
Abstract
To investigate the immune response and mechanisms associated with severe coronavirus disease 2019 (COVID-19), we performed single-cell RNA sequencing on nasopharyngeal and bronchial samples from 19 clinically well-characterized patients with moderate or critical disease and from five healthy controls. We identified airway epithelial cell types and states vulnerable to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. In patients with COVID-19, epithelial cells showed an average three-fold increase in expression of the SARS-CoV-2 entry receptor ACE2, which correlated with interferon signals by immune cells. Compared to moderate cases, critical cases exhibited stronger interactions between epithelial and immune cells, as indicated by ligand–receptor expression profiles, and activated immune cells, including inflammatory macrophages expressing CCL2, CCL3, CCL20, CXCL1, CXCL3, CXCL10, IL8, IL1B and TNF. The transcriptional differences in critical cases compared to moderate cases likely contribute to clinical observations of heightened inflammatory tissue damage, lung injury and respiratory failure. Our data suggest that pharmacologic inhibition of the CCR1 and/or CCR5 pathways might suppress immune hyperactivation in critical COVID-19.
Similar content being viewed by others


Main
Patients with COVID-19 exhibit a broad spectrum of disease progression, with 81% showing mild, moderate or no symptoms; 14% showing severe symptoms; and 5% experiencing critical disease with high mortality risk1. The risk of developing severe or critical disease has been associated with advanced age1,2, comorbidities1,2, hyperactivation of the immune system3,4, sex1,2 and other factors. However, an understanding of these risk factors at the molecular and cellular levels is in its infancy.
In this study, we investigated the immune response in patients with COVID-19 by single-cell RNA sequencing (scRNA-seq) of nasopharyngeal and bronchial samples to identify molecular correlates of disease severity. Two recent studies applied scRNA-seq to bronchioalveolar lavage fluid samples from patients with COVID-19 and provide an extensive characterization of the inflammatory immune phenotype in the lower respiratory tract5,6. However, SARS-CoV-2 and other coronaviruses infect and replicate in both the upper and lower respiratory tract. The upper airways are specialized in eliminating inhaled pathogens to prevent viral invasion in the lower respiratory tract and pulmonary injury7. Control of viral spread depends on interactions between epithelial cells and immune cells, mediated by cytokine signaling and cell–cell contacts7. After viral clearance, activated immune cells must be eliminated to avoid hyperactivation of the immune system and exacerbated tissue damage7,8. It has been suggested that the severe lung injury observed in some patients with critical COVID-19 is a consequence of the hyperactivated immune system rather than of inadequate viral clearance4,9. To investigate these questions, we conducted a comprehensive study of mucosal immune responses in patients with COVID-19 that includes samples from both the upper and lower respiratory tract and sequencing data on both immune and epithelial cells. We found that critical disease severity correlates with stronger interactions between epithelial and immune cells and hyperactivated inflammatory macrophages and cytotoxic T lymphocytes, likely contributing to exacerbated epithelial cell death.
Results
Cellular and molecular characterization of COVID-19 severity
We performed 3′ scRNA-seq on nasopharyngeal or pooled nasopharyngeal/pharyngeal swabs (NSs), bronchial protected specimen brushes (PSBs) and bronchial lavages (BLs) (Fig. 1a). We analyzed a dual-center cohort from Charité - Universitätsmedizin Berlin and University Hospital Leipzig, comprising 19 patients with COVID-19 (15 males and 4 females; age range, 21–76 years; median age, 61 years; Fig. 1b and Supplementary Table 1). Five SARS-CoV-2-negative donors with no signs of disease were included as controls (two males and three females; age range, 24–41 years; median age, 34 years). On the basis of World Health Organization (WHO) guidelines10, we classified eight patients as moderate and 11 patients as critical, two of whom died several days later in the course of the disease (Fig. 1b and Supplementary Table 1; patients BIH-CoV-01 and BIH-CoV-02).

a, Schematic representation of the experimental workflow. Depicted are the sampling sites (left) and the 3′ scRNA-seq library preparation using 10X Genomics (middle) followed by data analysis revealing cell type identity (right). b, Overview of the patient cohort. Given are age, sex and COVID-19 severity as well as onset of symptoms, hospitalization duration and sampling time points in days after onset of symptoms, with all patients being temporally aligned to the day of positive SARS-CoV-2 test. Admission to the ICU is also depicted if applicable. One patient required ECMO. We obtained NSs from all patients and, in addition, PSBs and BLs from patients BIH-CoV-01 and BIH-CoV-04 (marked with *). The sampling day relative to the onset of symptoms is given as a number in a square or triangle.
All patients and controls were tested by NS, which samples the primary infection site: the nasopharyngeal area. Five patients with COVID-19 were sampled longitudinally at 2–4 time points by NS to investigate cellular and transcriptional dynamics during the course of disease (patients BIH-CoV-06, BIH-CoV-07, BIH-CoV-12, BIH-CoV-14 and BIH-CoV-15). For two patients, multiple samples from both the upper and lower airways were collected (NS, PSB and BL), allowing comparison between compartments of the respiratory tract (patients BIH-CoV-01 and BIH-CoV-04).
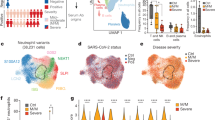
We provide transcriptional profiles of 160,528 cells obtained from a total of 36 samples taken from 24 individuals (Fig. 2a,b and Extended Data Fig. 1a). In NS samples, we identified 22 different cell types and states within the epithelial cell and immune cell populations (Fig. 2a,b and Extended Data Fig. 1a). We detected all previously described major epithelial cell types of the conducting airways, including basal, secretory and ciliated cells, as well as the recently discovered FOXN4+ cells and ionocytes (Fig. 2a,b, Extended Data Fig. 2a and Supplementary Table 2; see Methods for additional information)11,12,13. Notably, we identified a subpopulation of epithelial cells differentiating from immature secretory cells to ciliated cells with a distinctively strong interferon gamma (IFNG) response signature (IFNG-responsive cells; Fig. 2a,b and Extended Data Fig. 2a), suggesting their stimulation by the host immune system14,15. Within the immune cell population, we identified 13 different cell types and states, consisting of macrophages, dendritic cells, mast cells, neutrophils, B cells, T cells, natural killer (NK) cells and subsets thereof (Fig. 2a,b, Extended Data Fig. 2b and Supplementary Table 2; for further details, see Methods).

a, UMAP displaying all identified cell types and states. b, Table displaying the total contribution of each cell type aggregated for the controls. COVID-19 moderate and critical cases in percentage of total epithelial and immune cell types, respectively. Significant differences compared to control samples as calculated by logistic regression followed by Tukey’s post hoc test and Benjamini–Hochberg correction (P < 0.05; see Supplementary Table 2 for exact values) are indicated by *. c, Distribution of ACE2+, ACE2+/TMPRSS2+, ACE2+/FURIN+ and ACE2+/TMPRSS2+/FURIN+ cells across all cell types within the UMAP. d, UMAP depicting ACE2+ cells (dark gray) and virus RNA+ cells (red circles). e, Viral RNA reads within aggregated pseudo-bulks for each patient sample in CPM against days after symptoms. Typically, only very low numbers of viral RNA reads were detected 11 d after first symptoms and later. NS samples were used in this analysis: n = 5 controls, n = 8 patients with moderate COVID-19 and n = 11 patients with critical COVID-19.
Comparing the cellular landscapes, we observed that patients with critical COVID-19 showed a striking depletion of basal cells (P < 1.00 × 10−9) and a strong enrichment for neutrophils (P < 1.00 × 10−9) compared to both patients with moderate COVID-19 and controls (Fig. 2b, Extended Data Fig. 1a and Supplementary Table 2). This finding should be interpreted with caution as cell populations change dramatically over the course of active viral infections16,17,18 and are affected by clinical and genetic differences, sampling dates and the number of days since the onset of symptoms.
Dynamics of epithelial differentiation upon SARS-CoV-2 infection
As the stem cells of the airway epithelium, basal cells (TP63+/KRT5+) preferentially differentiate into secretory cells that further differentiate into ciliated cells19. This differentiation trajectory changes during an insult or injury of the airway epithelium. In such cases, basal cells are induced to directly differentiate into ciliated cells, thereby bypassing the intermediate secretory cell stage11,20,21. We inferred the differentiation pathways of basal cells from our scRNA-seq data by pseudo-time mapping using reversed graph embedding (Fig. 3)22. The prototypical differentiation of basal cells, through secretory cells to terminally differentiated ciliated cells, is mediated by FOXN4+ cells (path 1 in Fig. 3a,b,d). Genes driving this differentiation pathway include the master transcription factor for ciliated cells, FOXJ1, and a component for mature cilia, TCTEX1D2 (r(ref. 23,24,25). Moreover, FOXN4+ cells were indeed described to be in a state of multi-ciliated cell differentiation11. Thus, FOXN4+ cells represent the typical pathway of differentiation as shown by pseudo-time mapping and gene expression profiling.

a, UMAP displaying the subset of airway epithelial cells of all patients infected with SARS-CoV-2 used in this study (see Methods). b, Pseudo-time trajectory projected onto a UMAP of selected epithelial cells. Pseudo-time values are color coded. The red dot indicates the start point of the trajectory. Numbers in circles indicate three inferred differentiation paths c, Dot plot depicting the markers of the epithelial cell types. Expression levels are color coded; the percentage of cells expressing the respective gene is size coded. d, Cell density of airway epithelial cell clusters along inferred cellular trajectory reflecting epithelial cell differentiation. Cells are ordered by pseudo-time (cell-to-cell distance metric) as computed by reversed graph embedding approach of Monocle3 (left). Expression of specific genes along the cell trajectory important for the corresponding differentiation paths (right). The dots indicate the gene expression of individual cells colored by the epithelial cell type. The green lines approximate expression along the inferred trajectory by polynomial regression fits.
In addition to this classical differentiation pathway, we also identified basal cells initiating the wound healing response by differentiating directly toward the ciliated lineage (basal-diff). We detected multiple genes involved in this trajectory, such as cytokeratins (KRT4 and KRT7)26 and intraflagellar component genes (IFT43)25 (path 2 in Fig. 3a–d). This pathway resembles the described direct differentiation pathway from basal to ciliated cells upon chemical injury11.
Finally, our analysis predicted a new, alternative differentiation pathway (path 3 in Fig. 3b) leading from immature secretory cells directly into ciliated cells mediated by IFNG-responsive cells, suggesting that this direct differentiation pathway is dependent on the interferon response and marked by interferon-stimulated genes (ISGs), such as ISG15, IFIT1 and CXCL10 (Fig. 3d)27.
Variable ACE2 expression in the upper airway epithelium
SARS-CoV-2 is thought to initially infect the mucosa of the upper respiratory tract facilitated by viral binding to the host receptor ACE2 and S protein priming by specific proteases, such as TMPRSS2 or FURIN28,29,30,31,32. Expression levels of ACE2 with either one or both S-priming proteases were increased by three-fold in secretory cells in patients with COVID-19 compared to non-infected controls (Fig. 2c, Extended Data Fig. 1b,c and Supplementary Table 3). Although the number of ACE2+ ciliated cells was not increased after infection, this cell population had a high overall number of ACE2+ cells, suggesting a generally high susceptibility for infection. Indeed, this susceptibility was recently confirmed by preferential ACE2 protein localization on motile cilia and a strong infectivity of ciliated cells by SARS-CoV-2 in vitro33,34. Most ciliated cells were ACE2+/TMPRSS2+, whereas secretory and FOXN4+ cells were predominantly ACE2+/TMPRSS2+/FURIN+ (Fig. 2c, Extended Data Fig. 1b–d and Supplementary Table 3). Secretory and ciliated cells also contained the highest fraction of SARS-CoV-2-infected cells (Fig. 2d). In agreement with recent findings35, the number of detected viral transcripts as count per million (CPM) reads was rather low in all patients with COVID-19 and mostly detectable at considerable levels within the first 11 d after the onset of symptoms (Fig. 2e). ACE2+ cells were significantly overrepresented within the epithelial cells compared to the immune cells (58–98-fold enrichment, P < 0.001). Viral transcripts captured in a very low number of immune cells (Fig. 2d) were likely due to viral association (for example, phagocytosis or surface binding of viral particles) rather than productive infection. This view is in line with a study reporting virtual absence of viral reads in peripheral blood mononuclear cell samples from patients with COVID-1936.
Immune cell–epithelial cell interactions increase infectability of epithelial cells
A puzzling aspect of SARS-CoV-2 infection is the surprisingly small number of ACE2+ target cells in the human respiratory tract of healthy controls37,38. We found that, upon infection, ACE2 expression levels increased by three-fold in a subset of epithelial cells (Extended Data Fig. 1b,c and Supplementary Table 3). We observed a statistically significant correlation between the percentage of both ACE2+ ciliated and secretory cells in a given sample and the number of ligand–receptor interactions of those cells with cytotoxic T lymphocytes (CTLs) (P = 8.4 × 10−5; Fig. 4a and Methods). ACE2 expression has been identified as interferon responsive39. We found that STAT1, a central transcription factor of the interferon response40, was among the top predictors for ACE2 expression (Extended Data Fig. 3a), confirming ACE2 as an interferon-inducible gene in human airway epithelium. The preferential expression of IFNG by CTLs and of genes encoding its receptors, IFNGR1 and IFNGR2, by secretory and ciliated cells supports the notion that ACE2 is upregulated in epithelial cells at least partially through IFNG signaling by immune cells (Extended Data Fig. 3b).

a, Scatter plot of ACE2 expression in secretory and ciliated cells versus their interaction strength with CTLs in patients and controls, excluding samples with fewer than 20 cells per cell type (n = 19 patients and 27 samples). Spearmanʼs ρ2 and corresponding P values indicated for CTL interaction with secretory cells (green), ciliated cells (blue) and both combined (black). b, Heat map depicting cell–cell communications between all identified cell types derived from log-scaled ligand–receptor interaction counts in moderate (left, n = 8 patients and 14 samples) and critical (right, n = 11 patients and 13 samples) patients. Interaction strengths are color coded. c, Dot plots of pro-inflammatory and cytotoxic mediators in CTL, moMa and nrMa. Significantly altered expression (Benjamini–Hochberg adjusted two-tailed, negative-binomial P < 0.05) in critical versus moderate (circles around critical) and moderate versus controls (circles around moderate) is marked by a red circle. d, Dot plots of chemokine/receptor expression in the different immune cells of patients with critical COVID-19 (n = 11). Scaling according to Extended Data Fig. 4. e, Expression of apoptosis-related genes in epithelial cells comparing critical cases, moderate cases and controls (n = 5 controls, 8 moderate and 11 critical). Asterisks above critical indicate P values for critical versus control; asterisks above moderate indicate significance of moderate versus control. *P < 0.05, **P < 0.01, ***P < 0.001. Statistics as in c; exact P values in Extended Data Fig. 5b. f. Dot plots of inflammatory and cytotoxic mediators from NS, PSB and BL collected from patients with critical COVID-19 (BIH-CoV-01 and BIH-CoV-04). Visualization as in c. g, CASP3 expression in immune and epithelial cell subtypes of these patients. Comparing PSB versus NS (top) and BL versus NS (bottom). Significant decreases (nasal versus other sampling site) are indicated. Rel. Exp., relative gene expression; Pct. Exp., percentage of cells expressing the gene.
Immune-mediated epithelial damage in severe COVID-19
Infected epithelial cells secrete chemokines that recruit and activate different immune cell populations at the site of viral infection15. In patients with moderate COVID-19, secretory cells showed a significantly higher expression of the chemokine–ligand encoding genes CXCL1, CXCL3, CXCL6, CXCL16 and CXCL17 compared to the controls, likely promoting the recruitment of neutrophils, T cells (CTLs and CD4T/Treg) and mast cells, respectively (Extended Data Fig. 4, middle panel). Ciliated cells strongly expressed CCL15, which might contribute to an inflow of monocytes/macrophages and neutrophils via CCR1. The chemokine and chemokine receptor expression of the different cell populations increased markedly in the critical compared to the moderate cases (Extended Data Fig. 4, lower panel), suggesting an augmented recruitment of immune cells to the sites of inflammation.
To map the complex interactions of epithelial and immune cells, we inferred all possible intercellular communications by the expression of ligand–receptor pairs in both cell populations with CellPhoneDB41 (Fig. 4b). In moderate cases, the strong interactions among ciliated cells, secretory cells and different immune cell types, including CTLs, CD4T/Tregs, B cells, non-resident macrophages (nrMa), monocyte-derived macrophages (moMa) and neutrophils, might reflect the necessary and well-balanced immune response required for the efficient elimination of virus-infected epithelial cells (Fig. 4b, left). Interactions between the different cell types were higher in patients with critical COVID-19 compared to patients with moderate COVID-19, in particular for CTL, nrMa and moMa (Fig. 4b, right). Higher numbers of epithelium–immune cell interactions in patients with critical COVID-19 are consistent with a higher activation status of nrMa, moMa and CTL (Fig. 4c). In particular, nrMa showed a highly inflammatory profile characterized by the expression of the chemokine encoding genes CCL2 (encoding MCP1), CCL3 (encoding MIP1α), CCL20, CXCL1 and CXCL3 and the pro-inflammatory cytokines IL1B, IL8, IL18 and TNF. Except for CXCL3 and IL1B, each of these inflammatory mediators was expressed at significantly higher levels in patients with critical COVID-19 (Fig. 4c). moMa were mainly characterized by CCL2, CCL3 and CXCL10 expression, with higher CCL3 expression levels in the patients with critical COVID-19 (Fig. 4c). Notably, increased CCL2 and CCL3 expression corresponded to a significant induction of CCR1—encoding the receptor binding to MIP1α and, with a lower affinity, to MCP1 as well42—in neutrophils, CTLs and different macrophage populations (Fig. 4d and Extended Data Fig. 4).
In the patients with moderate COVID-19, CTL displayed the characteristic transcriptional profile of anti-viral CD8+ T lymphocytes with high expression of IFNG, TNF, CCL5 and PRF1 (encoding perforin) and GZMB and GZMA (encoding granzymes) together with genes encoding for cytotoxic receptors (KLRB1, KLRC1 and KLRD1) (Fig. 4c). In the patients with critical COVID-19, CTL expressed lower levels of CCL5, IFNG and TNF but strongly increased their cytotoxic potential (Fig. 4c). The potential resulting damage to epithelial cells is reflected by a statistically significantly increased death of ciliated and secretory cells in moderate and critical cases versus control cases (P values listed in Extended Data Fig. 5b). FOXN4+ cells, which were found only in patients with COVID-19, displayed the highest levels of effector caspase 3 (CASP3). This increased level of cell death is reflected by the marked upregulation of genes encoding cell death receptors (FAS and TNFRS1A) and pro-apoptotic factors, including the initiator caspase 8 (CASP8), CASP3 and cytochrome c (CYCS), in ciliated, FOXN4+ and secretory cells (Fig. 4e and Extended Data Fig. 5).
To assess whether the inflammatory macrophages and activated CTLs observed in the nasopharyngeal mucosa were also present in the lower airways, we collected multiple samples of two patients (BIH-CoV-01 and BIH-CoV-04) from the upper (NS) and lower (PSB and BL) airways. All epithelial and immune cell populations were present in the integrated scRNA-seq data set of all three sampled sites of the airways (Extended Data Fig. 6a,b). Furthermore, distribution of ACE2+ cells was spread across all three sites, whereas cells containing viral reads primarily appeared in neutrophils in BL samples, potentially reflecting the phagocytosis of apoptotic virus-infected epithelial cells (Extended Data Fig. 6c). This is in agreement with our observation that the viral load, as measured by total counts of viral reads and viral copy numbers, was higher in BL compared to the nasopharyngeal mucosa for both patients (Extended Data Fig. 6d,e).
Macrophages in the lower airways (PSB and BL) had a stronger inflammatory signature than those within the upper airways (NS) as both the expression level for genes encoding for inflammatory chemokines/cytokines and the number of positive macrophages were markedly elevated (Fig. 4f). The presence of highly inflammatory macrophages and CTLs (Fig. 4f) corresponded with an increased expression of effector CASP3 in the PSB as well as the BL samples. The potential for apoptosis in basal and ciliated cells was significantly higher in the lower (PSB or BL) compared to the upper (NS) airways, reflecting the presence of highly inflammatory macrophages and CTLs (Fig. 4g).
Patient-specific inflammatory profiles
To systematically compare the pronounced differences in the cytokine/chemokine expression profiles between individual cases, we compared the expression patterns within nrMa in longitudinal samples of patients with different degrees of disease severity (Fig. 5). Patient BIH-CoV-12 (male, 71 years old) is a typical patient with a moderate course of COVID-19. Patient BIH-CoV-07, a middle-aged male without any risk factors, suffered from critical COVID-19 with transient need for high-flow nasal oxygen ventilation and intensive care monitoring. He was admitted to the intensive care unit (ICU) with an isolated respiratory failure and bipulmonary infiltrates. He was discharged from the ICU after 2 d and left the hospital in good health. Patient BIH-CoV-06 is a 63-year-old male, with obesity and a history of smoking, who presented with critical COVID-19. He required mechanical ventilation and developed severe acute respiratory distress syndrome (ARDS) and multi-organ failure. After 2 weeks in the ICU, his condition gradually improved, and he could be extubated. Patient BIH-CoV-02 had critical COVID-19 with severe ARDS and died.

Dot plots depicting the average expression of specific cytokines/chemokines and extravasation cascade genes of patients with moderate (BIH-CoV-12) and critical (BIH-CoV-02, BIH-CoV-06 and BIH-CoV-07) COVID-19, ordered by severity as indicated by different clinical features. The sampling days per patient are reflected by ‘days after onset of symptoms’. Expression levels are color coded; the percentage of cells expressing the respective gene is size coded. Significant changes for each time point in the patients with critical COVID-19 versus an average of the three time points of the patient with modertate COVID-19 (BIH-CoV-12) (left panel) are marked by a red circle (Benjamini–Hochberg adjusted two-tailed, negative-binomial P < 0.05). Rel. Exp., relative gene expression; Pct. Exp., percentage of cells expressing the gene.
The expression patterns of genes encoding for cytokines/chemokines in patients BIH-CoV-12 and BIH-CoV-07 were similar. Only a slight increase of IL8 expressing nrMa over time was observed for patient BIH-CoV-07 (Fig. 5). Both mechanically ventilated patients with critical COVID-19 (BIH-CoV-02 and BIH-CoV-06) showed increased expression of CCL20, CXCL5, CXCL3, CXCL1, CCL3 and CCL2, along with an enhanced expression of TNF and IL1B compared to patients BIH-CoV-12 and BIH-CoV-07 (Fig. 5). In patient BIH-CoV-06, these changes increased over time, converging on a similar pattern as observed for patient BIH-CoV-02, who died shortly after the depicted observation time point. This would be in line with the clinical course of patient BIH-CoV-06, as he remained ill in critical condition for a prolonged period of time.
Extravasation of immune cells and the recruitment of blood monocytes to the infected or injured tissue and their migration across the endothelium are crucial events in early immune defense against viral infection, but they also promote capillary leakage and lung damage. A variety of molecules is involved in monocyte trafficking across the vessel wall (for example, integrins)43. Assessing the expression of markers along the extravasation cascade with respect to COVID-19 severity, we observed a strong increase of these genes in nrMa. ITGAL (encoding CD11a), ITGAM (encoding CD11b), ITGAX (encoding CD11c), ITGB1 (encoding CD29) and ITGB1 (encoding CD18) represent integrins forming the complexes LFA-1, Mac-1 and VLA-4, which are all described as essential for monocyte migration across the endothelium44.
Patient BIH-CoV-06 showed a marked increase of cells expressing ITGAM, ITGAX, ITGB2 and ITGB1 at both time points investigated compared to patient BIH-CoV-12 with moderate COVID-19 (Fig. 5). This is in line with the increased proportion of CD44, a crucial mediator of host defense, expressing cells in this patient45,46,47,48. We found that ICAM1 and VEGFA—attractant factors commonly expressed by endothelial (and epithelial) cells to recruit leukocytes to sites of infection—also expressed by monocytes themselves, suggesting that activated monocytes induced their own recruitment. The increased expression of these extravasation markers indicates an enhanced anti-viral immune response, which might damage the epithelial and endothelial barrier alike.
Patient BIH-CoV-02, with the most critical clinical course, showed a similar pattern of extravasation marker expression (Fig. 5). In general, a smaller proportion of nrMa expressed the extravasation markers in this patient.
Discussion
In this study, we provide the first comprehensive description of the transcriptional and cellular landscape of the upper and lower respiratory tract in patients with COVID-19. It has been repeatedly suggested that an excessive production of pro-inflammatory cytokines promotes the development of fatal pneumonia and acute lung injury in COVID-194,9. In agreement with recently published studies on bronchioalveolar lavage fluid cells of patients with mild and severe COVID-195,6, we show an inflammatory FCN1+ macrophage phenotype in patients with critical COVID-19, which shares similarities with our nrMa population. Expressing pro-inflammatory mediators such as CCL2, CCL3, CCL20, CXCL1, CXCL3, CXCL10, IL8, IL1B and TNF, this macrophage subpopulation might contribute to excessive inflammation by promoting monocyte recruitment and differentiation. Increased levels of those chemokines were found in plasma samples of patients with COVID-19 who died3.
Although recent studies have investigated the immune cell response in patients with COVID-19 in the lower respiratory tract5,6, our study adds fundamental understanding of the cellular response to SARS-CoV-2 infection in several aspects. First, we provide a cellular dissection of both the upper respiratory tract in the nasopharynx as the primary site of viral infection and the lower respiratory tract. The side-by-side comparison of the upper and lower respiratory tract in the same patients at the same time point showed that the earlier described hyperinflammatory phenotype5,6 is significantly enhanced in the bronchia compared to the nasopharynx.
Second, we provide a detailed characterization of the interaction between immune cells and epithelial cells that is likely to contribute to the clinical observations of heightened inflammatory tissue damage, lung injury and respiratory failure4,9. We demonstrated that infected epithelial cells upregulate the expression of the SARS-CoV-2 entry receptor ACE2, which correlates with interferon signals in immune cells. This upregulation of ACE2 as an ISG might counteract viral infection, which is in line with the anti-inflammatory and protective function of ACE2 in ARDS49,50,51. In patients with COVID-19, however, an increase in the number of ACE2+ cells potentially renders these patients even more vulnerable to SARS-CoV-2 infection. Together with the presently described IFNG-driven differentiation of immature secretory cells into the largely ACE2+ ciliated cells in patients with COVID-19, this host immune response pathway likely increases the number of ACE2+ cells and thus enhances the susceptibility of the airway epithelium to the SARS-CoV-2 infection.
Third, longitudinal samples of patients with moderate and critical COVID-19 allowed us to correlate the clinical course of disease with changes in the cellular and transcriptional landscape, suggesting pharmacological interventions for patients with critical COVID-19. Development of COVID-19 therapies for immune hyperactivation is currently focused on inhibitors of key immunological players, such as IL-6, TNF and interferon52 (NCT04359901, NCT04322773and NCT04311697). Our data suggest the possibility of targeting chemokine receptors. We found a significant induction of CCL2 and CCL3 expression in macrophages together with an increased expression of CCR1, the receptor for both chemokines, in patients with critical COVID-19. Because binding of CCL2 or CCL3 to CCR1, CCR2 or CCR5 can induce monocyte recruitment into the lung parenchyma with subsequent differentiation into inflammatory macrophages and consecutive recruitment and activation of additional immune cells and epithelial damage, CCR1, CCR2 and CCR5 might represent promising anti-inflammatory targets in COVID-19. Targeting the CCR2/CCL2 axis has been introduced in HIV and other viral infections53. However, we did not observe CCR2 expression in the respiratory tract of patients with COVID-19 (presumably because of its rapid downregulation in monocytes as they exit the bloodstream and enter tissues; Extended Data Fig. 7), leaving CCR1 and/or CCR5 as potential therapeutic targets.
A shortcoming of our study is that we were not able to stratify the patients included in our study, owing to a limited number of hospitalized patients during the COVID-19 pandemic. This impeded a more detailed analysis of potential contributing factors such as age, gender and further comorbidities. In addition, as we were not able to include patients with COVID-19 who showed mild symptoms and as cases without the need for hospitalization were lacking, future studies will have to assess the single-cell landscape of such patients, which, in comparison to our data, might lead to a better understanding of differences in the vulnerability to SARS-CoV-2 infection.
Methods
For a brief summary of the study design, applied statistics/software and data availability, refer to the Life Sciences Reporting Summary.
Material requests should be addressed to roland.eils@charite.de.
Patient recruitment and ethics approval
All patients were enrolled either in the prospective observational cohort study (Pa-COVID-19)54 at Charité - Universitätsmedizin Berlin or the prospective SC2-Study at University Hospital Leipzig, which both included all patients with COVID-19 who were hospitalized between March 11 and May 7, 2020, at either hospital. Both studies were approved by the respective institutional ethics committee of either Charité - Universitätsmedizin Berlin (EA2/066/20) or the Medical Faculty of the University of Leipzig (123/20-ek) and were conducted in accordance with the Declaration of Helsinki. We obtained signed informed consent from all patients before inclusion in our study.
Patient cohort
The classification of COVID-19 severity was based on WHO guidelines10. All patients with COVID-19 (median age, 61 years; range, 21–76 years; 15 males/4 females) described common cold symptoms such as dry cough and fever, followed by malaise, cephalalgia and myalgia at the onset of the disease. In addition, dyspnea was a common initial symptom of the patients with critical COVID-19. The median duration from first symptoms until hospital admission was 9 d for patients with moderate COVID-19 (range, 2–19 d) and 6 d for patients with critical COVID-19 (range, 1–9 d). Patients with moderate COVID-19 were hospitalized for a median duration of 8 d, whereas patients with critical COVID-19 were hospitalized for a median duration of 25 d, with some patients being hospitalized for more than 9 weeks. Most of the patients with critical COVID-19 required mechanical ventilation within the first 9 d after the onset of symptoms, with a median duration of ICU length of stay of 25 d. The only exception was patient BIH-CoV-07, who was admitted to the ICU for 2 d requiring nasal high-flow oxygen therapy. Patient BIH-CoV-08 was the only patient in this study cohort who required extracorporeal membrane oxygenation (ECMO) support. The sequential organ failure assessment (SOFA) scores as well as the respiratory system sub-score of SOFA of the patients in the ICU were used as an indicator for multi-organ dysfunction or lung injury, respectively (Supplementary Table 1). Sex, age, information on the onset of COVID-19 symptoms, diagnosis and hospitalization for each individual are provided in Fig. 1b and Supplementary Table 1. Two patients with critical COVID-19 (BIH-CoV-01 and BIH-CoV-02) died later in the disease course. We included samples of five control patients. These patients were hospitalized for an elective surgery (orthopedic/aesthetic), tested negative for SARS-CoV-2 and had no cold- or flu-like symptoms (BIH-Con-01 to BIH-Con-05; two males and three females; median age, 34 years; range, 24–41 years).
Notably, our cohort does not reflect the general patient distribution admitted to Charité or University Hospital Leipzig with regard to sex, age or COVID-19 severity, as the patients were randomly chosen based on their presence in the hospital and willingness to participate in this study. As a result, the critical and moderate patient groups are not matched with respect to days after onset of symptoms. Hence, in each statistical comparison, we considered ‘days after symptoms’ as a confounding factor in the statistical model (for details, see below) to correct for this imbalance.
We received NSs from all controls and patients with COVID-19 (for sampling days relative to the onset of symptoms, see Fig. 1b and Supplementary Table 1). In addition, we investigated samples obtained from PSBs and BLs from two patients: BIH-CoV-01 and BIH-CoV-04.
Isolation and preparation of single cells from human airway specimens
We used human airway specimens freshly procured using NSs, bronchial brushes or BLs for scRNA-seq. The NSs and bronchial brushes taken from donors were directly transferred into 500 µl of cold DMEM/F12 medium (Gibco, 11039). Processing of all samples started within 1 h after collection and under biosafety S3 conditions (non-infectious donor nasopharyngeal samples were processed under biosafety S1 and S2 conditions).
Nasopharyngeal and bronchial samples were pre-treated with 500 µl of 13 mM DTT (AppliChem, A2948), followed by dislodging the cells from the swabs/brushes by carefully pipetting the solution onto the swab/brush. The swab/brush was dipped 20 times in the solution to ensure the release of all cells and then discarded. BL samples were immediately treated with an equal volume of 13 mM DTT. Afterwards, all following steps were identical for NS, PSB and BL samples. All samples were incubated on a thermomixer at 37 °C at 500 r.p.m. for 10 min and then spun down at 350g at 4 °C for 5 min (but see next paragraph). The supernatant was carefully removed, followed by inspection of the cell pellet for any trace of blood. If so, cells were resuspended in 500 µl of 1× phosphate-buffered saline (PBS) (Sigma-Aldrich, D8537) and 1 ml of RBC Lysis Buffer (Roche, 11814389001), incubated at 25 °C for 10 min and subsequently spun down at 350g at 4 °C for 5 min. The cell pellet was resuspended in 500 µl of Accutase (Thermo Fisher, 00-4555-56) and incubated at room temperature for 10 min to achieve a single-cell suspension. Afterwards, 500 µl of DMEM/F12 supplemented with 10% fetal bovine serum (FBS) was mixed into the cell suspension, followed by centrifugation at 350g at 4 °C for 5 min. After removal of the supernatant, the cell pellet was resuspended in 100 µl of 1× PBS, and the suspension was passed through a 35-µm cell strainer (Falcon, 352235) to remove cellular aggregates, followed by cell counting using a disposable Neubauer chamber (NanoEnTek, DHC-N01). Cell and gel bead emulsions were generated by loading the cell suspension into the 10X Chromium Controller using the 10X Genomics Single Cell 3′ Library Kit v3.1 (10X Genomics: PN 1000223, PN 1000157, PN 1000213 and PN 1000122). The subsequent steps of reverse transcription, cDNA amplification and library preparation were performed according to the manufacturer’s protocol. Note that the incubation at 85 °C during reverse transcription was extended to 10 min to ensure virus inactivation. The final libraries were pooled (S2 flowcell: up to eight samples; S4 flowcell: up to 20 samples) and sequenced with the NovaSeq 6000 Sequencing System with either S2 or S4 flow cells (Illumina; paired end, single indexing).
Samples that were not immediately prepared for cell encapsulation were resuspended in cryopreservation medium (20% FBS (Gibco, 10500), 10% DMSO (Sigma-Aldrich, D8418) and 70% DMEM/F12) and stored at −80 °C. Frozen cells were thawed quickly at 37 °C, spun down at 350g at 4 °C for 5 min and proceeded with normal processing. In cases where satisfactory cell suspensions were achieved without protease treatment, the samples were filtered through a 20-µm cell strainer (pluriSelect, 43-50020-03) and directly loaded for encapsulation.
qRT–PCR
After DTT treatment of the patient samples, 140 µl of supernatant was used for the extraction of viral RNA with the QIAmp Viral RNA Mini Kit (Qiagen, 52904). Quantitative RT–PCR for the E gene of SARS-CoV-255 of the extracted RNA in technical triplicates along with three ten-fold dilutions of standardized SARS-CoV-2 genome equivalents was performed using the SuperScript III One-Step RT–PCR Kit (Thermo Fisher, 12574026) and the Roche LightCycler 480.
Pre-processing and data analysis
The raw 3′ scRNA-seq data were processed using CellRanger version 3.1.0 (10X Genomics). The transcripts were aligned to a customized reference genome in which the SARS-CoV-2 genome (Refseq-ID: NC_045512) was added as an additional chromosome to the human reference genome hg19 (10X Genomics, version 3.0.0). An entry summarizing the entire SARS-CoV-2 genome as one ‘gene’ was appended to the hg19 annotation gtf file, and the genome was indexed using ‘cellranger_mkref’.
After pre-processing, contaminating ambient RNA reads were filtered by using SoupX56 version 1.2.2 (https://github.com/constantAmateur/SoupX) and MUC1, MUC5AC and MUC5B as marker genes. The filtered expression matrices were loaded into R version 3.6.1 with Seurat version 3.1.4.9012 (https://github.com/satijalab/seurat), where further filtering was done to remove cells with fewer than 200 genes expressed or more than 15% mitochondrial transcripts. The quality of the scRNA-seq data set was assessed by plotting the number of unique molecular identifiers (UMIs) and genes per cell for each sample (Extended Data Fig. 8). The quality was consistent across samples, and differences in RNA and gene content could be ascribed to cell-type-specific effects.
An upper cutoff for the number of UMIs was manually determined for each sample based on a plot of gene count versus UMI count, with values ranging between 50,000 and 200,000 UMIs. After quality control filtering, the samples were normalized to 10,000 reads, scaled and centered. For integration, 3,000 shared highly variable genes were identified using Seurat’s ‘SelectIntegrationFeatures()’ function. Integration anchors were identified based on these genes using canonical correlation analysis57 with 90 dimensions as implemented in the ‘FindTransferAnchors()’ function. The data were then integrated using ‘IntegrateData()’ and scaled again using ‘ScaleData()’. Principal component analysis (PCA) and uniform manifold approximation and projection (UMAP) dimension reduction with 90 principal components were performed. A nearest-neighbor graph using the 90 dimensions of the PCA reduction was calculated using ‘FindNeighbors()’, followed by clustering using ‘FindClusters()’ with a resolution of 0.6.
Viral load was calculated on the raw data matrices output by CellRanger. All reads aligning to the SARS-CoV-2 genome per sample were aggregated and divided by the total number of reads in that sample. Multiplication by 106 then yielded the CPM viral load values.
Cell–cell interactions based on the expression of known ligand–receptor pairs in different cell types were calculated using CellPhoneDB41 version 2.1.2 (https://github.com/Teichlab/cellphonedb) on data sub-sampled according to the following strategy: to decrease the effect of samples with high cell numbers, all samples were randomly down-sampled to the size of the smallest sample. A maximum of 2,000 cells per cell type were kept to reduce calculation times. Transcription factor importance was scored using arboreto58 0.1.5 and pyscenic59 0.10.0.
To infer the cluster and lineage relationships between the different epithelial cell types identified, Monocle3 was used22,60 (https://github.com/cole-trapnell-lab/monocle3). A subset of the epithelial cells was used for trajectory inference: basal, secretory-diff, secretory, IFNG-responsive, FOXN4+, ciliated-diff and ciliated cells. After scaling and dimensional reduction by PCA, the UMAP was calculated for this subset. UMAP embeddings and cell clusters generated from Seurat were used as input, and trajectory graph learning and pseudo‐time measurement through reversed graph embedding were performed with Monocle3. To identify genes that are significantly regulated as the cells differentiate along the cell-to-cell distance trajectory, we used the differentialGeneTest() function implemented in Monocle222. This function fits a vector generalized additive model (VGAM) from the VGAM 1.1–2 package for each gene in our data set where the gene expression levels are modeled as a smooth nonlinear function of the pseudo-time value of each cell. Thereafter, genes were ranked by their order of importance based on q values (adjusted P values after Benjamini–Hochberg correction), where q values less than 0.05 were considered as pseudo-time-dependent genes. The expression change of genes, for which q < 1 × 10−250 and which are biologically informative for the corresponding differentiation pathways, was plotted along the cell-to-cell distance (pseudo-time). For the basal-diff trajectory, we performed a VGAM fit on basal, basal-diff, ciliated-diff and ciliated clusters. For the IFNG-responsive cell pathway, the basal, secretory-diff, IFNG responsive, ciliated-diff and ciliated cell types were extracted. Basal, secretory-diff, secretory, FOXN4+, ciliated-diff and ciliated cell clusters were considered when identifying critical genes for the FOXN4+ trajectory.
Statistics
For comparisons between expression values, the Seurat function ‘FindMarkers()’ was used with the ‘negbinom’ method and days after onset of symptoms as the confounder variable. Cell type markers were obtained using the ‘FindAllMarkers()’ function with a negative binomial test and the case severity as the latent variable. Cell type numbers were compared by logistic regression followed by Tukey’s test in multivariate cases and otherwise by Fisher’s exact test. Weights corresponding to the cell count per sample were introduced into the logistic regression to account for differences in information content. Correlation was assessed using Spearman’s Ρ, and significance of correlation was calculated using the corr.test function in R. All tests were two sided. P values were corrected using the Benjamini–Hochberg (false discovery rate) method. Homoskedasticity was assessed using Bartlett’s test. Cells were defined as positive for a gene if at least one UMI of that gene was found. Note that sample numbers in the figure legends refer to biological samples, and the n for statistical calculations is derived from the cell count in expression value comparisons.
Identification of cell type and state
Epithelial and immune cell types were primarily annotated based on their expression levels of their respective cell type markers (Extended Data Fig. 2). All previously described major epithelial cells of the conducting airways, including basal, secretory and ciliated cells, as well as the recently discovered FOXN4+ cells and ionocytes, were found. In addition, we identified a cell type from the most posterior region of the nasopharynx that we call ‘squamous’ cells that is characterized by a strong expression of SPRR genes essential to squamous cell cornification (Fig. 2a,b and Extended Data Fig. 2a)61,62. Basal, secretory and ciliated cells were present in different cell states from differentiating to terminally differentiated as shown by their intermediate expression levels of their respective cell type markers (Extended Data Fig. 2), inferred mitotic stages (Extended Data Fig. 9) and pseudo-time analysis (Fig. 3). In particular, secretory-diff and ciliated-diff cells represent the differentiating state, whereas secretory and ciliated cells are further differentiated. Notably, the FOXN4+ cells resemble the transient secretory cell type described as the most SARS-CoV-2-vulnerable bronchial cells in non-infected individuals by virtue of their function as transitionary cell types differentiating from the secretory to ciliated lineage37 (Figs. 2a, 3a).
We also identified small clusters of cells that are predominantly derived from single patients that we call ‘outliers epithelial’, which have a strong expression of ciliated markers. Similarly, a cluster of cells called ‘unknown epithelial’ is also identified (Extended Data Fig. 2a and Supplementary Table 2). These two cell types were excluded from any downstream analysis.
Within the immune cell population, we identified 13 different cell types, including different types of macrophages, dendritic cells, NK cells, NKT cells, mast cells, neutrophils, B cells and T cells (Fig. 2a,b, Extended Data Fig. 2b and Supplementary Table 2). In the monocyte-derived (MD) fraction, we distinguished resident macrophages (rMa), MD macrophages (moMas), MD dendritic cells (moDCs) and non-resident macrophages (nrMa)63,64. In the T lymphocyte population, we identified CD8+ cytotoxic (CTL) CD4+ T cells together with regulatory T cells (CD4T/Treg) and NKT cells65. Within the immune cell population, we also found NK cells (NCAM1, FCGR3A and KLRD1), distinguishable from the NKT population as they were missing the canonical T cell markers. In addition, proliferating NKT as described previously65 and plasma cells (CD27, SDC1 and CD79A) were observed. The CTL and NKT cell populations were similar to each other and mainly showed a quantitative difference in the expression of genes rather than a strong difference in characteristic markers.
By analyzing the epithelial compartment for trajectory inference, we were able to achieve a higher resolution of the epithelial cell types and identified a sub-cluster of basal cells differentiating to ciliated cells, called ‘basal-diff’. This cell type was defined by the low but mixed marker expression of basal and ciliated cells and by pseudo-time information (Fig. 3a–d).
Cell type identification was similarly done for samples from NSs, bronchial PSBs and BLs for two patients with critical COVID-19 (BIH-CoV-01 and BIH-CoV-04) (Extended Data Fig. 6). We used the same markers as shown in Extended Data Fig. 2 to determine cell types and states. As additional cell types in these samples, we characterized secretory (Secretory LYPD2) and ciliated (Ciliated IFN) cell clusters with a heterogeneous expression of ISGs (IFI6, IFI16, IFI44, IFIT3 and IFITM3). For one particular patient (BIH-CoV-04), we identified a ‘hybrid’ cell type that expresses mixed markers for secretory and ciliated cells (SCGB1A1, MUC5AC, MSMB, TFF3, AGR2, CAPS and SNTN).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Because of the potential risk of de-identification of pseudonymized RNA sequencing data, the raw data are available under controlled access and require a Data Transfer Agreement in the European Genome-phenome Archive repository: EGAS00001004481. In addition, count and metadata tables containing patient identification, sex, age, cell type and quality control metrics for each cell are available at FigShare: https://doi.org/10.6084/m9.figshare.12436517. All count and metadata are integrated for further visualization and analysis in Magellan COVID-19 data explorer at https://digital.bihealth.org.
References
Wu, Z. & McGoogan, J. M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA 323, 1239–1242 (2020).
Guan, W. J. et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 382, 1708–1720 (2020).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Mehta, P. et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395, 1033–1034 (2020).
Bost, P. et al. Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell https://doi.org/10.1016/j.cell.2020.05.006 (2020).
Liao, M. et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 26, 842–844 (2020).
Branchett, W. J. & Lloyd, C. M. Regulatory cytokine function in the respiratory tract. Mucosal Immunol. 12, 589–600 (2019).
Tisoncik, J. R. et al. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 76, 16–32 (2012).
Wen, W. et al. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell Discov. 6, 31 (2020).
Report of the WHO-China Joint Mission on Coronavirus Disease 2019 (COVID-19) https://www.who.int/docs/default-source/coronaviruse/who-china-joint-mission-on-covid-19-final-report.pdf (2020).
Montoro, D. T. et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324 (2018).
Plasschaert, L. W. et al. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560, 377–381 (2018).
Vieira Braga, F. A. et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 25, 1153–1163 (2019).
Schneider, W. M., Chevillotte, M. D. & Rice, C. M. Interferon-stimulated genes: a complex web of host defenses. Annu. Rev. Immunol. 32, 513–545 (2014).
Newton, A. H., Cardani, A. & Braciale, T. J. The host immune response in respiratory virus infection: balancing virus clearance and immunopathology. Semin. Immunopathol. 38, 471–482 (2016).
Ibricevic, A. et al. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J. Virol. 80, 7469–7480 (2006).
Lam, W. Y. et al. Avian influenza virus A/HK/483/97(H5N1) NS1 protein induces apoptosis in human airway epithelial cells. J. Virol. 82, 2741–2751 (2008).
Steuerman, Y. et al. Dissection of influenza infection in vivo by single-cell RNA sequencing. Cell Syst. 6, 679–691 (2018).
Pardo-Saganta, A. et al. Injury induces direct lineage segregation of functionally distinct airway basal stem/progenitor cell subpopulations. Cell Stem Cell 16, 184–197 (2015).
Vaughan, A. E. et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 517, 621–625 (2015).
Zuo, W. et al. p63(+)Krt5(+) distal airway stem cells are essential for lung regeneration. Nature 517, 616–620 (2015).
Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017).
Yu, X., Ng, C. P., Habacher, H. & Roy, S. Foxj1 transcription factors are master regulators of the motile ciliogenic program. Nat. Genet. 40, 1445–1453 (2008).
Cruz, C. et al. Foxj1 regulates floor plate cilia architecture and modifies the response of cells to sonic hedgehog signalling. Development 137, 4271–4282 (2010).
Hirano, T., Katoh, Y. & Nakayama, K. Intraflagellar transport-A complex mediates ciliary entry and retrograde trafficking of ciliary G protein-coupled receptors. Mol. Biol. Cell 28, 429–439 (2017).
Ruiz Garcia, S. et al. Novel dynamics of human mucociliary differentiation revealed by single-cell RNA sequencing of nasal epithelial cultures. Development 146, dev177428 (2019).
Rusinova, I. et al. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 41, D1040–D1046 (2013).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280 (2020).
Walls, A. C. et al. Structure, function and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292 (2020).
Yan, R. et al. Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448 (2020).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Coutard, B. et al. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 176, 104742 (2020).
Ravindra, N. G. et al. Single-cell longitudinal analysis of SARS-CoV-2 infection in human bronchial epithelial cells. Preprint at https://www.biorxiv.org/content/10.1101/2020.05.06.081695v1 (2020).
Lee, I. T. et al. Robust ACE2 protein expression localizes to the motile cilia of the respiratory tract epithelia and is not increased by ACE inhibitors or angiotensin receptor blockers. Preprint at https://www.medrxiv.org/content/10.1101/2020.05.08.20092866v1 (2020).
Wolfel, R. et al. Virological assessment of hospitalized patients with COVID-2019. Nature 581, 465–469 (2020).
Xiong, Y. et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 9, 761–770 (2020).
Lukassen, S. et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 39, e105114 (2020).
Sungnak, W. et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 26, 681–687 (2020).
Ziegler, C. G. K. et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 181, 1016–1035 (2020).
Krause, C. D., He, W., Kotenko, S. & Pestka, S. Modulation of the activation of Stat1 by the interferon-gamma receptor complex. Cell Res. 16, 113–123 (2006).
Efremova, M., Vento-Tormo, M., Teichmann, S. A. & Vento-Tormo, R. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 15, 1484–1506 (2020).
Neote, K., DiGregorio, D., Mak, J. Y., Horuk, R. & Schall, T. J. Molecular cloning, functional expression, and signaling characteristics of a C-C chemokine receptor. Cell 72, 415–425 (1993).
Gerhardt, T. & Ley, K. Monocyte trafficking across the vessel wall. Cardiovasc. Res. 107, 321–330 (2015).
Meerschaert, J. & Furie, M. B. The adhesion molecules used by monocytes for migration across endothelium include CD11a/CD18, CD11b/CD18, and VLA-4 on monocytes and ICAM-1, VCAM-1, and other ligands on endothelium. J. Immunol. 154, 4099–4112 (1995).
Rivadeneira, E. D., Sauls, D. L., Yu, Y., Haynes, B. F. & Weinberg, J. B. Inhibition of HIV type 1 infection of mononuclear phagocytes by anti-CD44 antibodies. AIDS Res. Hum. Retroviruses 11, 541–546 (1995).
Vachon, E. et al. CD44-mediated phagocytosis induces inside-out activation of complement receptor-3 in murine macrophages. Blood 110, 4492–4502 (2007).
Xu, H., Manivannan, A., Crane, I., Dawson, R. & Liversidge, J. Critical but divergent roles for CD62L and CD44 in directing blood monocyte trafficking in vivo during inflammation. Blood 112, 1166–1174 (2008).
He, X. et al. CD44-mediated monocyte transmigration across Cryptococcus neoformans-infected brain microvascular endothelial cells is enhanced by HIV-1 gp41-I90 ectodomain. J. Biomed. Sci. 23, 28 (2016).
Rodrigues Prestes, T. R., Rocha, N. P., Miranda, A. S., Teixeira, A. L. & Simoes, E. S. A. C. The anti-inflammatory potential of ACE2/angiotensin-(1-7)/Mas receptor axis: evidence from basic and clinical research. Curr. Drug Targets 18, 1301–1313 (2017).
Verdecchia, P., Cavallini, C., Spanevello, A. & Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 76, 14–20 (2020).
Imai, Y. et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436, 112–116 (2005).
Coomes, E. A., & Haghbayan, H. Interleukin-6 in COVID-19: a systematic review and meta-analysis. Preprint at https://www.medrxiv.org/content/10.1101/2020.03.30.20048058v1 (2020).
Covino, D. A., Sabbatucci, M. & Fantuzzi, L. The CCL2/CCR2 axis in the pathogenesis of HIV-1 infection: a new cellular target for therapy? Curr. Drug Targets 17, 76–110 (2016).
Kurth, F. et al. Studying the pathophysiology of coronavirus disease 2019 - a protocol for the Berlin prospective COVID-19 patient cohort (Pa- COVID-19). Preprint at https://www.medrxiv.org/content/10.1101/2020.05.06.20092833v1 (2020).
Corman, V. M. et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT–PCR. Euro. Surveill. 25, 2000045 (2020).
Young, M. & Behjati, S. SoupX removes ambient RNA contamination from droplet based single-cell RNA sequencing data. Preprint at https://www.biorxiv.org/content/10.1101/303727v1 (2020).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Moerman, T. et al. GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics 35, 2159–2161 (2019).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Steinert, P. M., Candi, E., Kartasova, T. & Marekov, L. Small proline-rich proteins are cross-bridging proteins in the cornified cell envelopes of stratified squamous epithelia. J. Struct. Biol. 122, 76–85 (1998).
Kalinin, A. E., Kajava, A. V. & Steinert, P. M. Epithelial barrier function: assembly and structural features of the cornified cell envelope. Bioessays 24, 789–800 (2002).
Martinez, F. O., Gordon, S., Locati, M. & Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J. Immunol. 177, 7303–7311 (2006).
Gren, S. T. et al. A single-cell gene-expression profile reveals inter-cellular heterogeneity within human monocyte subsets. PLoS ONE 10, e0144351 (2015).
Travaglini, K. J. et al. A molecular cell atlas of the human lung from single cell RNA sequencing. Preprint at https://www.biorxiv.org/content/10.1101/742320v1 (2019).
Acknowledgements
We thank our patients for kindly donating samples and data to this study. The German Bioinformatics Network de.NBI (031A537B, 031A533A, 031A538A, 031A533B, 031A535A, 031A537C, 031A534A and 031A532B, to R.E.) supported this study in technical aspects regarding the efficient processing of sequencing data. We thank Illumina GmbH for financial support via the allocation of reagents and sequencing flow cells, as well as M. Vossmann, M. Allgaier and O. Krätke for the realization of the sequencing runs at the Illumina Solutions Center Berlin. We further wish to thank Intel Germany GmbH for generously donating computational infrastructure. M.T.V. currently holds a scholarship from the Clinician Scientist Program of the University of Leipzig, Medical Faculty. This study was supported by the COVID-19 research program of the Berlin Institute of Health (to A.H., C.v.K., I.L., L.E.S. and R.E.), the German Research Foundation, SFB-TR84 (C8 to L.E.S, C6 and C9 to M.W. and Z2 to N.S.), the German Ministry of Education and Research in the framework of the CAPSyS project (grant 01ZX1304B to M.W. and N.S.) and the PROVID project (FKZ 01KI20160A to L.E.S., M.W. and N.S.), the Berlin-University Alliance (PreEP-Corona grant to L.E.S), the Juergen Manchot Foundation (to D.W.) and the European Commission (ESPACE, 874710, Horizon 2020 to R.E.). This publication is part of the Human Cell Atlas—www.humancellatlas.org/publications.
Author information
Authors and Affiliations
Contributions
C.C., I.L., L.E.S. and R.E. conceived, designed and supervised the project. R.L.C, Jo.L., F.P., D.W., J.K. and Je.L. performed experiments. R.L.C., So.L., Sa.T., B.P.H., F.P., O.D., L.T., N.I., C.C, I.L. and R.E. analyzed data. D.W., F.K., F.M., H.M.-R., M.T.V., M.M., U.G.L., A.K., Se.S., F.B., C.v.K., N.S., M.W., Sv.L. and L.E.S. provided the human specimens, clinical data and annotation of the patients. A.H., C.G., C.D. and Sv.L. contributed with discussion of the results. B.T., Sv.T., St.S. and J.E. provided technical and experimental support. Sv.T., St.S. and J.E. developed Magellan. R.L.C., So.L., Sa.T., B.P.H., Sv.L., C.C., L.E.S., I.L. and R.E. wrote the manuscript. All authors read, revised and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Distribution of cell types and their ACE2 expression status from the upper respiratory tract.
a, UMAP displaying all identified cell types and states split between controls, moderate and critical COVID-19 patients. b, Distribution of ACE2+, ACE2+/TMPRSS2+, ACE2+/FURIN+, and ACE2+/TMPRSS2+/FURIN+ cells across all cell types within the UMAP split between controls, moderate, and critical COVID-19 patients. c, Percentage of ACE2+ cells across control, moderate, and critical COVID-19 patients. * - indicates a significant difference compared to control samples (Benjamini-Hochberg adjusted p-value obtained from logistic regression followed by Tukey’s post-hoc test < 0.05, see Supplementary Table 2). d, Composition of ACE2+ cells in terms of double or triple positive status for TMPRSS2 and FURIN. Nasopharyngeal swab samples were used in this analysis: n = 5 control samples, n = 14 moderate COVID-19 samples, and n = 13 critical COVID-19 samples for all panels.
Extended Data Fig. 2 Markers used to identify and stratify the different cells of the upper respiratory tract.
a, Dot plot depicting the markers of the epithelial cell types. b, Dot plot depicting the markers used to identify the different immune cell types. Expression levels are color coded, the percentage of cells expressing the respective gene is size coded. For details on cell types and markers refer to Methods. Nasopharyngeal swab samples were used in this analysis: n = 5 control samples, n = 14 moderate COVID-19 samples, and n = 13 critical COVID-19 samples. Rel. Exp. = relative gene expression, Pct. Exp. = percentage of cells expressing the gene.
Extended Data Fig. 3 Identified regulons that are activated in ACE2+ cells.
a, Transcription factors ranked according to association strength with ACE2 expression (for details see Methods). Top 10 hits are highlighted. b, Expression profile of the interferon signaling mediators in all cell types. Please note that IFNG is strongly expressed in CTL. Expression levels are color coded, the percentage of cells expressing the respective gene is size coded. Rel. Exp. = relative gene expression, Pct. Exp. = percentage of cells expressing the gene.
Extended Data Fig. 4 Interaction of chemokines with their respective receptors comparing controls, to moderate and critical COVID-19 cases.
Expression of chemokines and their respective receptors in the different cellular subpopulations is depicted by dot plots. Connecting lines represent receptor/ligand pairs as obtained from the molecular cell atlas of the human lung65 complemented by literature research. Expression levels are color coded while the percentage of cells expressing the gene are depicted by the different dot sizes. Red circles show significance (Benjamini-Hochberg adjusted two-tailed negative-binomial p-value < 0.05) of expression when comparing critical and moderate patients (critical panels) or moderate patients and controls (moderate panels) as calculated by a two-sided significance test. (n = 5 moderate and 6 critical COVID-19 patients, 10 samples each). Nasopharyngeal swab samples were used in this analysis: n = 5 control samples, n = 14 moderate COVID-19 samples, and n = 13 critical COVID-19 samples. Rel. Exp. = relative gene expression, Pct. Exp. = percentage of cells expressing the gene.
Extended Data Fig. 5 Apoptotic marker expression in epithelial cells.
a, Comparison of apoptosis marker gene expression in the different epithelial cell types of critical and moderate COVID-19 cases as well as controls. Asterisks above critical violins indicate p-values for the comparison of critical vs. control samples, asterisks above moderate indicate the comparison moderate vs. control. *: p < 0.05, **: p < 0.01, ***: p < 0.001. p-Values were obtained from a negative-binomial test and Benjamini-Hochberg corrected. Exact p-values are shown in b. Only upregulation is shown, no significant downregulation was observed. b, Table indicating p-values and log fold changes of markers of the extrinsic apoptotic cascade (FAS, CASP8) in epithelial cell populations (for other apoptotic markers see Fig. 3e). The respective comparison, critical vs. control and moderate vs. control, is indicated (n = 5 control, 14 moderate, 13 critical samples).
Extended Data Fig. 6 Cell types/states throughout the upper (NS) and lower (PBS, BL) respiratory tract in the two critical COVID-19 patients BIH-CoV-01 and BIH-CoV-04.
a, UMAP depicting all identified cell types across the upper (NS) and lower (PSB, BL) respiratory tract of the two critical patients BIH-CoV-01 and BIH-CoV-04 (sampling days +17 and +13 after onset of symptoms, respectively). Please note that the majority of cell types correspond to the cell types identified in the upper respiratory tract (cf. Figure 2). For identification and description of the additional cell types including secretory LYPD2, ciliated IFN, basophil, NK and hybrid cells please refer to Methods. b, UMAPs depicting the cell types in the NS (left), PSB (middle) or BL (right) of patients BIH-CoV-01 and BIH-CoV-04. c, UMAP depicting ACE2+ cells and virus+ cells across NS, PBS, or BL samples. d, Viral RNA reads within aggregated pseudo-bulks for patient BIH-CoV-01 and BIH-CoV-04 in counts per million (CPM) detected in either NS, PSB or BL. e. Viral genome equivalents/ml of patient BIH-CoV-01 cell-free sample as measured by quantitative RT-PCR. NS: nasopharyngeal swab, PSB: protected specimen brush, BL: bronchial lavage.
Extended Data Fig. 7 Expression levels of CCL2, CLL3 and their receptors CCR1, CCR2, and CCR5 in immune cells of the lower respiratory tract.
Dot plot depicting the average expression of CCL3/CCR1, CCR5 and CCL2/CCR2, CCR1 in different immune cells in PSB and BL samples for patients BIH-CoV-01 and BIH-CoV-04. Connecting lines represent receptor/ligand pairs. Expression levels are color coded while the percentage of cells expressing the gene are depicted by different dot sizes. PSB - protected specimen brush, BL - bronchial lavage. Rel. Exp. = relative gene expression, Pct. Exp. = percentage of cells expressing the gene.
Extended Data Fig. 8 Quality control metrics.
a, Violin plot depicting Log10 values of total RNA transcripts detected per sample. b, Violin plot showing Log10 values of total genes detected per sample. c, UMAP representing Log10 values of total RNA transcripts detected per cell across all samples. d, UMAP depicting Log10 values of total genes detected per cell for across all samples. Nasopharyngeal swab samples were used in this analysis: n = 5 control samples, n = 14 moderate COVID-19 samples, and n = 13 critical COVID-19 samples.
Extended Data Fig. 9 Inferred cell cycle stages of cells from the upper respiratory tract.
a, Proportion of cells in G1, G2/M, or S phase for each cell type. b, UMAP depicting cells in either G1, G2/M, or S of a subsample of the entire dataset (10,000 cells). Nasopharyngeal swab samples were used in this analysis: n = 5 control samples, n = 14 moderate COVID-19 samples, and n = 13 critical COVID-19 samples.
Supplementary information
Supplementary Information
Supplementary Table 1: Clinical annotation of the cohort of patients with COVID-19.
Supplementary Table 2
Contribution of each cell type from each patient in raw numbers and in percentage for each patient. Confidence intervals and P values were computed.
Supplementary Table 3
Contribution of ACE2+/FURIN+/TMPRSS2+ cells for each cell type from all patients in raw numbers and in percentage for each patient. Confidence intervals and P values were computed using logistic multinomial regression (cell type and disease-state-specific differences) or logistic binomial regression (epithelial–immune comparison).
Rights and permissions
About this article

Cite this article
Chua, R.L., Lukassen, S., Trump, S. et al. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat Biotechnol 38, 970–979 (2020). https://doi.org/10.1038/s41587-020-0602-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-020-0602-4
