
Abstract
Humans have infected a wide range of animals with SARS-CoV-21,2,3,4,5, but the establishment of a new natural animal reservoir has not been observed. Here we document that free-ranging white-tailed deer (Odocoileus virginianus) are highly susceptible to infection with SARS-CoV-2, are exposed to multiple SARS-CoV-2 variants from humans and are capable of sustaining transmission in nature. Using real-time PCR with reverse transcription, we detected SARS-CoV-2 in more than one-third (129 out of 360, 35.8%) of nasal swabs obtained from O. virginianus in northeast Ohio in the USA during January to March 2021. Deer in six locations were infected with three SARS-CoV-2 lineages (B.1.2, B.1.582 and B.1.596). The B.1.2 viruses, dominant in humans in Ohio at the time, infected deer in four locations. We detected probable deer-to-deer transmission of B.1.2, B.1.582 and B.1.596 viruses, enabling the virus to acquire amino acid substitutions in the spike protein (including the receptor-binding domain) and ORF1 that are observed infrequently in humans. No spillback to humans was observed, but these findings demonstrate that SARS-CoV-2 viruses have been transmitted in wildlife in the USA, potentially opening new pathways for evolution. There is an urgent need to establish comprehensive ‘One Health’ programmes to monitor the environment, deer and other wildlife hosts globally.
Similar content being viewed by others


Main
As of 9 November 2021, SARS-CoV-2, the virus responsible for coronavirus disease 2019 (COVID-19), has caused more than 5 million deaths globally6. The zoonotic origins of SARS-CoV-2 are not fully resolved7, exposing large gaps in our knowledge of susceptible host species and potential new reservoirs. Natural infections of SARS-CoV-2 linked to human exposure have been reported in domestic animals such as cats, dogs and ferrets, and in wildlife under human care, including several species of big cats, Asian small-clawed otters, western lowland gorillas and mink1. Detection of SARS-CoV-2 by PCR in free-ranging wildlife has been limited to small numbers of mink in Spain and in Utah in the USA, which were thought to have escaped from nearby farms8,9. An in silico study modelling SARS-CoV-2 binding sites on the angiotensin-converting enzyme 2 (ACE2) receptor across host species predicted that cetaceans, rodents, primates and several species of deer are at high risk of infection10. Experimental infections have identified additional animal species susceptible to SARS-CoV-2, including hamsters, North American raccoons, striped skunks, white-tailed deer, raccoon dogs, fruit bats, deer mice, domestic European rabbits, bushy-tailed woodrats, tree shrews and multiple non-human primate species11,12,13,14,15,16,17,18,19,20. Moreover, several species are capable of intraspecies SARS-CoV-2 transmission13,14,15,17,21,22,23, including cats, ferrets, fruit bats, hamsters, raccoon dogs, deer mice and white-tailed deer. Vertical transmission has also been documented in experimentally infected white-tailed deer23. In July 2021, antibodies for SARS-CoV-2 were reported in 152 free-ranging white-tailed deer (seroprevalence 40%) sampled across Michigan, Pennsylvania, Illinois and New York in the USA24, raising the possibility that SARS-CoV-2 has infected deer in the Midwest and northeast regions.
In this study, we report the detection of SARS-CoV-2 in 129 out of 360 (35.8%) free-ranging white tailed deer (O. virginianus) from northeast Ohio using real-time PCR with reverse transcription (rRT–PCR) between January and March 2021. SARS-CoV-2 is a reportable disease in animals, and per international health regulations, these results were reported immediately to the World Organisation for Animal Health (OIE) on 31 August 2021 (Report ID: FUR_151387, Outbreak ID: 89973)25; this was the first PCR-confirmed report of natural infection of SARS-CoV-2 in a cervid globally. Whole-genome sequences of 14 SARS-CoV-2 viruses were deposited in GISAID on 5 October 2021 (Extended Data Table 4). Additionally, we recovered two viable SARS-CoV-2 isolates from our samples, providing evidence that naturally infected deer shed infectious SARS-CoV-2 virus. We used genetic sequence data to estimate the number of human-to-deer transmission events, characterize the genetic diversity of the virus in deer and identify phylogenetic clades of deer-only viruses arising from deer-to-deer transmission.
High infection rate of SARS-CoV-2
We sampled 360 free-ranging white-tailed deer across from 9 locations (Fig. 1a) in northeast Ohio between January and March 2021. Across all sites, SARS-CoV-2 was detected by rRT–PCR in 35.8% of nasal swabs from white-tailed deer (129 out of 360, 95% confidence interval 30.9–41.0%) (Supplementary Table 1). Each site was sampled 1–3 times during the study period, for a total of 18 collection dates (Extended Data Table 1). At least 1 rRT–PCR-positive sample was identified from 17 out of 18 collection dates, with the majority of positive sample Ct values being below 31 (Fig. 1b). Prevalence estimates varied from 13.5% to 70% across the nine sites (Fig. 1c). The highest prevalence estimates of SARS-CoV-2 were observed at four sites (sites 2, 5, 7 and 9) situated in the northern section of the sampled area, adjacent to urban areas with high human population densities (Fig. 1a, c). Male deer (χ2 = 25.45, P < 0.0005) and heavier deer (Wilcoxon–Mann–Whitney P = 0.0056) were more likely to test positive for SARS-CoV-2 (Extended Data Table 2).

a, The nine study sites were spread across a 1,000-km2 landscape of varying population density in northeast Ohio. Darker shading corresponds to higher human population density (people per square mile). Sampling sites 1, 2, 5, 7 and 9 are in close proximity to human populations and are indicated as urban sites with an asterisk in b, c. b, Nasal swabs from white-tailed deer were tested for the presence of SARS-CoV-2 viral RNA using real-time reverse transcriptase PCR (rRT–PCR). Estimates of amount of SARS-CoV-2 viral RNA are represented by (40 − Ct value of the N1 rRT–PCR target). Negative samples are represented with a value of zero. c, The prevalence of SARS-CoV-2 in the white-tailed deer at each study site was estimated using rRT–PCR. The proportion of positive samples is shown with Clopper–Pearson exact 95% confidence interval bars. The number of samples collected at each site is indicated in parentheses. Map created with ArcMap v.10.8.1, using base layers and data from Esri, Garmin, OpenStreetMap, GIS user community, Infogroup and the US Census Bureau.
Three SARS-CoV-2 lineages identified
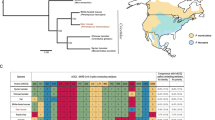
We sequenced the complete genome of 14 viruses collected from 6 of the 9 sites, collected at 7 time points spanning from 26 January 2021 to 25 February 2021 (Supplementary Table 1). The deer samples were collected approximately six weeks after the peak of Ohio’s 2020–2021 winter epidemic of SARS-CoV-2 in humans, which was dominated by B.1.2 viruses (more than 50% of human viruses) (Fig. 2a, Extended Data Table 3). B.1.2 viruses genetically similar to human viruses were detected in deer at sites 4, 7, 8 and 9 (Fig. 2b). B.1.596, a minor lineage (accounting for around 11% of human viruses), was identified in seven deer samples at site 1, spanning two collection times (2 February and 25 February 2021). A rarer lineage, B.1.582 (present in approximately 1% of human samples), was identified in two deer samples at site 6. No sequences belonging to the Alpha (B.1.1.7) or Delta (B.1.617.2) lineages were identified in the deer samples, as these variants became widespread in the human population only after February 2021.

a, The number of weekly COVID-19 cases in humans from October 2020 to September 2021 in Ohio. Shading indicates the proportion of viruses sequenced each week in Ohio that belong to one of five Pango lineages (or ‘other’). b, Summary of six human-to-deer transmission events observed in Ohio, with putative deer-to-deer transmission. c, Maximum-likelihood tree inferred for SARS-CoV-2 viruses in humans and white-tailed deer in Ohio during January to March 2021. Tips are shaded by Pango lineage and major lineages are boxed, labelled and shaded similar to Fig. 2b. Viruses found in white-tailed deer (clusters or singletons) are shaded red and labelled by location (the B.1.2 virus identified at site 4 is not shown owing to lower sequence coverage). All branch lengths are drawn to scale.
Six human-to-deer transmission events
Although B.1.2 was identified in deer at 4 sites, our phylogenetic analysis found no evidence of B.1.2 viruses transmitting in deer across sites. Rather, each site experienced a separate human-to-deer transmission event of a slightly genetically different B.1.2 virus positioned in a different section of the B.1.2 clade on the phylogenetic tree (Fig. 2c). In total, six human-to-deer transmission events were observed: B.1.582 (site 6), B.1.596 (site 1) and B.1.2 (sites 4, 7, 8 and 9). There is a degree of uncertainty about the timing of each viral entry into the deer population, owing to long branch lengths that separate the deer viruses from the ancestral human viruses on the phylogenetic tree. To estimate the timing and location of human-to-deer transmission for the larger cluster of B.1.596 deer viruses, a time-scale Bayesian maximum clade credibility (MCC) tree was inferred using a phylogeographic approach (Fig. 3a). The MCC tree is consistent with human-to-deer transmission occurring geographically in Ohio (posterior probability = 0.98) and temporally during the winter epidemic when viral loads in humans (and the environment) would be peaking. The MCC tree indicates that B.1.596 viruses were introduced into humans in Ohio multiple times from other US states during the autumn of 2020 and winter of 2020–2021, forming three co-circulating Ohio clades in humans. The largest Ohio clade then seeded the deer outbreak. Deer viruses in this cluster were collected on 2 February and 25 February 2021, and the MCC tree estimates that human-to-deer transmission occurred several weeks, or possibly months, earlier (Fig. 3a). Gaps in sampling in both humans and deer make it difficult to narrow this time estimate further.

a, Bayesian time-scale MCC tree inferred for the cluster of 7 B.1.596 viruses identified in white-tailed deer at site 1, the 46 most closely related human B.1.596 viruses, and a random sampling of other B.1.596 viruses observed in the USA during November 2020 to March 2021. Tips are shaded by location state (host species and geography). Branches are shaded by the location state inferred from an ancestral reconstruction. Posterior probabilities are provided for key nodes. Cartoons indicate the host-switch branch where human-to-deer transmission may have occurred, followed by putative deer-to-deer transmission within site 1. The estimated timing and location state probability is provided for key nodes defining the host-switch branch. b, Clade-defining amino acid changes observed in all 7 B.1.596 deer viruses are listed. c, The E484D substitution in the spike protein’s receptor-binding motif (RBM) is shown in one of the B.1.596 deer viruses (OH-OSU-340). NTD, N-terminal domain; RBD, receptor-binding domain.
Deer-to-deer transmission and evolution
Viable SARS-CoV-2 virus was recovered from two of the deer samples (Extended Data Table 4). Deer-to-deer transmission may have occurred within the three study sites where more than 1 deer sample was sequenced: site 1 (B.1.596), site 6 (B.1.582) and site 9 (B.1.2). Only two viruses were collected from sites 6 and 9, both from the same sampling date (Fig. 2b), limiting what can be inferred about transmission. Instead, our analysis of deer-to-deer transmission and evolution focused on the larger deer cluster of 7 B.1.596 viruses observed in site 1 that spans two collection dates (Fig. 3a). A number of uncommon amino acid substitutions were observed in all seven deer viruses in this clade (that is, all site 1 sequences) that were not observed in the most closely related human viruses. Five clade-defining mutations were observed in ORF1ab: a five-residue deletion in nsp1 (∆82–86), nsp2 T434I, nsp2 P597L, nsp12 A382V and nsp13 M474I (numbering in Fig. 3b refers to ORF1a and ORF1b). A clade-defining deletion (∆141–144) was also observed in the S1 domain of the spike protein in the 7 deer viruses. All 6 clade-defining mutations observed in these deer are uncommon among human viruses (<0.05% frequency globally; https://outbreak.info/).
Uncommon amino acid changes in the spike protein S1 domain also were observed in singleton deer viruses. A B.1.2 virus from site 7 (hCoV-19/deer/USA/OH-OSU-0212/2021) has a substitution in the N-terminal domain of the spike protein (H245Y). A single B.1.596 virus from site 1 (hCoV-19/deer/USA/OH-OSU-0340/2021) has a substitution in the spike protein receptor-binding motif (E484D) (Fig. 3c). Both mutations are relatively rare in humans, being found in less than 0.5% of all SARS-CoV-2 viruses sequenced globally. In experimental studies, viruses with the E484D substitution are less sensitive to neutralization by convalescent sera26. The E484D substitution has been detected only in 201 SARS-CoV-2 sequences from humans globally, 71 of which were in the USA, but none of the B.1.596 viruses in humans that were most closely related to the deer virus have this mutation. It is therefore impossible to differentiate whether the E484D mutation arose in an unsampled human virus and was transmitted to deer or arose de novo in deer. Additionally, owing to low availability of sequence data from deer it is not possible to determine whether these spike mutations have been transmitted to other deer.
Discussion
Our finding that white-tailed deer are frequently infected with SARS-CoV-2 viruses raises profound questions about the future trajectory of SARS-CoV-2. The potential establishment of a new reservoir of SARS-CoV-2 viruses in white-tailed deer could open new pathways for evolution, transmission to other wildlife species and potential spillback of novel variants to humans that the human immune system has not previously encountered. SARS-CoV-2 viruses have a high capacity for adaptive evolution when infection rates are high in a community or population. It is therefore concerning that more than one-third of deer in our study tested positive for SARS-CoV-2 by PCR, suggesting an active or recent infection during the major wave the previous winter. A number of mutations were observed in white-tailed deer that occur at very low frequency in humans, including a mutation in the receptor-binding motif. Such mutations could potentially be amplified in a new reservoir host with high infection rates and different constraints on evolution. There is an urgent need to expand monitoring of SARS-CoV-2 viruses in potential wildlife hosts to document the breadth of the problem in white-tailed deer nationally, understand the ecology of transmission and track evolutionary trajectories going forward, including in other potential host species.
The impact of urban sprawl on disease ecology is well documented for Lyme disease and other multihost zoonotic systems that include white-tailed deer, rodents and other species that have become ubiquitous and well adapted in expanding US urban and semi-urban environments, creating opportunities for pathogen exchange. Approximately 30 million free-ranging white-tailed deer are distributed broadly across urban, suburban, and rural environments in the USA, and can live at densities of greater than 45 deer per square mile in some areas27. Ohio is home to more than 700,000 free-ranging white-tailed deer28 and another 440 commercial deer farms29. Estimates of deer density in and around our sites range from approximately 8 km−2 to upwards of 30 km−2. There are no deer farms in the study area and public feeding of deer is prohibited. There is ample forage available around urban and suburban residences in gardens and plantings, drawing deer into close proximity with humans and their companion animals. Therefore, is unsurprising that deer in urban sites were at higher risk for infection in our study. Urban settings provide ample opportunities for deer to have direct and indirect contact with human-contaminated sources (for example, trash, backyard feeders, bait stations and wildlife hospitals) that could serve as a pathway for viral spillover into wildlife. Additionally, urban and suburban environments include waterways that could be contaminated by multiple sources18,30. Viable SARS-CoV-2 is shed in human stool. SARS-CoV-2 RNA and has also been detected in wastewater31,32 and urban runoff33, although the infectivity of SARS-CoV-2 from these sources is undetermined. The recent detection of genetically distinct SARS-CoV-2 virus fragments in New York City wastewater introduces an intriguing hypothesis that SARS-CoV-2 could be transmitting cryptically in rodents34. However, although sensitive techniques for detecting viral RNA in wastewater have vastly improved, providing a potentially useful tool for early detection of outbreaks, isolating or whole-genome sequencing viruses to characterize their genetic diversity remains challenging.
A major outstanding question is how the virus is transmitted between deer. Deer are social animals that live in small herds and frequently touch noses. It is unclear whether baiting the deer before collection contributed the increased frequency of SARS-CoV-2 in this study, but concentrating deer with bait could potentially have facilitated pathogen transmission through a population. However, baiting is regularly used in deer population-management programmes and the practice is commonly employed by deer hunters, which makes understanding the effect of baiting on SARS-CoV-2 transmission in free-ranging deer paramount for future studies. The increased rate of infection in males in this study could reflect sex-linked differences in behaviour that increase disease transmission. The higher prevalence of chronic wasting disease and tuberculosis in male white-tailed deer is attributed to larger male home ranges, increased movement and contact with other deer during breeding season (autumn–winter), and dynamic male social group composition and size35. Deer may experience high levels of viraemia and shedding that may be conducive to environmental or aerosol transmission. Another question is whether deer experience clinical disease and whether clinical signs such as sneezing or nasal discharge increase the risk of transmission. Two previous experimental studies reported only subclinical infections in white-tailed deer challenged with SARS-CoV-2, but these studies had very small sample sizes14,23.
Although extensive measures were taken to prevent cross-contamination during sample collection and testing, the nature of field work makes it impossible to completely exclude the possibility. Cross-contamination during sample collection would not invalidate the detection of SARS-CoV-2 in white-tailed deer, but would artificially inflate prevalence estimates. However, the extent of genomic diversity among the sequences recovered on a single sampling day (for example, site 1, sampling 2) indicates cross-contamination during sample collection was probably minimal.
Although our study was limited to northeast Ohio, these findings have implications for other US states, including Michigan, Pennsylvania, New York and Illinois, where high rates of exposure to SARS-CoV-2 in white-tailed deer have been reported on the basis of serology24. Serological assays are notoriously difficult to interpret and many animal health experts had hoped that those results were an artefact. Moreover, the detection of antibodies does not prove active infection. However, the present study suggests that the antibodies observed in deer in other US states may have arisen from active infection, and that the true extent of infections may remain underestimated.
Moreover, it is worth noting that white-tailed deer are a relatively convenient surveillance target because of their abundance and accessibility. The detection of SARS-CoV-2 in free-ranging white-tailed deer naturally raises the question of whether less accessible species are also being infected through viral spillover from humans, which calls for broader surveillance efforts.
Methods
Sample collection
Between January and March 2021, 360 free-ranging white-tailed deer originating from 9 study sites in northeast Ohio (USA) were euthanized as part of a deer population-management programme. Collection occurred at locations that were baited with whole kernel corn for up to two weeks prior to each culling session, and additional deer were collected opportunistically when they were observed away from the bait on a culling session day. In the field, once a deer was collected, the head was wrapped in a plastic bag and an identification tag was attached to a leg. Each day of the programme, collected deer carcasses were transported to a central processing point where samples were collected. All samples were collected by one experienced veterinarian who wore a facemask and gloves that were changed or washed between each sample. A nasal swab was collected from each deer and placed into a tube with brain heart infusion broth (BHIB). After collection, samples were immediately chilled on ice packs then transferred into a −80 °C freezer within 12 h, where they remained until testing was initiated. Samples were collected post mortem, which was exempt from oversight by The Ohio State University Institutional Animal Care and Use Committee.
Diagnostic testing
Samples were initially tested using the Charité/Berlin (WHO) assay36. Viral RNA was extracted from 200 µl of BHIB using Omega Bio-tek Mag-Bind Viral DNA/RNA kit (catalogue (cat.) no. M6246-03). Xeno Internal Control (Life Technologies cat. no. A29763) was included in the extraction to ensure the accuracy of negative results. Five microlitres of extracted RNA was added to Path-ID MPX One-Step Kit master mix (Life Technologies cat. no. 4442135) containing 12.5 µl 2× Multiplex RT-PCR buffer, 2.5 µl enzyme mix, 1.5 µl nuclease free water, 4.5 µl E assay primer/probe panel (Integrated DNA Technologies cat. no. 1006804), and 1 µl XENO VIC Internal Control Assay (Life Technologies cat. no. A29765) for each sample. The cycling parameters for the rRT–PCR were 48 °C for 10 min, 95 °C for 10 min, 45 cycles of 95 °C 15 s and 58 °C 45 s. Samples with a cycle threshold (Ct) of ≤40 were considered positive. If the E assay was positive, the RdRp confirmatory and discriminatory assays were completed using the above master mix formulation and thermocycler parameters, replacing the E assay primer–probe panel with the confirmatory and discriminatory primer–probe panel (Integrated DNA Technologies cat. no. 10006805 and 10006806). The RNA from all samples that tested positive with the E assay, was retested with the CDC rRT–PCR protocol37. Samples that were 2019-nCoV N1 and N2 positive were classified as presumptive positive. A subset of presumptive positive samples was selected for retesting, in which RNA was re-extracted from original samples to verify the rRT–PCR result.
Genomic sequencing
Original sample material for 76 representative presumptive positive samples were sent to the National Veterinary Services Laboratories (NVSL) for confirmatory rRT–PCR testing using the CDC protocol and whole-genome sequencing. Viral RNA was amplified by PCR38 and cDNA libraries were prepared using the Nextera XT DNA Sample Preparation Kit according to manufacturer instructions. Sequencing was performed using the 500-cycle MiSeq Reagent Kit v2. Sequences were assembled using IRMA v.0.6.7 and DNAStar SeqMan NGen v.14.0.1. Additional sequencing was attempted at Ohio State’s Applied Microbiology Services Laboratory using a modified ARTIC V3 method (https://www.protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bh42j8ye). Extracted RNA was reverse transcribed and amplified by PCR with the ARTIC SARS-CoV-2 FS Library Prep Kit (New England Biolabs) according to the manufacturer’s recommended protocol. Amplified products were converted to Illumina sequencing libraries using the RNA Prep with Enrichment (L) Tagmentation Kit protocol (Illumina) with unique dual indexes and 10 cycles of TagPCR. Sequencing libraries were pooled and quantified using ProNex NGS Library Quant Kit (NG1201, Promega). The 650 pM libraries were loaded on P2 sequencing cartridges and analysed with the NextSeq2000 (lllumina) with 2 × 101 bp cycles. Data were transmitted to the BaseSpace Cloud platform (Illumina) and converted to FASTQ file format using DRAGEN FASTQ Generation v3.8.4 (Illumina). DRAGEN COVID Lineage app v.3.5.3 (Illumina) was used to align sequence data and produce quality metrics and consensus genome sequences.
Data analysis
Pangolin v.3.1.11, 2021-09-17 was used to assign lineage39,40. Prevalence was estimated using the number of presumptive positive nasal swabs based upon the final CDC rRT–PCR results. Prevalence estimates, confidence intervals, and other descriptive statistics were calculated using STATA 14.2 (StataCorp).
Virus isolation
In brief, at the NVSL, the samples were diluted between 1:2 and 1:3 in minimum essential medium with Earle’s balanced salt solution (MEM-E). Vero 76 cells that were mycoplasma-free were inoculated with 1.5 ml diluted sample material and adsorbed for 1 h at 37 oC. After adsorption, a replacement medium containing 2 μg ml−1 N-p-tosyl-l-phenylalanine chloromethyl ketone-treated trypsin was added, and cells were incubated at 37 oC for up to 7 days. Cell cultures with exhibiting no cytopathic effects (CPE) were frozen, thawed, and subjected to two blind passages, inoculating the fresh cultures with those lysates as described above. At the end of two blind passages or upon observation of CPE, cell culture material was tested by rRT–PCR for SARS-CoV-2 using the CDC N1 and N2 primer and probe sets.
Phylogenetic analysis
First, a background dataset was compiled from GISAID that included all SARS-CoV-2 sequences available from humans in Ohio, USA during the study period (1 January to 31 March 2021), downloaded on 27 September 2021 (n = 4,801 sequences). To our knowledge, these are the first SARS-CoV-2 viruses sequenced from white-tailed deer globally and no additional sequences from white-tailed deer were available in any public repository for comparison. Pangolin was used to assign a lineage to each human virus. In total, 102 lineages were identified in this dataset, with the most common being B.1.2 (n = 1,766), B.1.1.7 (n = 833), B.1.1.519 (n = 411), B.1.429 (n = 307) and B.1.596 (n = 274). The dataset was aligned using NextClade with Wuhan-Hu-1 as a reference. The alignment was manually trimmed at the 5′ and 3′ ends. The final alignment included only coding regions and was manually edited to be in frame, with stop codons present only at the terminus of genes. A phylogenetic tree was inferred from this dataset using maximum-likelihood methods available in IQ-TREE version 1.6.12 with a GTR + G model of nucleotide substitution and 1,000 bootstrap replicates, using the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health (http://biowulf.nih.gov). The inferred tree was visualized in FigTree v.1.4.4. Outlier sequences were removed with long branch lengths and incongruence between genetic divergence and sampling date, as assessed using TempEst v.1.5.3, typically arising from poor sequence coverage. One of the 14 sequences obtained from deer in our study (hCoV-19/deer/USA/OH-OSU-0025/2021, site 4) was lower in coverage and had a very long branch length and was excluded from the final phylogenetic analysis. To examine the evolutionary origins of the cluster of 7 B.1.596 viruses obtained from deer at site 1 in more granular detail, a second phylogenetic tree was inferred that included all B.1.596 sequences available globally from NCBI’s GenBank (n = 5,586), nearly all (99.8%) from the USA, using similar methods as above. For purposes of visualization a separate phylogenetic tree was inferred that was limited to the sub-clade of B.1.596 viruses (n = 46) most closely related to the 7 deer viruses. This clade, plus 100 viruses randomly sampled from other sections of the tree as background, was used in a subsequent Bayesian phylogeographic analysis. A time-scaled Bayesian analysis using the Markov chain Monte Carlo (MCMC) method was performed the BEAST v.1.10.4 package41, again using the Biowulf Linux cluster. A relaxed uncorrelated lognormal (UCLN) molecular clock was used with a flexible Bayesian skyline population, and a general-time reversible (GTR) model of nucleotide substitution with gamma-distributed rate variation among sites. Each sample was assigned to one of three categories based on host and geography: (1) viruses collected in humans in all US states except Ohio, (2) viruses collected in humans in Ohio, and (3) viruses collected in deer in Ohio. The MCMC chain was run separately three times for each of the datasets for at least 100 million iterations with subsampling every 10,000 iterations, using the BEAGLE 3 library to improve computational performance42. All parameters reached convergence, as assessed visually using Tracer v.1.7.1, with statistical uncertainty reflected in values of the 95% highest posterior density (HPD). At least 10% of the chain was removed as burn-in and runs were combined using LogCombiner v1.10.4 and a MCC tree was summarized using TreeAnnotator v.1.10.4 and visualized in FigTree v.1.4.4. The NVSL vSNP pipeline ((https://github.com/USDA-VS/vSNP) was applied for SNP based phylogenetic analysis using Wuhan-Hu-1 (NC_045512) as a reference.
Epidemiological data
The epidemiological curve of SARS-CoV-2 cases in Ohio from April 2020 to September 2020 was generated using the number of daily reported COVID-19 cases in the state of Ohio (all age groups), available from the US Centers for Disease Control and Prevention (https://data.cdc.gov/Case-Surveillance/COVID-19-Case-Surveillance-Public-Use-Data-with-Ge/n8mc-b4w4). All SARS-CoV-2 genetic sequences from Ohio were downloaded from GISAID on 8 October 2021 (n = 18,052) to estimate the proportion of viruses belonging to different Pango lineages during each week of the epidemic. To account for the intensity of surveillance not being even over time the number of viruses per lineage per week was normalized against the epidemiological curve derived from COVID-19 case counts and visualized using R. To further minimize biases only sequences categorized in the GISAID submission as obtained using a ‘baseline surveillance’ sampling strategy were included in the analysis. The dataset was further trimmed to include only submissions with complete collection dates and sufficient coverage to assign a Pango lineage, resulting in a final dataset of 9,947 sequences from Ohio. For simplicity sub-lineages of B.1.617.2 (for example, AY.3) were consolidated into the Delta category and sub-lineages of B.1.1.7 (for example, Q.3) were consolidated into the Alpha category. Baseline surveillance data before 20 December 2020 was too thinly sampled to reliably estimate the proportion of viruses from different lineages from this time period, so a second figure was generated using all available sequence data. As the proportions of Pango lineages over time proved to be very similar in the baseline data and the complete dataset, the larger dataset that dated back to October 2020 was used in the final figure.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Sequence data from the 14 SARS-CoV-2 viruses from white-tailed deer sequenced in this study are available at NCBI Sequence Read Archive (SRA) under accessions SRR17187542, SRR17187543, SRR17187544, SRR17187545, SRR17187546, SRR17187547, SRR17187548, SRR17187549, SRR17187550, SRR17187551, SRR17187552, SRR17187553, SRR17187554 and SRR17187555 and assembled genome sequences are on GISAID (Extended Data Table 4).
References
Confirmed Cases of SARS-CoV-2 in Animals in the United States, https://www.aphis.usda.gov/aphis/dashboards/tableau/sars-dashboard (USDA APHIS, 2021).
McAloose. D. et al. From people to Panthera: natural SARS-CoV-2 infection in tigers and lions at the Bronx Zoo. mBio 11, e02220 (2020).
Oude Munnink, B. B. et al. Transmission of SARS-CoV-2 on mink farms between humans and mink and back to humans. Science 371, 172–177 (2021).
Schulz, C. et al. SARS-CoV-2-specific antibodies in domestic cats during first COVID-19 wave, Europe. Emerg. Infect. Dis. 27, 3115–3118 (2021).
Sit, T. H. C., et al. Infection of dogs with SARS-CoV-2. Nature 586, 776–778 (2020).
World Health Organization coronavirus (COVID-19) dashboard. WHO https://covid19.who.int/ (2021).
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C. & Garry, R. F. The proximal origin of SARS-CoV-2. Nat. Med. 26, 450–452 (2020).
Shriner, S. A. et al. SARS-CoV-2 exposure in escaped mink, Utah, USA. Emerg. Infect. Dis. 27, 988–990 (2021).
Aguiló-Gisbert, J. et al. First description of SARS-CoV-2 infection in two feral American mink (Neovison vison) caught in the wild. Animals 11, 1422 (2021).
Damas, J. et al. Broad host range of SARS-CoV-2 predicted by comparative and structural analysis of ACE2 in vertebrates. Proc. Natl Acad. Sci. USA 117, 22311–22322 (2020).
Francisco, R. et al. Experimental susceptibility of North American raccoons (Procyon lotor) and striped skunks (Mephitis mephitis) to SARS-CoV-2. Preprint at https://doi.org/10.1101/2021.03.06.434226 (2021).
Schlottau, K. et al. SARS-CoV-2 in fruit bats, ferrets, pigs, and chickens: an experimental transmission study. Lancet Microbe 1, e218–e225 (2020).
Freuling, C. et al. Susceptibility of raccoon dogs for experimental SARS-CoV-2 infection. Emerg. Infect. Dis. 26, 2982–2985 (2020).
Palmer, M. V. et al. Susceptibility of white-tailed deer (Odocoileus virginianus) to SARS-CoV-2. J. Virol. 95, e00083 (2021).
Fagre, A. et al. SARS-CoV-2 infection, neuropathogenesis and transmission among deer mice: implications for spillback to New World rodents. PLoS Pathog. 17, e1009585 (2021).
Lu, S. et al. Comparison of nonhuman primates identified the suitable model for COVID-19. Signal Transduct. Target. Ther. 5, 157 (2020).
Sia, S. F. et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature 583, 834–838 (2020).
Bosco-Lauth, A. et al. Peridomestic mammal susceptibility to severe acute respiratory syndrome coronavirus 2 infection. Emerg. Infect. Dis. 27, 2073–2080 (2021).
Mykytyn, A. Z. et al. Susceptibility of rabbits to SARS-CoV-2. Emerg. Microbes Infect. 10, 1–7 (2021).
Zhao, Y. et al. Susceptibility of tree shrew to SARS-CoV-2 infection. Sci. Rep. 10, 16007 (2020).
Kim, Y.-I. et al. Infection and rapid transmission of SARS-CoV-2 in ferrets. Cell Host Microbe 27, 704–709.e702 (2020).
Shi, J. et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS–coronavirus 2. Science 368,1016–1020 (2020).
Cool, K. et al. Infection and transmission of ancestral SARS-CoV-2 and its Alpha variant in pregnant white-tailed deer. Preprint at https://doi.org/10.1101/2021.08.15.456341 (2021).
Chandler, J. C. et al. SARS-CoV-2 exposure in wild white-tailed deer (Odocoileus virginianus). Proc. Natl Acad. Sci. USA 118, e2114828118 (2021).
Follow-Up Report 18, SARS-CoV-2 in Animals, United States of America, FUR_151387, https://wahis.oie.int/#/report-info?reportId=38714 (World Organisation for Animal Health (OIE), 2021).
Liu, Z. et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 29, 477–488.e474 (2021).
Walters, B. F., Woodall, C. W. & Russell, M. B. White-tailed deer density estimates across the eastern United States, 2008. Data Repository for the University of Minnesota (2016).
ODNR. Managing Ohio’s Deer Herd https://ohiodnr.gov/static/documents/wildlife/wildlife-management/Managing%20Ohio's%20Deer%20Herd%20pub087.pdf
Shepstone Management Company (ed. Inc. Whitetail Deer Farmers of Ohio) (2010).
Franklin, A. B. & Bevins, S. N. Spillover of SARS-CoV-2 into novel wild hosts in North America: a conceptual model for perpetuation of the pathogen. Sci. Total Environ. 733, 139358 (2020).
Bivins, A. et al. Persistence of SARS-CoV-2 in water and wastewater. Environ. Sci. Technol. Lett. 7, 937–942 (2020).
Ai, Y. et al. Wastewater-based epidemiology for tracking COVID-19 trend and variants of concern in Ohio, United States. Preprint at https://doi.org/10.1101/2021.06.08.21258421 (2021).
Bernard, K. et al. Detection of SARS-CoV-2 in urban stormwater: An environmental reservoir and potential interface between human and animal sources. Sci. Total Environ. 807, 151046 (2021).
Smyth, D. S. et al. Tracking cryptic SARS-CoV-2 lineages detected in NYC wastewater. Preprint at https://doi.org/10.1101/2021.07.26.21261142 (2021).
O’Brien, D. J. et al. Epidemiology of Mycobacterium bovis in free-ranging white-tailed deer, Michigan, USA, 1995–2000. Prev. Vet. Med. 54, 47–63 (2002).
Corman, V. M. et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT–PCR. Euro Surveill. 25, 2000045 (2020).
CDC 2019-Novel Coronavirus (2019-nCoV) Real-Time RT-PCR Diagnostic Panel, https://www.fda.gov/media/134922/download (CDC, 2021).
Paden, C. R. et al. Rapid, sensitive, full-genome sequencing of severe acute respiratory syndrome coronavirus 2. Emerg. Infect. Dis. 26, 2401–2405 (2020).
O’Toole, A. et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 7, veab064 (2021).
Rambaut, A. et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 5, 1403–1407 (2020).
Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016 (2018).
Ayres, D. L. et al. BEAGLE 3: improved performance, scaling, and usability for a high-performance computing library for statistical phylogenetics. Syst. Biol. 68, 1052–1061 (2019).
Acknowledgements
This work was supported by The Ohio State University Infectious Diseases Institute and The Ohio State University Center of Microbiome Science (Targeted Investment: eSCOUT – Environmental Surveillance for COVID-19 in Ohio: Understanding Transmission), Centers of Excellence for Influenza Research and Surveillance, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under contract HHSN272201400006C and HHSN272201400008C, and the NIAID Intramural Research Program. We thank H. Cochran, E. Ohl, A. Cleggett, S. Treglia, A. M. Williams, F. Savona, J. W. Smith, D. Sizemore and the Cleveland Metroparks Deer Management Team. The content does not necessarily reflect the views or policies of the U.S. Government or imply endorsement of any products.
Author information
Authors and Affiliations
Contributions
Conceptualization: V.L.H., P.M.D., J.M.N., D.L., S.G., L.S., S.A.F. and A.S.B. Methodology: V.L.H., P.M.D., J.M.N., S.A.F., M.I.N. and A.S.B. Investigation: P.M.D., J.G., C.M., D.H., M.E., R.M.T., M.L.K., K.L., S.R.-A., M.T. and A.S.B. Visualization: M.I.N., D.S.M. and A.S.B. Funding acquisition: V.L.H., J.M.N., J.W., S.A.F. and A.S.B. Project administration: V.L.H., P.M.D., J.M.N. and A.S.B. Supervision: V.L.H., P.M.D., J.G., M.T. and A.S.B. Writing, original draft: V.L.H., M.I.N., D.S.M., M.L.K., S.A.F. and A.S.B. Writing, review and editing: P.M.D., C.M., D.H., J.G., J.M.N., D.L., J.W., S.G., L.S., M.E., R.T., K.L., S.R.-A. and M.T.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Malik Peiris and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Supplementary information
Supplementary Table 1
rRT–PCR testing results. The cycle threshold (Ct) value results for the E assay screen, N1 and N2 rRT–PCR targets are shown for the 360 nasal swabs collected from white-tailed deer as a part of this study. Final SARS-CoV-2 rRT–PCR result was considered presumptive positive if Ct values for all three targets were < 40. For the 76 samples sent to NVSL, confirmatory rRT–PCR results are listed in addition to the samples for which whole-genome sequencing was successfully completed. See Extended Data Table 4 for GISAID accession numbers for the 14 sequences generated.
Rights and permissions
About this article

Cite this article
Hale, V.L., Dennis, P.M., McBride, D.S. et al. SARS-CoV-2 infection in free-ranging white-tailed deer. Nature 602, 481–486 (2022). https://doi.org/10.1038/s41586-021-04353-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-04353-x
This article is cited by
-
Unveiling drivers of fecundity in an urban white-tailed deer population over 20 years of active management
Urban Ecosystems (2025)
-
Study on sentinel hosts for surveillance of future COVID-19-like outbreaks
Scientific Reports (2024)
-
Widespread exposure to SARS-CoV-2 in wildlife communities
Nature Communications (2024)
-
Resistance to SARS-CoV-2 infection in camelid nasal organoids is associated with lack of ACE2 expression
npj Viruses (2024)
-
Immunogenetic-pathogen networks shrink in Tome’s spiny rat, a generalist rodent inhabiting disturbed landscapes
Communications Biology (2024)
