e). We found more specific V(D)J usage in the severe condition compared with the other three conditions, indicating that B cells might have undergone unique and specific V(D)J rearrangements in severe patients (Fig. 7g). We also discovered comprehensive usage of IGHJ4 in all HDs and patients (Fig. 7f), but the paired IGHV genes of IGHJ4 were different in severe patients compared with patients in the other three conditions (Fig. 7g). We observed over-representation of IGHV3-7 in severe patients (Fig. 7f). Moreover, the top two paired V-J frequencies in severe patients were IGHV3-7/IGHJ4 and IGKV3-15/IGKJ3 (Fig. 7g). Taken together, increased B cell clonality and skewed usage of the IGHV and IGKJ genes in severe patients suggest that SARS-CoV-2 infection is associated with V(D)J rearrangements in B cells of the host. Notably, selective usage of dominant IGV genes, especially IGHV3-7 and IGKV3-15 in severe patients, may facilitate the design of vaccines.
Discussion
COVID-19 is usually considered as an acute self-limited viral disease29, although it remains unknown whether SARS-CoV-2 infection can lead to chronic disease in asymptomatic carriers. Host immune response against acute SARS-CoV-2 infection not only plays an antiviral role, but also leads to simultaneous pathogenic injury of organs and tissues, especially in the lungs of patients with COVID-19, which determines the disease severity, progression and outcome. Studies have reported the characteristics of innate and adaptive immune responses15,18,24, which have helped us understand the potential pathogenesis of SARS-CoV-2 infection. However, it is difficult to obtain an integrated scenario of the cellular and molecular immune responses upon SARS-CoV-2 infection. To address this issue, here we have profiled the immunological response landscape in patients with COVID-19 at single-cell resolution, which illustrates the dynamic nature of cellular responses during disease progression and reveals the critical factors responsible for antiviral immunity and pathogenesis in moderate and severe patients.
Our study provides an unbiased visualization of the immunological hallmarks for patients with COVID-19. First, patients with COVID-19 showed a concerted and strong IFN-α response, an overall acute inflammatory response and an enhanced migration ability, which peaked in patients with severe disease in most major cell types in the PBMCs. Second, broad immune activation was observed in patients with COVID-19, evidenced by increased proportions of activated T, pro T and plasma B cells and decreased proportions of naive T and mono DC compartments. Third, the proportions of active state T cell clusters were significantly higher in patients with COVID-19 and with a preferential enrichment of effector T cell subsets, such as CD4+ effector-GNLY, CD8+ effector-GNLY and NKT CD160 cells in moderate patients and an NKT CD56 subset in severe patients. T cells showed higher cytotoxicity and more robust expansion in moderate patients, whereas higher exhaustion levels and less specific clonal expansion were seen in severe patients. Fourth, B cells experienced unique and specific V(D)J rearrangements in severe patients, indicated by an increase of B cell clonality and a skewed use of the IGHV and IGKJ genes. Finally, though most of the clinical parameters recovered to a normal range in patients at the early phase of convalescence, the state of the immune system was not fully restored, exemplified by the ratios of naive T and Treg subsets. A long-term follow-up study is needed to investigate how long it takes to achieve full immune recovery in patients with COVID-19.
An effective antiviral immune response in moderate patients was characterized by moderate and broad activation of innate immune signals as well as expansion of highly cytotoxic effector T cell subsets. The expanded effector T clusters, including CD4+ effector-GNLY, CD8+ effector-GNLY, NKT CD56 and NKT CD160, share features of high expression of NKG7, GZMA, GZMB, GZMH and GNLY and may promote rapid resolution of SARS-CoV-2 infection through their direct cytotoxicity. The CD4+ effector-GNLY cluster resembles classical CD4+ cytotoxic T cells30. CD4+ cytotoxic T cells with major histocompatibility complex class II-restricted cytotoxic activity play an important role in viral infections31, autoimmune diseases32 and malignancies33. Further, it is of great interest to identify immune factors that may predict or prevent progression to severe illness. Notably, the expansion of an NKT CD160 cluster in moderate patients is almost absent in the severe condition. The NKT CD160 cluster refers to a previously described γδ NKT or Vδ1 T cell subset that shows phenotypical and functional similarity to traditional NK cells34. Moreover, FCGR3A was also among the enriched genes in the NKT CD160 cluster, suggesting that it may mediate the antibody-dependent cell-mediated cytotoxicity. Furthermore, Vδ1 T cells are implicated in immune responses to viral infection, particularly to cytomegalovirus35,36, Epstein–Barr virus37, and Vδ1 T cells can also recognize a broad range of cancer cells38,39. As in COVID-19, the preferential expansion of NKT CD160 cells might promote rapid control of the disease through direct cytotoxicity as well as mediating the antibody-dependent cell-mediated cytotoxicity effect. Mechanisms underlying the expansion and function of NKT CD160 cells in COVID-19 warrant future studies. It will be valuable to investigate whether Vδ1 T cells could be used in adoptive cellular therapies to curb overt COVID-19-associated tissue and organ damages.
The immunopathogenesis of disease deterioration in severe patients was characterized by a deranged IFN response, profound immune exhaustion with skewed TCR repertoire and broad T cell expansion. Notably, previous studies on SARS-CoV showed that the virus can harness multiple mechanisms to antagonize IFN responses in host cells12. On the basis of the results of in vitro cell infection, SARS-CoV-2 was recognized as a weak inducer of type I IFNs40,41. However, here we observed strong IFN-α responses in almost all cells types in the PBMCs from severe patients in comparison to moderate patients. Considering that the virus burden peaked soon after disease onset and then decreased gradually7,8, it seems that the systematic IFN-α signal activation in severe patients might be induced by factors other than the virus alone. Overactivation of IFN pathways may contribute to immune dysfunction and immune injury in patients with severe COVID-19. Particularly, IFN-α2b nebulization was widely applied in SARS-CoV-2 infection, which was developed from treating Middle East respiratory syndrome (MERS) and SARS42. The use of IFN for treatment may need a careful reconsideration and reexamination, especially in severe COVID-19 cases.
There are several limitations in this study. For example, it was very difficult to obtain immune cells in bronchoalveolar lavage fluid due to biosafety reasons during the outbreak of COVID-19 when we performed this study. Also, the sample size is comparatively small. Therefore, future studies with longitudinal samples from more patients with COVID-19 may help to determine the cause-and-effect relationships between immune characteristic of different cell types and disease outcome.
Taken together, this integrated, multicellular description in our study lays the foundation for future characterization of the complex, dynamic immune responses to SARS-CoV-2 infection. The transcriptomic data, coupled with detailed TCR- and BCR-based lineage information, can serve as a rich resource for deeper understanding of peripheral lymphocytes in patients with COVID-19 and pave the way for rationally designed therapies as well as development of SARS-CoV-2-specific vaccines.
Methods
Patient cohort and clinical characteristics
Thirteen patients with COVID-19 were admitted at the Fifth Medical Center of PLA General Hospital and enrolled in the study from 23 January to 15 February 2020. The categorization of three clinical groups (moderate, severe and conv) was based on Guidelines for Diagnosis and Treatment of Corona Virus Disease 2019 issued by the National Health Commission of China (7th edition) (http://www.chinacdc.cn/jkzt/crb/zl/szkb_11803/jszl_11815/202003/t20200305_214142.html). The moderate group included nonpneumonia and mild pneumonia cases. The severe group included severe patients who met one of the following criteria: (1) Respiratory distress, respiratory rate ≥30 breaths min−1; (2) Pulse oxygen saturation (SpO2) ≤93% without inhalation of oxygen support at quiet resting state; (3) Arterial partial pressure of oxygen (PaO2)/oxygen concentration (FiO2) ≤300 mm Hg; (4) Computed tomography (CT) image shows there is more than 50% increase of lung infiltrating change within 24 to 48 h. Critically ill patients who were grouped in the severe condition in this study generally required mechanical ventilation and exhibited respiratory failure, septic shock, and/or multiple organ dysfunction/failure that required monitoring and treatment in the intensive care unit. One patient in the severe group died during the study period. The patients in the convalescent group met the discharge criteria as follows: afebrile for more than 3 d, resolution of respiratory symptoms, substantial improvement of chest CT images and two consecutive negative RT–qPCR tests for viral RNA in respiratory tract swab samples obtained at least 24 h apart. This study was approved by the ethics committee of the hospital and written informed consents or telephone call permissions were obtained from each patient or their guardian in the very difficult conditions of the early COVID-19 pandemic.
The clinical data and disease course of the 13 patients are shown in Supplementary Table 1 and Fig. 1b, respectively. Blood sampling for scRNA-seq was usually performed at the time of admission or discharge. CT images for one moderate and one severe patient exhibited bilateral ground-glass opacities (Supplementary Fig. 1).
RT–qPCR
The throat swab, sputum from the upper respiratory tract and blood were collected from patients at various time points after hospitalization. Sample collection, processing and laboratory testing complied with World Health Organization guidance. Viral RNA was extracted from samples using the QIAamp RNA Viral kit (Qiagen) according to the manufacturer’s instructions. Patients infected with SARS-CoV-2 were confirmed using a RT–qPCR kit (TaKaRa) as recommended by the China CDC.
Preparation of single-cell suspensions
Peripheral venous blood samples were obtained on admission of 13 patients with COVID-19 and 5 HDs within 24 h, placed into vacutainer tubes and centrifuged at 400g for 5 min at 4 °C. The time of sampling relative to the onset of symptoms was recorded. Plasma samples were collected and stored at −80 °C until use. For each sample, cell viability exceeded 90%.
Droplet-based single-cell sequencing
Using a Single Cell 5′ Library and Gel Bead kit (10X Genomics, 1000006) and Chromium Single Cell A Chip kit (10X Genomics, 120236), the cell suspension (300–600 living cells per ml as determined by Count Star) was loaded onto a Chromium single cell controller (10X Genomics) to generate single-cell gel beads in the emulsion (GEMs) according to the manufacturer’s protocol. Briefly, single cells were suspended in PBS containing 0.04% BSA. Approximately 10,000 cells were added to each channel and approximately 6,000 target cells were recovered. Captured cells were lysed and the released RNA was barcoded through reverse transcription in individual GEMs. Reverse transcription was performed on a S1000TM Touch Thermal Cycler (Bio Rad) at 53 °C for 45 min, followed by 85 °C for 5 min and a hold at 4 °C. Complementary DNA was generated and amplified, after which, quality was assessed using an Agilent 4200 (performed by CapitalBio Technology). According to the manufacturer’s introduction, scRNA-seq libraries were constructed using a Single Cell 5′ Library and Gel Bead kit, Single Cell V(D)J Enrichment kit, Human T Cell (1000005) and a Single Cell V(D)J Enrichment kit, Human B Cell (1000016). The libraries were sequenced using an Illumina Novaseq6000 sequencer with a paired-end 150-bp (PE150) reading strategy (performed by CapitalBio Technology).
Single cell RNA-seq data processing
Raw gene expression matrices were generated for each sample by the Cell Ranger (v.3.0.2) Pipeline coupled with human reference version GRCh38. The output filtered gene expression matrices were analyzed by R software (v.3.5.3) with the Seurat43 package (v.3.0.0). In brief, genes expressed at a proportion >0.1% of the data and cells with >200 genes detected were selected for further analyses. Low-quality cells were removed if they met the following criteria: (1) <800 unique molecular identifiers (UMIs); (2) <500 genes; or (3) >10% UMIs derived from the mitochondrial genome. After removal of low-quality cells, gene expression matrices were normalized by the NormalizeData function and 2,000 features with high cell-to-cell variation were calculated using the FindVariableFeatures function. To reduce dimensionality of the datasets, the RunPCA function was conducted with default parameters on linear-transformation scaled data generated by the ScaleData function. Next, the ElbowPlot, DimHeatmap and JackStrawPlot functions were used to identify the true dimensionality of each dataset, as recommended by the Seurat developers. Finally, we clustered cells using the FindNeighbors and FindClusters functions and performed nonlinear dimensional reduction with the RunUMAP function with default settings. All details regarding the Seurat analyses performed in this work can be found in the website tutorial (https://satijalab.org/seurat/v3.0/pbmc3k_tutorial.html).
Multiple dataset integration
To compare cell types and proportions across four conditions, we employed the integration methods described at https://satijalab.org/seurat/v3.0/integration.html44. The Seurat package (v.3.0.0) was used to assemble multiple distinct scRNA-seq datasets into an integrated and unbatched dataset. In brief, we identified 2,000 features with high cell-to-cell variation as described above. Second, we identified ‘anchors’ between individual datasets with the FindIntegrationAnchors function and inputted these anchors into the IntegrateData function to create a batch-corrected expression matrix of all cells, which allowed cells from different datasets to be integrated and analyzed together.
Subclustering of B cells and T cells
B cells and plasma cells were extracted from PBMCs. Next, these major cell types were integrated for further subclustering. After integration, genes were scaled to unit variance. Scaling, principal component analysis and clustering were performed as described above. Naive and activated T cells in PBMCs were also extracted and subclustered using the procedure used for B cells.
Cell-type annotation and cluster marker identification
After nonlinear dimensional reduction and projection of all cells into two-dimensional space by UMAP, cells were clustered together according to common features. The FindAllMarkers function in Seurat was used to find markers for each of the identified clusters. Clusters were then classified and annotated based on expressions of canonical markers of particular cell types. Clusters expressing two or more canonical cell-type markers were classified as doublet cells and excluded from further analysis.
DEG identification and functional enrichment
Differential gene expression testing was performed using the FindMarkers function in Seurat with parameter ‘test.use=wilcox’ by default and the Benjamini–Hochberg method was used to estimate the false discovery rate (FDR). DEGs were filtered using a minimum log2(fold change) of 0.5 and a maximum FDR value of 0.01. Enrichment analysis for the functions of the DEGs was conducted using the Metascape webtool (www.metascape.org). Gene sets were derived from the GO Biological Process Ontology.
Defining cell state scores
We used cell scores to evaluate the degree to which individual cells expressed a certain predefined expression gene set45,46,47. The cell scores were initially based on the average expression of the genes from the predefined gene set in the respective cell. For a given cell i and a gene set j (Gj), the cell score SCj (i) quantifying the relative expression of Gj in cell i as the average relative expression (Er) of the genes in Gj compared to the average relative expression of a control gene set (Gjcont): SCj (i) = average(Er(Gj,i)) – average(Er(Gjcont,i)). The control gene set was randomly selected based on aggregate expression levels bins, which yield a comparable distribution of expression levels and over size to that of the considered gene set. The AddModuleScore function in Seurat was used to implement the method with default settings. We used RESPONSE TO INTERFERON ALPHA (GO:0035455), RESPONSE TO INTERFERON BETA (GO:0035456), ACUTE INFLAMMATORY RESPONSE (GO:0002526), APOPTOTIC SIGNALING PATHWAY (GO:0097190), LEUKOCYTE MIGRATION (GO:0050900), 4 well-defined naive markers (CCR7, TCF7, LEF1 and SELL), 12 cytotoxicity-associated genes (PRF1, IFNG, GNLY, NKG7, GZMB, GZMA, GZMH, KLRK1, KLRB1, KLRD1, CTSW and CST7) and 6 well-defined exhaustion markers (LAG3, TIGIT, PDCD1, CTLA4, HAVCR2 and TOX) to define the IFN-α/β response, inflammatory response, apoptosis, migration, naive state, cytotoxicity and exhaustion score, respectively.
TCR and BCR V(D)J sequencing and analysis
Full-length TCR/BCR V(D)J segments were enriched from amplified cDNA from 5′ libraries via PCR amplification using a Chromium Single-Cell V(D)J Enrichment kit according to the manufacturer’s protocol (10X Genomics). Demultiplexing, gene quantification and TCR/BCR clonotype assignment were performed using Cell Ranger (v.3.0.2) vdj pipeline with GRCh38 as reference. In brief, a TCR/BCR diversity metric, containing clonotype frequency and barcode information, was obtained. For the TCR, only cells with at least one productive TCR α-chain (TRA) and one productive TCR β-chain (TRB) were kept for further analysis. Each unique TRA(s)-TRB(s) pair was defined as a clonotype. For the BCR, only cells with at least one productive heavy chain (IGH) and one productive light chain (IGK or IGL) were kept for further analysis. Each unique IGH(s)-IGK/IGL(s) pair was defined as a clonotype. If one clonotype was present in at least two cells, cells harboring this clonotype were considered to be clonal and the number of cells with such pairs indicated the degree of clonality of the clonotype. Using barcode information, T cells with prevalent TCR clonotypes and B cells with prevalent BCR clonotypes were projected on UMAP plots.
Plasma cytokine detection
Plasma levels of IFN-γ, interleukin (IL)-6, IL-18, monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein (MIP)-1α and IP-10 were evaluated by using an Aimplex kit (Beijing Quantobio) following the manufacturer’s instructions. Type I IFN, represented by IFN-β, was detected by ELISA kit (cat. no. EK1236, Multi Sciences) according to the manufacture’s protocols. All samples were performed in duplicate.
Statistics
The statistical tools, methods and threshold for each analysis are explicitly described with the results or detailed in the figure legends or Methods sections.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive of the Beijing Institute of Genomics (BIG) Data Center, BIG, Chinese Academy of Sciences, under accession code HRA000150 and are publicly accessible at http://bigd.big.ac.cn/gsa-human. Other supporting raw data are available from the corresponding author upon reasonable request. Source data are provided with this paper.
Code availability
Experimental protocols and the data analysis pipeline used in our work follow the 10X Genomics and Seurat official websites. The analysis steps, functions and parameters used are described in detail in the Methods section. Custom scripts for analyzing data are available upon reasonable request. Source data are provided with this paper.
References
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 5, 536–544 (2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280 (2020).
Hoehl, S. et al. Evidence of SARS-CoV-2 infection in returning travelers from Wuhan, China. N. Engl. J. Med. 382, 1278–1280 (2020).
Rothe, C. et al. Transmission of 2019-nCoV infection from an asymptomatic contact in Germany. N. Engl. J. Med. 382, 970–971 (2020).
He, X. et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. 26, 672–675 (2020).
Zou, L. et al. SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N. Engl. J. Med. 382, 1177–1179 (2020).
Wölfel, R. et al. Virological assessment of hospitalized patients with COVID-2019. Nature 581, 465–469 (2020).
Pan, Y., Zhang, D., Yang, P., Poon, L. L. & Wang, Q. Viral load of SARS-CoV-2 in clinical samples. Lancet Infect. Dis. 20, 411–412 (2020).
Peiris, J. S. M. et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 361, 1767–1772 (2003).
Cao, X. COVID-19: immunopathology and its implications for therapy. Nat. Rev. Immunol. 20, 269–270 (2020).
Tay, M. Z., Poh, C. M., Rénia, L., MacAry, P. A. & Ng, L. F. P. The trinity of COVID-19: immunity, inflammation and intervention. Nat. Rev. Immunol. 20, 363–374 (2020).
Wang, D. et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 323, 1061–1069 (2020).
Zhang, C., Shi, L. & Wang, F.-S. Liver injury in COVID-19: management and challenges. Lancet Gastroenterol. Hepatol. 5, 428–430 (2020).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Chen, N. et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395, 507–513 (2020).
Thevarajan, I. et al. Breadth of concomitant immune responses prior to patient recovery: a case report of non-severe COVID-19. Nat. Med. 26, 453–455 (2020).
Chen, G. et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 130, 2620–2629 (2020).
Tan, L. et al. Lymphopenia predicts disease severity of COVID-19: a descriptive and predictive study. Signal Transduct. Target Ther. 5, 33 (2020).
Xu, Z. et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 8, 420–422 (2020).
Liao, M. et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 26, 842–844 (2020).
Zhou, Y. G. et al. Pathogenic T cells and inflammatory monocytes incite inflammatory storm in severe COVID-19 patients. Natl Sci. Rev. 7, 998–1002 (2020).
Zheng, H. Y. et al. Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID-19 patients. Cell Mol. Immunol. 17, 541–543 (2020).
Qin, C. et al. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 71, 762–768 (2020).
Zhao, J. et al. Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa344 (2020).
Okba, N. M. A. et al. Severe acute respiratory syndrome coronavirus 2-specific antibody responses in coronavirus disease 2019 patients. Emerg. Infect. Dis. 26, 1478–1488 (2020).
Xiang, F. et al. Antibody detection and dynamic characteristics in patients with COVID-19. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa461 (2020).
Iwasaki, A. & Yang, Y. The potential danger of suboptimal antibody responses in COVID-19. Nat. Rev. Immunol. 20, 339–341 (2020).
Wang, F. S. & Zhang, C. What to do next to control the 2019-nCoV epidemic? Lancet 395, 391–393 (2020).
Zheng, C. et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169, 1342–1356 (2017).
Fu, J. et al. Impairment of CD4+ cytotoxic T cells predicts poor survival and high recurrence rates in patients with hepatocellular carcinoma. Hepatology 58, 139–149 (2013).
Raveney, B. J. et al. Eomesodermin-expressing T-helper cells are essential for chronic neuroinflammation. Nat. Commun. 6, 8437 (2015).
Brown, D. M. Cytolytic CD4 cells: direct mediators in infectious disease and malignancy. Cell Immunol. 262, 89–95 (2010).
Pizzolato, G. et al. Single-cell RNA sequencing unveils the shared and the distinct cytotoxic hallmarks of human TCRVδ1 and TCRVδ2 γδ T lymphocytes. Proc. Natl Acad. Sci. USA 116, 11906–11915 (2019).
Couzi, L. et al. Common features of γδ T cells and CD8+ αβ T cells responding to human cytomegalovirus infection in kidney transplant recipients. J. Infect. Dis. 200, 1415–1424 (2009).
Déchanet, J. et al. Implication of γδ T cells in the human immune response to cytomegalovirus. J. Clin. Invest. 103, 1437–1449 (1999).
Farnault, L. et al. Clinical evidence implicating γδ T cells in EBV control following cord blood transplantation. Bone Marrow Transpl. 48, 1478–1479 (2013).
Fujishima, N. et al. Skewed T cell receptor repertoire of Vδ1+ γδ T lymphocytes after human allogeneic haematopoietic stem cell transplantation and the potential role for Epstein–Barr virus-infected B cells in clonal restriction. Clin. Exp. Immunol. 149, 70–79 (2007).
Maeurer, M. et al. Human intestinal Vδ1+ lymphocytes recognize tumor cells of epithelial origin. J. Exp. Med. 183, 1681–1696 (1996).
Chu, H. et al. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID-19. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa410 (2020).
O’Brien, T. R. et al. Weak induction of interferon expression by severe acute respiratory syndrome coronavirus 2 supports clinical trials of interferon-λ to treat early COVID-19. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa453 (2020).
de Wit, E., van Doremalen, N., Falzarano, D. & Munster, V. J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 14, 523–534 (2016).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Venteicher, A. S. et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355, eaai8478 (2017).
Puram, S. V. et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171, 1611–1624 (2017).
Acknowledgements
We thank the study participants who provided blood samples and C.-B. Zhou and J.-H. Yuan for flow cytometry analysis. J. Hou provided guidance for safely handling samples from patients with COVID-19. This work was supported by the National Key Research and Development Program of China (grant nos. 2020YFC0841900 and 2020YFC0844000) to F.-S.W. from the Ministry of Science and Technology of China. A.Z. is co-PI of the Pan-African Network on Emerging and Re-emerging Infections (PANDORA-ID-NET) funded by the European and Developing Countries Clinical Trials Partnership. A.Z. is in receipt of a UK NIHR Senior Investigator Award.
Author information
Authors and Affiliations
Contributions
F.-S.W. and F.B. conceived and designed the study; J.-Y.Z., X.F., J.-W.S., X.-P.D. and C.Z. performed the experiments; X.-M.W. and X.X. led the bioinformatic analyses; X.-M.W., X.X., J.-Y.Z., X.F., J.-W.S., C.Z., F.B. and F.-S.W. wrote the manuscript; J.-L.F., P.X., S.-Y.W., L.S., Z.X., L.H. and T.-J.J. took care of patients and provided the clinical information; R.-N.X., M.S., Y.Z., A.Z. and M.M. provided intellectual input into the experiments throughout the study, provided comments and helped edit the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Zoltan Fehervari was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–6.
Supplementary Table 1
Supplementary Tables 1–3.
Source data
Source Data Fig. 1
Statistical Source Data.
Source Data Fig. 2
Statistical Source Data.
Source Data Fig. 3
Statistical Source Data.
Source Data Fig. 4
Statistical Source Data.
Source Data Fig. 5
Statistical Source Data.
Source Data Fig. 6
Statistical Source Data.
Source Data Fig. 7
Statistical Source Data.
Rights and permissions
About this article

Cite this article
Zhang, JY., Wang, XM., Xing, X. et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat Immunol 21, 1107–1118 (2020). https://doi.org/10.1038/s41590-020-0762-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41590-020-0762-x
