
Abstract
Links between T cell clonotypes, as defined by T cell receptor (TCR) sequences, and phenotype, as reflected in gene expression (GEX) profiles, surface protein expression and peptide:major histocompatibility complex binding, can reveal functional relationships beyond the features shared by clonally related cells. Here we present clonotype neighbor graph analysis (CoNGA), a graph theoretic approach that identifies correlations between GEX profile and TCR sequence through statistical analysis of GEX and TCR similarity graphs. Using CoNGA, we uncovered associations between TCR sequence and GEX profiles that include a previously undescribed ‘natural lymphocyte’ population of human circulating CD8+ T cells and a set of TCR sequence determinants of differentiation in thymocytes. These examples show that CoNGA might help elucidate complex relationships between TCR sequence and T cell phenotype in large, heterogeneous, single-cell datasets.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

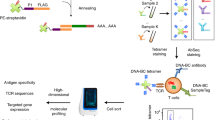

Data availability
All datasets analyzed here are openly available and accessible at https://www.10xgenomics.com/resources/datasets/ and https://developmentcellatlas.ncl.ac.uk/ (human thymic T cell data) (see Supplementary Table 1 for details). Source data are provided with this paper.
Code availability
The CoNGA software repository is available on GitHub (https://github.com/phbradley/conga).
References
Yost, K. E. et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 25, 1251–1259 (2019).
Wu, T. D. et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 579, 274–278 (2020).
Guo, X. et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 24, 978–985 (2018).
Emmi, J., Huuhtanen, J., Mustjoki, S., Heinonen, M. & Lähdesmäki, H. Predicting recognition between T cell receptors and epitopes with TCRGP. PLoS Comput. Biol. 17, e1008814 (2021).
Zheng, C. et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169, 1342–1356 (2017).
Zhang, L. et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564, 268–272 (2018).
Gueguen, P. et al. Contribution of resident and circulating precursors to tumor-infiltrating CD8+ T cell populations in lung cancer. Sci. Immunol. 6, eabd5778 (2021).
Azizi, E. et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 174, 1293–1308 (2018).
Minervina, A. A. et al. Primary and secondary anti-viral response captured by the dynamics and phenotype of individual T cell clones. eLife 9, e53704 (2020).
Zemmour, D. et al. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat. Immunol. 19, 291–301 (2018).
Godfrey, D. I., Stankovic, S. & Baxter, A. G. Raising the NKT cell family. Nat. Immunol. 11, 197–206 (2010).
Toubal, A., Nel, I., Lotersztajn, S. & Lehuen, A. Mucosal-associated invariant T cells and disease. Nat. Rev. Immunol. 19, 643–657 (2019).
Schattgen, S. A. & Thomas, P. G. Bohemian T cell receptors: sketching the repertoires of unconventional lymphocytes. Immunol. Rev. 284, 79–90 (2018).
Dash, P. et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 547, 89–93 (2017).
Glanville, J. et al. Identifying specificity groups in the T cell receptor repertoire. Nature 547, 94–98 (2017).
Zhang, H. et al. Investigation of antigen-specific T-cell receptor clusters in human cancers. Clin. Cancer Res. 26, 1359–1371 (2020).
Tubo, N. J. et al. Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell 153, 785–796 (2013).
Khatun, A. et al. Single-cell lineage mapping of a diverse virus-specific naive CD4 T cell repertoire. J. Exp. Med. 218, e20200650 (2021).
McInnes, L., Healy, J., Saul, N. & Großberger, L. UMAP: uniform manifold approximation and projection. J. Open Source Softw. 3, 861 (2018).
Blondel, V. D., Guillaume, J.-L., Lambiotte, R. & Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. 2008, P10008 (2008).
Traag, V. louvain-igraph: v0.5.3. https://zenodo.org/record/35117#.YN8BzruSlPY (2015).
Schneider, T. D. & Stephens, R. M. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 18, 6097–6100 (1990).
Godfrey, D. I., Koay, H.-F., McCluskey, J. & Gherardin, N. A. The biology and functional importance of MAIT cells. Nat. Immunol. 20, 1110–1128 (2019).
10x Genomics. A new way of exploring immunity: linking highly multiplexed antigen recognition to immune repertoire and phenotype (Application Note). https://pages.10xgenomics.com/rs/446-PBO-704/images/10x_AN047_IP_A_New_Way_of_Exploring_Immunity_Digital.pdf (2020).
Lu, J. et al. Molecular constraints on CDR3 for thymic selection of MHC-restricted TCRs from a random pre-selection repertoire. Nat. Commun. 10, 1019 (2019).
Elhanati, Y., Murugan, A., Callan, C. G. Jr, Mora, T. & Walczak, A. M. Quantifying selection in immune receptor repertoires. Proc. Natl. Acad. Sci. USA 111, 9875–9880 (2014).
Krovi, S. H., Kappler, J. W., Marrack, P. & Gapin, L. Inherent reactivity of unselected TCR repertoires to peptide-MHC molecules. Proc. Natl Acad. Sci. USA 116, 22252–22261 (2019).
Stadinski, B. D. et al. Hydrophobic CDR3 residues promote the development of self-reactive T cells. Nat. Immunol. 17, 946–955 (2016).
Wirasinha, R. C. et al. αβ T-cell receptors with a central CDR3 cysteine are enriched in CD8αα intraepithelial lymphocytes and their thymic precursors. Immunol. Cell Biol. 96, 553–561 (2018).
Schattgen, S. A. et al. Intestinal intraepithelial lymphocyte repertoires are imprinted clonal structures selected for MHC reactivity. Immunity https://papers.ssrn.com/sol3/papers.cfm?abstract_id=3467160 (2019).
Park, J.-E. et al. A cell atlas of human thymic development defines T cell repertoire formation. Science 367, eaay3224 (2020).
Carter, J. A. et al. Single T cell sequencing demonstrates the functional role of αβ TCR pairing in cell lineage and antigen specificity. Front. Immunol. 10, 1516 (2019).
Klarenbeek, P. L. et al. Somatic variation of T-cell receptor genes strongly associate with HLA class restriction. PLoS ONE 10, e0140815 (2015).
Emerson, R. et al. Estimating the ratio of CD4+ to CD8+ T cells using high-throughput sequence data. J. Immunol. Methods 391, 14–21 (2013).
Li, H. M. et al. TCRβ repertoire of CD4+ and CD8+ T cells is distinct in richness, distribution, and CDR3 amino acid composition. J. Leukoc. Biol. 99, 505–513 (2016).
Majumder, K., Bassing, C. H. & Oltz, E. M. Regulation of Tcrb gene assembly by genetic, epigenetic, and topological mechanisms. Adv. Immunol. 128, 273–306 (2015).
Luo, H., Yu, G., Wu, Y. & Wu, J. EphB6 crosslinking results in costimulation of T cells. J. Clin. Invest. 110, 1141–1150 (2002).
Luo, H., Yu, G., Tremblay, J. & Wu, J. EphB6-null mutation results in compromised T cell function. J. Clin. Invest. 114, 1762–1773 (2004).
Huang, H., Wang, C., Rubelt, F., Scriba, T. J. & Davis, M. M. Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nat. Biotechnol. 38, 1194–1202 (2020).
Jameson, S. C. & Masopust, D. Understanding subset diversity in T cell memory. Immunity 48, 214–226 (2018).
Fischer, D. S. et al. Predicting antigen specificity of single T cells based on TCR CDR3 regions. Mol. Systems Biol. 16, e9416 (2020).
Thomas, P. G. & Crawford, J. C. Selected before selection: a case for inherent antigen bias in the T-cell receptor repertoire. Curr. Opin. Syst. Biol. 18, 36–43 (2019).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Berg, J. M., Tymoczko, J. L. & Stryer, L. Biochemistry (W. H. Freeman, 2002).
Miyazawa, S. & Jernigan, R. L. Residue–residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J. Mol. Biol. 256, 623–644 (1996).
Kosmrlj, A., Jha, A. K., Huseby, E. S., Kardar, M. & Chakraborty, A. K. How the thymus designs antigen-specific and self-tolerant T cell receptor sequences. Proc. Natl Acad. Sci. USA 105, 16671–16676 (2008).
Martin, J. & Lavery, R. Arbitrary protein−protein docking targets biologically relevant interfaces. BMC Biophys. 5, 7 (2012).
Dunker, A. K. et al. Intrinsically disordered protein. J. Mol. Graph. Model. 19, 26–59 (2001).
Kidera, A., Konishi, Y., Oka, M., Ooi, T. & Scheraga, H. A. Statistical analysis of the physical properties of the 20 naturally occurring amino acids. J. Protein Chem. 4, 23–55 (1985).
Shugay, M. et al. VDJtools: unifying post-analysis of T cell receptor repertoires. PLoS Comput. Biol. 11, e1004503 (2015).
Atchley, W. R., Zhao, J., Fernandes, A. D. & Drüke, T. Solving the protein sequence metric problem. Proc. Natl Acad. Sci. USA 102, 6395–6400 (2005).
Emerson, R. O. et al. Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire. Nat. Genet. 49, 659–665 (2017).
Murugan, A., Mora, T., Walczak, A. M. & Callan, C. G. Statistical inference of the generation probability of T-cell receptors from sequence repertoires. Proc. Natl Acad. Sci. USA 109, 16161–16166 (2012).
Horn, H. S. Measurement of ‘overlap’ in comparative ecological studies. Amer. Naturalist 100, 419–424 (1966).
Shugay, M. et al. VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res. 46, D419–D427 (2018).
Tickotsky, N., Sagiv, T., Prilusky, J., Shifrut, E. & Friedman, N. McPAS-TCR: a manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics 33, 2924–2929 (2017).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Godec, J. et al. Compendium of immune signatures identifies conserved and species-specific biology in response to inflammation. Immunity 44, 194–206 (2016).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7 (2013).
Hillje, R., Pelicci, P. G. & Luzi, L. Cerebro: interactive visualization of scRNA-seq data. Bioinformatics 36, 2311–2313 (2020).
Acknowledgements
The authors would like to thank J. Park and S. Teichmann for assistance with the thymus atlas T cell dataset, E. Matsen for comments and suggestions on an earlier version of this manuscript, E. Newell and T. Bi for helpful discussions and N. Bradley for suggesting the use of kernel principal components analysis. We would also like to thank the developers of the scanpy single-cell analysis package, which provides the framework on which the CoNGA software is built. This research was supported by National Institutes of Health (NIH) grant R01 AI136514 to P.T., NIH ORIP S10OD028685 to support high-performance computing at the Fred Hutchinson Cancer Research Center, the St. Jude Neoma Boadway Postdoctoral Fellowship to S.S. and the American Lebanese Syrian Associated Charities to P.T.
Author information
Authors and Affiliations
Contributions
S.S. designed, conducted and interpreted experiments, analyzed data and helped prepare the manuscript. K.G. and J.C.C. analyzed data and helped prepare the manuscript. A.S. conducted experiments. A.M.B. and M.J.T.S. provided technical expertise and advice. P.T. designed and interpreted experiments and helped prepare the manuscript. P.B. conceptualized and coded the software, analyzed and interpreted data and prepared the manuscript.
Corresponding authors
Ethics declarations
Competing interests
M.J.T.S. is employed by 10x Genomics. M.J.T.S., A.M.B. and J.C.C. are option or shareholders of 10x Genomics. P.B., P.G.T. and J.C.C. served as unpaid consultants for 10x Genomics on the initial data analysis of the 10x_200k dataset. P.G.T. has filed patents related to the cloning, expression and characterization of T cell receptors. P.G.T. has received travel or speaking expenses from 10x Genomics, Illumina and PACT Pharma.
Additional information
Peer review information Nature Biotechnology thanks Benny Chain, Dmitriy Chudakov and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 T cells belonging to the same clonotype have similar gene expression profiles.
Gene expression UMAP projections of the 10x_200k_donor2a dataset before condensing to a single cell per clonotype, with the 16 largest clonotypes shown in blue (one per panel) and the remainder of the dataset in gray.
Extended Data Fig. 2 CoNGA graph-vs-graph analysis of human and mouse peripheral blood T cells.
CoNGA graph-vs-graph results for PBMC T cell datasets: (a-c) human CD4 and CD8 T cells (human_pbmc1); (d-f) human CD4 and CD8 T cells (human_pbmc2); (g-i) mouse CD4 and CD8 T cells (mouse_pbmc). Same arrangement of plots as in main text Fig. 3.
Extended Data Fig. 3 Matching of CoNGA cluster TCR sequences to bulk repertoires.
TCRβ sequences from human CoNGA clusters were matched to bulk TCRβ repertoires using TCRdist. To score the overlap between the set of TCR sequences in a CoNGA cluster and the set of sequences in a bulk repertoire, we developed a variant of the Morisita-Horn (MH) overlap index that accounts for sequence similarity in addition to exact identity (see Methods for further details). (a) The MH overlaps (y-axis) are plotted against subject age (x-axis) for the two CoNGA clusters indicated in the panel titles. The first cluster (a MAIT cluster) appears to decline with subject age, while the second one (a HOBIT cluster) appears to increase (R value and 2-sided P value in legend). (b) The distribution of MH overlaps for a set of CD4+ repertoires is compared with the distribution of MH overlaps for a set of CD8+ repertoires for two different clusters from the thymus_atlas dataset. (c) The distribution of MH overlaps for a set of memory repertoires is compared with the distribution of MH overlaps for a set of naive repertoires for the two clusters indicated in the panel titles. Boxes in panels b and c show quartiles with whiskers extending to 1.5*IQR. (d) All-vs-all scatter plots (with kernel density estimates along the diagonal) for the following CoNGA cluster features (see Methods for feature calculation details): log10_Pgen, the average log10 generation probability of the cluster TCRβ chains; log10_publicity, the average log10 rate of occurrence in a large (N = 666) dataset of PBMC repertoires; age_correlation, the linear correlation coefficient between MH overlap and subject age (see panel (a)); CD8_vs_CD4, t-statistic comparing MH overlaps for CD8 and CD4 repertoires (higher indicates greater preference for CD8 repertoires; see panel (b)); memory_vs_naive, t-statistic comparing MH overlaps for memory and naive repertoires (higher indicates greater preference for memory repertoires; see panel (c)). The CoNGA clusters are grouped according to the discussion in the main text; ‘pre_hobit’ refers to the two clusters in the thymus_atlas dataset that may be precursors of the HOBIT+ population, (CD8αα(I):2) and (CD8αα(II):2).
Extended Data Fig. 4 Specific versus non-specific binding in the 10x_200k dataset.
Comparison of binding data for four ‘specific’ pMHC multimers (A02_GIL, A02_ELA, B08_RAK, A02_GLC) and four ‘sticky’ pMHC multimers (A03_KLG, A03_RLR, A03_RIA, A11_AVF) in the 10x_200k_donor2 dataset. (a) GEX landscapes colored by pMHC binding signal (log(1+ UMI read count)). (b) TCR landscapes colored by pMHC binding signal. The ‘specific’ pMHCs show binding that is focused in certain areas of the landscapes, whereas the binding of the putative ‘sticky’ pMHCs is dispersed across the landscapes. (c) The Pearson correlation between binding profiles for different pMHCs is shown in matrix form according to the indicated color mapping. The specific pMHCs show little correlation whereas the sticky pMHCs are significantly correlated in their binding, suggesting that a shared cellular property (TCR or CD8 surface expression, expression of other HLA-interacting molecules, general level of activation) is jointly influencing their binding. Note that A11_AVF (and A11_IVT) show additional specific binding in donor 1, who is A*11:01 positive; the A*03:01 pMHC multimers appear non-specific regardless of donor HLA type.
Extended Data Fig. 5 Flow cytometry gating strategies for HOBIT/HELIOS CD8 T cells in Fig. 2.
(a) Gating strategy for KLRC2+ KIR2Dmix and KLRC2-KIR2D+ CD8 T cells in panels (b+ c). After gating on single lymphocytes the gating is Ghost510-CD14-CD19-CD3+ CD8B+ CCR7-CD45RA+. (b) Representative example of CD1d:PBS-57 and MR1:5-OP-RU tetramer labeling of KLRC2+ KIR2Dmix, KLRC2-KIR2D+, and CCR7-CD45RO+ CD8 T cells. (c) Frequency of CD1d and MR1-labelled KLRC2+ KIR2Dmix, KLRC2-KIR2D+, and CCR7-CD45RO+ CD8 T cells (n = 12; Supplementary Note 3). P values calculated by 1-sided t-test. The lower limit of the box corresponds to the 1st quartile, center line the median, and upper limit the 3rd quartile (d) Gating strategy for HELIOS intracellular staining of KLRC2+ KIR2Dmix and KLRC2-KIR2D+ CD8 T cells in panels. Single lymphocytes were gated on Ghost510-CD14-CD19-CD3+ CD8B+ CD248-CCR7-CD45RO-CD45RA+.
Extended Data Fig. 6 Detection of GEX neighborhoods with elevated iMHC scores across multiple donors.
2D GEX projection of the 10x_200k_donor1 (a), 10x_200k_donor2 (b), 10x_200k_donor3 (c), and 10x_200k_donor4 (d) datasets colored by P values for iMHC enrichment in each clonotype’s graph neighborhood (the set of iMHC scores in each clonotype’s neighborhood are compared to the remainder of the iMHC scores using an unpaired, 1-sided Mann-Whitney-Wilcoxon test). (e) Top 10 DEGs for the clonotypes with significant iMHC enrichment in the 10x_200k_donor1 dataset. (f) Top 10 DEGs for the clonotypes with significant iMHC enrichment in the 10x_200k_donor3 dataset. (g) Top 10 DEGs for the clonotypes with significant iMHC enrichment in the 10x_200k_donor4 dataset. (There were too few clonotypes with significant iMHC enrichment in the 10x_200k_donor2 dataset to identify differentially expressed genes). (h) Graph-vs-feature correlation between a TCR feature, iMHC score (left panel), and 2 scores derived from the GEX profile (right panels, ZNF683 and KLRC3 expression) is illustrated by mapping the scores onto the 2D UMAP GEX landscape for the 10x_200k_donor1 dataset (after Z-score normalization and averaging over graph neighborhoods).
Extended Data Fig. 7
Gating strategy for assessment of EPHB6 protein levels in TRBV30 ± CD4+ and CD8+ T cells in Fig. 5f.
Extended Data Fig. 8 Matching of pMHC-positive TCR sequences to bulk repertoires and epitope-specific TCR sequences from the literature.
(a) TCRβ sequences from the pMHC-positive clonotypes in the 10x_200k dataset were matched to bulk TCRβ repertoires using TCRdist. To score the overlap between the set of TCR sequences in a pMHC-positive repertoire and the set of sequences in a bulk repertoire, we developed a variant of the Morisita-Horn (MH) overlap index that accounts for sequence similarity in addition to exact identity (see Methods for further details). All-vs-all scatter plots (with kernel density estimates along the diagonal) are shown for the following pMHC-positive repertoire features (see Methods for feature calculation details): log10_Pgen, the average log10 generation probability of the repertoire TCRβ chains; log10_publicity, the average log10 rate of occurrence in a large (N = 666) dataset of PBMC repertoires; age_correlation, the linear correlation coefficient between MH overlap and subject age in the N = 666 PBMC repertoire dataset (see Extended Data Fig. 3a); CD8_vs_CD4, t-statistic comparing MH overlaps for CD8 and CD4 repertoires (higher indicates greater preference for CD8 repertoires; see Extended Data Fig. 3b); memory_vs_naive, t-statistic comparing MH overlaps for memory and naive repertoires (higher indicates greater preference for memory repertoires; see Extended Data Fig. 3c). (b) The pMHC-positive repertoires were matched against one another and against a set of literature-derived TCR sequences taken primarily from the VDJdb55 and McPAS56 databases (excluding those TCRs in the VDJdb that were themselves derived from the 10x_200k dataset). The heatmap shows MH overlaps calculated using paired-chain TCRdist distances. Reasonable concordance between repertoires positive for the same pMHC from different donors and between pMHC-positive and literature-derived repertoires can be seen.
Extended Data Fig. 9 Epitope-specific T cell populations differ in activation status.
(a) Log-transformed read counts for DNA-barcoded anti-CD45RA (x-axis) and anti-CD45RO (y-axis) antibodies, averaged over pMHC+ clonotypes, are plotted for the pMHCs shown in Fig. 6. In the panel on the left, clonotypes are weighted equally, while in the panel on the right, larger clonotypes are given more weight (proportional to the logarithm of the clone size) to better reflect the underlying distribution of cells (particularly for the d1_A11 pMHCs, both of which have a relatively large number of positive cells distributed unevenly among a small number of clonotypes). (b) Heatmap of gene set variation analysis (GSVA) scores for pMHC-specific clonotypes by donor. Significant hits (P values < 0.05 after multiple hypothesis correction using the Benjamini-Hochberg method) from the MSigDB (https://www.gsea-msigdb.org/gsea/msigdb) C7 collection57,58 are shown. Analysis performed using Seurat59, GSVA60, and Cerebro61 R packages.
Extended Data Fig. 10 CoNGA’s ability to recover invariant T cell subsets depends on their frequency in the dataset.
To assess the sensitivity of CoNGA’s graph-vs-graph algorithm in detecting a known GEX/TCR correlation, we created artificial datasets by subsampling the MAIT cell clonotypes (iNKT cell clonotypes in mouse) down to specified levels within the context of five datasets in which those clonotypes could be clearly identified both as a distinct GEX cluster and by virtue of their invariant TCR sequences. (a) The fraction of MAIT or iNKT clonotypes recovered as CoNGA hits (y-axis) is plotted against the frequency to which these clonotypes were downsampled in the dataset. (b) The fraction of recovered clonotypes is plotted against the absolute number of downsampled clonotypes present in the dataset. Recovery rate appears to depend more strongly on the number of downsampled clonotypes than their fraction in the total dataset.
Supplementary information
Supplementary Information
Supplementary Figs. 1–9, Notes 1–3, Tables 1–8 and References.
Supplementary Data 1
TCR sequence information on all CoNGA clusters for 10x_200k_donors.
Supplementary Data 2
TCR sequence information for pMHC-specific CD8 T cells from 10x_200k_donors used for analysis
Supplementary Data 3
Source Data for Supplementary Fig. 3
Supplementary Data 4
Source Data for Supplementary Fig. 4
Supplementary Data 5
Source Data for Supplementary Fig. 5
Source data
Source Data Fig. 1
Statistical Source Data
Source Data Fig. 2
Statistical Source Data
Source Data Fig. 3
Statistical Source Data
Source Data Fig. 5
Statistical Source Data
Source Data Extended Data Fig. 2
Statistical Source Data
Source Data Extended Data Fig. 3
Statistical Source Data
Source Data Extended Data Fig. 5
Statistical Source Data
Source Data Extended Data Fig. 8
Statistical Source Data
Source Data Extended Data Fig. 9
Statistical Source Data
Rights and permissions
About this article

Cite this article
Schattgen, S.A., Guion, K., Crawford, J.C. et al. Integrating T cell receptor sequences and transcriptional profiles by clonotype neighbor graph analysis (CoNGA). Nat Biotechnol 40, 54–63 (2022). https://doi.org/10.1038/s41587-021-00989-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-021-00989-2
