
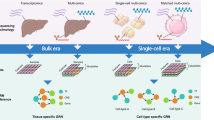
Abstract
The interplay between chromatin, transcription factors and genes generates complex regulatory circuits that can be represented as gene regulatory networks (GRNs). The study of GRNs is useful to understand how cellular identity is established, maintained and disrupted in disease. GRNs can be inferred from experimental data — historically, bulk omics data — and/or from the literature. The advent of single-cell multi-omics technologies has led to the development of novel computational methods that leverage genomic, transcriptomic and chromatin accessibility information to infer GRNs at an unprecedented resolution. Here, we review the key principles of inferring GRNs that encompass transcription factor–gene interactions from transcriptomics and chromatin accessibility data. We focus on the comparison and classification of methods that use single-cell multimodal data. We highlight challenges in GRN inference, in particular with respect to benchmarking, and potential further developments using additional data modalities.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


References
Kim, S. & Wysocka, J. Deciphering the multi-scale, quantitative cis-regulatory code. Mol. Cell 83, 373–392 (2023). This extensive review covers the molecular basis of the cis-regulatory code.
Zaret, K. S. & Carroll, J. S. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 25, 2227–2241 (2011).
Lai, X., Wolkenhauer, O. & Vera, J. Understanding microRNA-mediated gene regulatory networks through mathematical modelling. Nucleic Acids Res. 44, 6019–6035 (2016).
Du, J.-X. et al. Splicing factors: insights into their regulatory network in alternative splicing in cancer. Cancer Lett. 501, 83–104 (2021).
Statello, L., Guo, C.-J., Chen, L.-L. & Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 22, 96–118 (2021).
Carthew, R. W. Gene regulation and cellular metabolism: an essential partnership. Trends Genet. 37, 389–400 (2021).
Davidson, E. H. & Erwin, D. H. Gene regulatory networks and the evolution of animal body plans. Science 311, 796–800 (2006).
Su, E. Y., Spangler, A., Bian, Q., Kasamoto, J. Y. & Cahan, P. Reconstruction of dynamic regulatory networks reveals signaling-induced topology changes associated with germ layer specification. Stem Cell Rep. 17, 427–442 (2022).
Claringbould, A. & Zaugg, J. B. Enhancers in disease: molecular basis and emerging treatment strategies. Trends Mol. Med. 27, 1060–1073 (2021).
Jacob, F. & Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 3, 318–356 (1961). This seminal study delineates a gene regulatory system.
Ideker, T., Galitski, T. & Hood, L. A new approach to decoding life: systems biology. Annu. Rev. Genomics Hum. Genet. 2, 343–372 (2001).
Davidson, E. H. et al. A genomic regulatory network for development. Science 295, 1669–1678 (2002).
Snyder, M. & Gallagher, J. E. G. Systems biology from a yeast omics perspective. FEBS Lett. 583, 3895–3899 (2009).
Han, H. et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 46, D380–D386 (2018).
Garcia-Alonso, L., Holland, C. H., Ibrahim, M. M., Turei, D. & Saez-Rodriguez, J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 29, 1363–1375 (2019).
Liu, Z.-P., Wu, C., Miao, H. & Wu, H. RegNetwork: an integrated database of transcriptional and post-transcriptional regulatory networks in human and mouse. Database 2015, bav095 (2015).
Keenan, A. B. et al. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 47, W212–W224 (2019).
Margolin, A. A. et al. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinforma. 7, S7 (2006).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 9, 559 (2008).
Huynh-Thu, V. A., Irrthum, A., Wehenkel, L. & Geurts, P. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 5, e12776 (2010).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Fiers, M. W. E. J. et al. Mapping gene regulatory networks from single-cell omics data. Brief. Funct. Genomics 17, 246–254 (2018).
Cha, J. & Lee, I. Single-cell network biology for resolving cellular heterogeneity in human diseases. Exp. Mol. Med. 52, 1798–1808 (2020).
Klein, A. M. et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015).
Macosko, E. Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Chen, S., Lake, B. B. & Zhang, K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol. 37, 1452–1457 (2019).
Liu, L. et al. Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity. Nat. Commun. 10, 470 (2019).
Ma, S. et al. Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell 183, 1103–1116.e20 (2020).
Mercatelli, D., Scalambra, L., Triboli, L., Ray, F. & Giorgi, F. M. Gene regulatory network inference resources: a practical overview. Biochim. Biophys. Acta Gene Regul. Mech. 1863, 194430 (2020).
Moerman, T. et al. GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics 35, 2159–2161 (2019).
Lambert, S. A. et al. The human transcription factors. Cell 175, 598–599 (2018).
Holland, C. H. et al. Robustness and applicability of transcription factor and pathway analysis tools on single-cell RNA-seq data. Genome Biol. 21, 36 (2020).
Marbach, D. et al. Wisdom of crowds for robust gene network inference. Nat. Methods 9, 796–804 (2012). This work is a crowdsourced benchmark for GRN inference from bulk transcriptomics data.
Pratapa, A., Jalihal, A. P., Law, J. N., Bharadwaj, A. & Murali, T. M. Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data. Nat. Methods 17, 147–154 (2020).
McCalla, S. G. et al. Identifying strengths and weaknesses of methods for computational network inference from single-cell RNA-seq data. G3 3, jkad004 (2023).
Johnson, D. S., Mortazavi, A., Myers, R. M. & Wold, B. Genome-wide mapping of in vivo protein–DNA interactions. Science 316, 1497–1502 (2007).
Kaya-Okur, H. S. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930 (2019).
Lee, T. I. et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298, 799–804 (2002).
Grosselin, K. et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet. 51, 1060–1066 (2019).
Bartosovic, M., Kabbe, M. & Castelo-Branco, G. Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues. Nat. Biotechnol. 39, 825–835 (2021).
Bartosovic, M. & Castelo-Branco, G. Multimodal chromatin profiling using nanobody-based single-cell CUT&Tag. Nat. Biotechnol. https://doi.org/10.1038/s41587-022-01535-4 (2022).
Qin, J., Hu, Y., Xu, F., Yalamanchili, H. K. & Wang, J. Inferring gene regulatory networks by integrating ChIP-seq/chip and transcriptome data via LASSO-type regularization methods. Methods 67, 294–303 (2014).
Boyle, A. P. et al. High-resolution mapping and characterization of open chromatin across the genome. Cell 132, 311–322 (2008).
Kelly, T. K. et al. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 22, 2497–2506 (2012).
Minnoye, L. et al. Chromatin accessibility profiling methods. Nat. Rev. Methods Prim. 1, 1–24 (2021).
Pranzatelli, T. J. F., Michael, D. G. & Chiorini, J. A. ATAC2GRN: optimized ATAC-seq and DNase1-seq pipelines for rapid and accurate genome regulatory network inference. BMC Genom. 19, 563 (2018).
Qin, Q. et al. Lisa: inferring transcriptional regulators through integrative modeling of public chromatin accessibility and ChIP-seq data. Genome Biol. 21, 32 (2020).
Sonawane, A. R., DeMeo, D. L., Quackenbush, J. & Glass, K. Constructing gene regulatory networks using epigenetic data. NPJ Syst. Biol. Appl. 7, 45 (2021).
Tabula Sapiens Consortium et al. The Tabula Sapiens: a multiple-organ, single-cell transcriptomic atlas of humans. Science 376, eabl4896 (2022).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017). This work presents the first bespoke method to infer GRNs at the single-cell level, introducing the use of TF binding motif information for the estimation of GRNs.
Herring, C. A., Chen, B., McKinley, E. T. & Lau, K. S. Single-cell computational strategies for lineage reconstruction in tissue systems. Cell Mol. Gastroenterol. Hepatol. 5, 539–548 (2018).
Wagner, A., Regev, A. & Yosef, N. Revealing the vectors of cellular identity with single-cell genomics. Nat. Biotechnol. 34, 1145–1160 (2016).
Specht, A. T. & Li, J. LEAP: constructing gene co-expression networks for single-cell RNA-sequencing data using pseudotime ordering. Bioinformatics 33, 764–766 (2017).
Papili Gao, N., Ud-Dean, S. M. M., Gandrillon, O. & Gunawan, R. SINCERITIES: inferring gene regulatory networks from time-stamped single cell transcriptional expression profiles. Bioinformatics 34, 258–266 (2018).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Buenrostro, J. D. et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). This paper introduces single-cell assay for transpose-accessible chromatin (scATAC) technology.
Ramirez, R. N. et al. Dynamic gene regulatory networks of human myeloid differentiation. Cell Syst. 4, 416–429.e3 (2017).
Starks, R. R., Biswas, A., Jain, A. & Tuteja, G. Combined analysis of dissimilar promoter accessibility and gene expression profiles identifies tissue-specific genes and actively repressed networks. Epigenetics Chromatin 12, 16 (2019).
Johnson, J. S. et al. A comprehensive map of the monocyte-derived dendritic cell transcriptional network engaged upon innate sensing of HIV. Cell Rep. 30, 914–931.e9 (2020).
Argelaguet, R., Cuomo, A. S. E., Stegle, O. & Marioni, J. C. Computational principles and challenges in single-cell data integration. Nat. Biotechnol. 39, 1202–1215 (2021).
Ma, A. et al. Single-cell biological network inference using a heterogeneous graph transformer. Nat. Commun. 14, 964 (2023).
Kartha, V. K. et al. Functional inference of gene regulation using single-cell multi-omics. Cell Genom. 2, 100166 (2022). This paper introduces FigR, which has a novel integration strategy for scRNA-seq and scATAC-seq data that can enhance GRN inference.
Cao, Z.-J. & Gao, G. Multi-omics single-cell data integration and regulatory inference with graph-linked embedding. Nat. Biotechnol. 40, 1458–1466 (2022).
Jin, S., Zhang, L. & Nie, Q. scAI: an unsupervised approach for the integrative analysis of parallel single-cell transcriptomic and epigenomic profiles. Genome Biol. 21, 25 (2020).
Jansen, C. et al. Building gene regulatory networks from scATAC-seq and scRNA-seq using linked self organizing maps. PLoS Comput. Biol. 15, e1006555 (2019).
González-Blas, C. B. et al. SCENIC+: single-cell multiomic inference of enhancers and gene regulatory networks. Preprint at bioRxiv https://doi.org/10.1101/2022.08.19.504505 (2022). This study presents a large, curated collection of TF binding motifs and introduces a novel GRN inference method.
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Gasperini, M. et al. A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell 176, 1516 (2019).
Zuin, J. et al. Nonlinear control of transcription through enhancer–promoter interactions. Nature 604, 571–577 (2022).
Kamal, A. et al. GRaNIE and GRaNPA: inference and evaluation of enhancer-mediated gene regulatory networks. Mol. Syst. Biol. https://doi.org/10.15252/msb.202311627 (2023).
Zhang, L., Zhang, J. & Nie, Q. DIRECT-NET: an efficient method to discover cis-regulatory elements and construct regulatory networks from single-cell multiomics data. Sci. Adv. 8, eabl7393 (2022).
Duren, Z., Chen, X., Jiang, R., Wang, Y. & Wong, W. H. Modeling gene regulation from paired expression and chromatin accessibility data. Proc. Natl Acad. Sci. USA 114, E4914–E4923 (2017).
Burdziak, C., Azizi, E., Prabhakaran, S. & Pe’er, D. A nonparametric multi-view model for estimating cell type-specific gene regulatory networks. Preprint at arXiv https://doi.org/10.48550/arXiv.1902.08138 (2019).
Bachireddy, P. et al. Mapping the evolution of T cell states during response and resistance to adoptive cellular therapy. Cell Rep. 37, 109992 (2021).
Kamimoto, K. et al. Dissecting cell identity via network inference and in silico gene perturbation. Nature 614, 742–751 (2023). This work presents a novel GRN inference method from scRNA-seq and scATAC-seq data that also introduces an in silico TF perturbation strategy.
Skok Gibbs, C. et al. High-performance single-cell gene regulatory network inference at scale: the Inferelator 3.0. Bioinformatics 38, 2519–2528 (2022).
Fleck, J. S. et al. Inferring and perturbing cell fate regulomes in human brain organoids. Nature https://doi.org/10.1038/s41586-022-05279-8 (2022).
Li, Z., Nagai, J. S., Kuppe, C., Kramann, R. & Costa, I. G. scMEGA: single-cell multi-omic enhancer-based gene regulatory network inference. Bioinform. Adv. 3, vbad003 (2023).
Jiang, J. et al. IReNA: integrated regulatory network analysis of single-cell transcriptomes and chromatin accessibility profiles. iScience 25, 105359 (2022).
Wang, L. et al. Dictys: dynamic gene regulatory network dissects developmental continuum with single-cell multi-omics. Preprint at bioRxiv https://doi.org/10.1101/2022.09.14.508036 (2022).
Zhang, S. et al. Inference of cell type-specific gene regulatory networks on cell lineages from single cell omic datasets. Nat. Commun. 14, 3064 (2023).
Duren, Z., Chen, X., Xin, J., Wang, Y. & Wong, W. H. Time course regulatory analysis based on paired expression and chromatin accessibility data. Genome Res. 30, 622–634 (2020).
Xu, Q. et al. ANANSE: an enhancer network-based computational approach for predicting key transcription factors in cell fate determination. Nucleic Acids Res. 49, 7966–7985 (2021).
Duren, Z. et al. sc-compReg enables the comparison of gene regulatory networks between conditions using single-cell data. Nat. Commun. 12, 4763 (2021).
Kuppe, C. et al. Spatial multi-omic map of human myocardial infarction. Nature 608, 766–777 (2022).
Argelaguet, R. et al. Decoding gene regulation in the mouse embryo using single-cell multi-omics. Preprint at bioRxiv https://doi.org/10.1101/2022.06.15.496239 (2022).
Zeng, W. et al. DC3 is a method for deconvolution and coupled clustering from bulk and single-cell genomics data. Nat. Commun. 10, 4613 (2019).
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Anderson, A. G. et al. Single nucleus multiomics identifies ZEB1 and MAFB as candidate regulators of Alzheimer’s disease-specific cis-regulatory elements. Cell Genomics 3, 100263 (2023).
Thompson, D., Regev, A. & Roy, S. Comparative analysis of gene regulatory networks: from network reconstruction to evolution. Annu. Rev. Cell Dev. Biol. 31, 399–428 (2015).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Lou, S. et al. TopicNet: a framework for measuring transcriptional regulatory network change. Bioinformatics 36, i474–i481 (2020).
Alvarez, M. J. et al. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 48, 838–847 (2016).
Badia-i-Mompel, P. et al. decoupleR: ensemble of computational methods to infer biological activities from omics data. Bioinforma. Adv. 2, vbac016 (2022).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Garcia-Alonso, L. et al. Transcription factor activities enhance markers of drug sensitivity in cancer. Cancer Res. 78, 769–780 (2018).
Walsh, L. A. et al. An integrated systems biology approach identifies TRIM25 as a key determinant of breast cancer metastasis. Cell Rep. 20, 1623–1640 (2017).
Guan, X. et al. Androgen receptor activity in T cells limits checkpoint blockade efficacy. Nature 606, 791–796 (2022).
Melms, J. C. et al. A molecular single-cell lung atlas of lethal COVID-19. Nature 595, 114–119 (2021).
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
de Sousa Abreu, R., Penalva, L. O., Marcotte, E. M. & Vogel, C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 5, 1512–1526 (2009).
Chung, H. et al. Joint single-cell measurements of nuclear proteins and RNA in vivo. Nat. Methods 18, 1204–1212 (2021).
Bennett, H. M., Stephenson, W., Rose, C. M. & Darmanis, S. Single-cell proteomics enabled by next-generation sequencing or mass spectrometry. Nat. Methods 20, 363–374 (2023).
Uhlén, M. et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteom. 4, 1920–1932 (2005).
Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Weidemüller, P., Kholmatov, M., Petsalaki, E. & Zaugg, J. B. Transcription factors: bridge between cell signaling and gene regulation. Proteomics 21, e2000034 (2021).
Sousa, A. et al. Pan-cancer landscape of protein activities identifies drivers of signalling dysregulation and patient survival. Mol. Syst. Biol. 19, e10631 (2023).
Inukai, S., Kock, K. H. & Bulyk, M. L. Transcription factor–DNA binding: beyond binding site motifs. Curr. Opin. Genet. Dev. 43, 110–119 (2017).
Oki, S. et al. ChIP-Atlas: a data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 19, e46255 (2018).
Boix, C. A., James, B. T., Park, Y. P., Meuleman, W. & Kellis, M. Regulatory genomic circuitry of human disease loci by integrative epigenomics. Nature 590, 300–307 (2021).
Puig, R. R., Boddie, P., Khan, A., Castro-Mondragon, J. A. & Mathelier, A. UniBind: maps of high-confidence direct TF–DNA interactions across nine species. BMC Genom. 22, 482 (2021).
Krebs, A. R. et al. Genome-wide single-molecule footprinting reveals high RNA polymerase II turnover at paused promoters. Mol. Cell 67, 411–422.e4 (2017).
Sönmezer, C. et al. Molecular co-occupancy identifies transcription factor binding cooperativity in vivo. Mol. Cell 81, 255–267.e6 (2021).
Gasperini, M., Tome, J. M. & Shendure, J. Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. Genet. 21, 292–310 (2020).
Ibarra, I. L. et al. Mechanistic insights into transcription factor cooperativity and its impact on protein–phenotype interactions. Nat. Commun. 11, 124 (2020).
Jolma, A. et al. DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature 527, 384–388 (2015).
Lu, H. et al. Recent advances in the development of protein–protein interactions modulators: mechanisms and clinical trials. Signal. Transduct. Target. Ther. 5, 1–23 (2020).
Orchard, S. et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 42, D358–D363 (2014).
Patwardhan, R. P. et al. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol. 27, 1173–1175 (2009).
Ren, X. et al. Parallel characterization of cis-regulatory elements for multiple genes using CRISPRpath. Sci. Adv. 7, eabi4360 (2021).
ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 306, 636–640 (2004).
Thurman, R. E. et al. The accessible chromatin landscape of the human genome. Nature 489, 75–82 (2012).
Hardison, R. C., Oeltjen, J. & Miller, W. Long human–mouse sequence alignments reveal novel regulatory elements: a reason to sequence the mouse genome. Genome Res. 7, 959–966 (1997).
Pennacchio, L. A. et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature 444, 499–502 (2006).
Wang, S. K. et al. Single-cell multiome of the human retina and deep learning nominate causal variants in complex eye diseases. Cell Genom. 2, 100164 (2022).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Mumbach, M. R. et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919–922 (2016).
Ramani, V. et al. Massively multiplex single-cell Hi-C. Nat. Methods 14, 263–266 (2017).
Dixon, J. R. et al. Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336 (2015).
Chen, H. et al. Dynamic interplay between enhancer–promoter topology and gene activity. Nat. Genet. 50, 1296–1303 (2018).
Fukaya, T., Lim, B. & Levine, M. Enhancer control of transcriptional bursting. Cell 166, 358–368 (2016).
Xie, S., Duan, J., Li, B., Zhou, P. & Hon, G. C. Multiplexed engineering and analysis of combinatorial enhancer activity in single cells. Mol. Cell 66, 285–299.e5 (2017).
GTEx Consortium et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
van der Wijst, M. G. P. et al. Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat. Genet. 50, 493–497 (2018).
Kerimov, N. et al. A compendium of uniformly processed human gene expression and splicing quantitative trait loci. Nat. Genet. 53, 1290–1299 (2021).
Dixit, A. et al. Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167, 1853–1866.e17 (2016).
Datlinger, P. et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 14, 297–301 (2017).
Schraivogel, D. et al. Targeted Perturb-seq enables genome-scale genetic screens in single cells. Nat. Methods 17, 629–635 (2020).
Ng, A. H. M. et al. A comprehensive library of human transcription factors for cell fate engineering. Nat. Biotechnol. 39, 510–519 (2021).
Joung, J. et al. A transcription factor atlas of directed differentiation. Cell 186, 209–229.e26 (2023).
Littman, R., Wang, N., Peng, C. & Yang, X. SCING: single cell integrative gene regulatory network inference elucidates robust, interpretable gene regulatory networks. Preprint at bioRxiv https://doi.org/10.1101/2022.09.07.506959 (2022).
Yurkovsky, E. & Nachman, I. Event timing at the single-cell level. Brief. Funct. Genomics 12, 90–98 (2013).
Co, A. D., Lagomarsino, M. C., Caselle, M. & Osella, M. Stochastic timing in gene expression for simple regulatory strategies. Nucleic Acids Res. 45, 1069–1078 (2017).
Lee, M. Y. Y., Kaestner, K. H. & Li, M. Benchmarking algorithms for joint integration of unpaired and paired single-cell RNA-seq and ATAC-seq data. Preprint at bioRxiv https://doi.org/10.1101/2023.02.01.526609 (2023).
Squair, J. W. et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 12, 5692 (2021).
Kuijjer, M. L., Tung, M. G., Yuan, G., Quackenbush, J. & Glass, K. Estimating sample-specific regulatory networks. iScience 14, 226–240 (2019).
Luecken, M. D. & Theis, F. J. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 15, e8746 (2019).
Yan, F., Powell, D. R., Curtis, D. J. & Wong, N. C. From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis. Genome Biol. 21, 22 (2020).
Vandereyken, K., Sifrim, A., Thienpont, B. & Voet, T. Methods and applications for single-cell and spatial multi-omics. Nat. Rev. Genet. https://doi.org/10.1038/s41576-023-00580-2 (2023).
Blencowe, M. et al. Network modeling of single-cell omics data: challenges, opportunities, and progresses. Emerg. Top. Life Sci. 3, 379–398 (2019).
Lähnemann, D. et al. Eleven grand challenges in single-cell data science. Genome Biol. 21, 31 (2020).
van Dijk, D. et al. Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729.e27 (2018).
Bravo González-Blas, C. et al. cisTopic: cis-regulatory topic modeling on single-cell ATAC-seq data. Nat. Methods 16, 397–400 (2019).
Li, Z. et al. Chromatin-accessibility estimation from single-cell ATAC-seq data with scOpen. Nat. Commun. 12, 6386 (2021).
Ly, L.-H. & Vingron, M. Effect of imputation on gene network reconstruction from single-cell RNA-seq data. Patterns 3, 100414 (2022).
Baran, Y. et al. MetaCell: analysis of single-cell RNA-seq data using K–nn graph partitions. Genome Biol. 20, 206 (2019).
Persad, S. et al. SEACells infers transcriptional and epigenomic cellular states from single-cell genomics data. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01716-9 (2023).
Klemm, S. L., Shipony, Z. & Greenleaf, W. J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 20, 207–220 (2019).
Miao, Z. & Kim, J. Is single nucleus ATAC-seq accessibility a qualitative or quantitative measurement? Preprint at bioRxiv https://doi.org/10.1101/2022.04.20.488960 (2022).
Martens, L. D., Fischer, D. S., Theis, F. J. & Gagneur, J. Modeling fragment counts improves single-cell ATAC-seq analysis. Preprint at bioRxiv https://doi.org/10.1101/2022.05.04.490536 (2022).
Stuart, T., Srivastava, A., Madad, S., Lareau, C. A. & Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021).
Granja, J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411 (2021).
Bahr, C. et al. Author Correction: a Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 558, E4 (2018).
Monahan, K., Horta, A. & Lomvardas, S. LHX2- and LDB1-mediated trans interactions regulate olfactory receptor choice. Nature 565, 448–453 (2019).
Chen, X. et al. Mapping disease regulatory circuits at cell-type resolution from single-cell multiomics data. Preprint at medRxiv https://doi.org/10.1101/2022.12.06.22282077 (2022).
Stevens, T. J. et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544, 59–64 (2017).
Flyamer, I. M. et al. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 544, 110–114 (2017).
Zhang, R., Zhou, T. & Ma, J. Multiscale and integrative single-cell Hi-C analysis with Higashi. Nat. Biotechnol. 40, 254–261 (2022).
Yu, M. & Ren, B. The three-dimensional organization of mammalian genomes. Annu. Rev. Cell Dev. Biol. 33, 265–289 (2017).
Marti-Renom, M. A. et al. Challenges and guidelines toward 4D nucleome data and model standards. Nat. Genet. 50, 1352–1358 (2018).
Rossini, R., Kumar, V., Mathelier, A., Rognes, T. & Paulsen, J. MoDLE: high-performance stochastic modeling of DNA loop extrusion interactions. Genome Biol. 23, 247 (2022).
Tan, J. et al. Cell-type-specific prediction of 3D chromatin organization enables high-throughput in silico genetic screening. Nat. Biotechnol. https://doi.org/10.1038/s41587-022-01612-8 (2023). This work demonstrates that the prediction of Hi-C data from chromatin accessibility is a promising strategy to replace the use of genomic distance thresholds.
Yuan, H. & Kelley, D. R. scBasset: sequence-based modeling of single-cell ATAC-seq using convolutional neural networks. Nat. Methods 19, 1088–1096 (2022). This work introduces the concept of in silico mutagenesis to contextualize TF binding motifs to cell types.
Taskiran, I. I., Spanier, K. I., Christiaens, V., Mauduit, D. & Aerts, S. Cell type directed design of synthetic enhancers. Preprint at bioRxiv https://doi.org/10.1101/2022.07.26.501466 (2022).
Lundberg, S. M. & Lee, S.-I. A unified approach to interpreting model predictions. Adv. Neural Inf. Process. Syst. 30, 4765–4774 (2017).
Regev, A. et al. The human cell atlas. eLife 6, e27041 (2017).
HuBMAP Consortium. The human body at cellular resolution: the NIH Human Biomolecular Atlas Program. Nature 574, 187–192 (2019).
Neikes, H. K. et al. Quantification of absolute transcription factor binding affinities in the native chromatin context using BANC-seq. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01715-w (2023).
Xu, W. et al. ISSAAC-seq enables sensitive and flexible multimodal profiling of chromatin accessibility and gene expression in single cells. Nat. Methods 19, 1243–1249 (2022).
Ogbeide, S., Giannese, F., Mincarelli, L. & Macaulay, I. C. Into the multiverse: advances in single-cell multiomic profiling. Trends Genet. 38, 831–843 (2022).
Chen, A. F. et al. NEAT-seq: simultaneous profiling of intra-nuclear proteins, chromatin accessibility and gene expression in single cells. Nat. Methods 19, 547–553 (2022).
Yan, R., Cheng, X. & Guo, F. Protocol for scChaRM-seq: simultaneous profiling of gene expression, DNA methylation, and chromatin accessibility in single cells. STAR Protoc. 2, 100972 (2021).
Hansen, T. J. & Hodges, E. ATAC-STARR-seq reveals transcription factor-bound activators and silencers within chromatin-accessible regions of the human genome. Genome Res. 32, 1529–1541 (2022).
Budnik, B., Levy, E., Harmange, G. & Slavov, N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 19, 161 (2018).
Blair, J. D. et al. Phospho-seq: integrated, multi-modal profiling of intracellular protein dynamics in single cells. Preprint at bioRxiv https://doi.org/10.1101/2023.03.27.534442 (2023).
Uffelmann, E. et al. Genome-wide association studies. Nat. Rev. Methods Prim. 1, 1–21 (2021).
Tedesco, M. et al. Chromatin velocity reveals epigenetic dynamics by single-cell profiling of heterochromatin and euchromatin. Nat. Biotechnol. 40, 235–244 (2022).
Jerkovic, I. & Cavalli, G. Understanding 3D genome organization by multidisciplinary methods. Nat. Rev. Mol. Cell Biol. 22, 511–528 (2021).
Hsieh, T.-H. S. et al. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell 162, 108–119 (2015).
Zhang, Y. et al. Enhancing Hi-C data resolution with deep convolutional neural network HiCPlus. Nat. Commun. 9, 750 (2018).
Lance, C. et al. Multimodal single cell data integration challenge: Results and lessons learned. In Proc. NeurIPS 2021 Competitions and Demonstrations Track (eds. Kiela, D., Ciccone, M. & Caputo, B.) Vol. 176, 162–176 (PMLR, 2022).
Rappez, L. et al. SpaceM reveals metabolic states of single cells. Nat. Methods 18, 799–805 (2021).
Garrido-Rodriguez, M., Zirngibl, K., Ivanova, O., Lobentanzer, S. & Saez-Rodriguez, J. Integrating knowledge and omics to decipher mechanisms via large-scale models of signaling networks. Mol. Syst. Biol. 18, e11036 (2022).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020).
Armingol, E., Officer, A., Harismendy, O. & Lewis, N. E. Deciphering cell–cell interactions and communication from gene expression. Nat. Rev. Genet. 22, 71–88 (2021).
Dimitrov, D. et al. Comparison of methods and resources for cell–cell communication inference from single-cell RNA-seq data. Nat. Commun. 13, 3224 (2022).
Tanevski, J., Flores, R. O. R., Gabor, A., Schapiro, D. & Saez-Rodriguez, J. Explainable multiview framework for dissecting spatial relationships from highly multiplexed data. Genome Biol. 23, 97 (2022).
Fischer, D. S., Schaar, A. C. & Theis, F. J. Modeling intercellular communication in tissues using spatial graphs of cells. Nat. Biotechnol. 41, 332–336 (2023).
Zhang, D. et al. Spatial epigenome-transcriptome co-profiling of mammalian tissues. Nature 616, 113–122 (2023).
Avsec, Ž. et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods 18, 1196–1203 (2021).
Li, Z. et al. Applications of deep learning in understanding gene regulation. Cell Rep. Methods 3, 100384 (2023).
Miraldi, E. R. et al. Leveraging chromatin accessibility for transcriptional regulatory network inference in T helper 17 cells. Genome Res. 29, 449–463 (2019).
Alanis-Lobato, G. et al. MICA: a multi-omics method to predict gene regulatory networks in early human embryos. Preprint at bioRxiv https://doi.org/10.1101/2023.02.03.527081 (2023).
Zenere, A., Rundquist, O., Gustafsson, M. & Altafini, C. Using high-throughput multi-omics data to investigate structural balance in elementary gene regulatory network motifs. Bioinformatics 38, 173–178 (2021).
Ledru, N. et al. Predicting regulators of epithelial cell state through regularized regression analysis of single cell multiomic sequencing. Preprint at bioRxiv https://doi.org/10.1101/2022.12.29.522232 (2022).
Jiang, Y. et al. Nonparametric single-cell multiomic characterization of trio relationships between transcription factors, target genes, and cis-regulatory regions. Cell Syst. 13, 737–751.e4 (2022).
Zambelli, F., Pesole, G. & Pavesi, G. Motif discovery and transcription factor binding sites before and after the next-generation sequencing era. Brief. Bioinform 14, 225–237 (2013).
Weirauch, M. T. et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443 (2014).
Kheradpour, P. & Kellis, M. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res. 42, 2976–2987 (2014).
Kulakovskiy, I. V. et al. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-seq analysis. Nucleic Acids Res. 46, D252–D259 (2018).
Castro-Mondragon, J. A. et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 50, D165–D173 (2022).
Matys, V. TRANSFAC® and its module TRANSCompel®: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34, D108–D110 (2006).
Newburger, D. E. & Bulyk, M. L. UniPROBE: an online database of protein binding microarray data on protein–DNA interactions. Nucleic Acids Res. 37, D77–D82 (2009).
Grant, C. E., Bailey, T. L. & Noble, W. S. FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011).
Bruse, N. & van Heeringen, S. J. GimmeMotifs: an analysis framework for transcription factor motif analysis. Preprint at bioRxiv https://doi.org/10.1101/474403 (2018).
Korhonen, J. H., Palin, K., Taipale, J. & Ukkonen, E. Fast motif matching revisited: high-order PWMs, SNPs and indels. Bioinformatics 33, 514–521 (2017).
Schep, A. N., Wu, B., Buenrostro, J. D. & Greenleaf, W. J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017).
Li, Z. et al. RGT: a toolbox for the integrative analysis of high throughput regulatory genomics data. BMC Bioinforma. 24, 79 (2023).
Sherwood, R. I. et al. Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat. Biotechnol. 32, 171–178 (2014).
Ambrosini, G., Groux, R. & Bucher, P. PWMScan: a fast tool for scanning entire genomes with a position-specific weight matrix. Bioinformatics 34, 2483–2484 (2018).
Janssens, J. et al. Decoding gene regulation in the fly brain. Nature 601, 630–636 (2022).
Wayman, J. A. et al. An atlas of gene regulatory networks for memory CD4+ T cells in youth and old age. Preprint at bioRxiv https://doi.org/10.1101/2023.03.07.531590 (2023).
Acknowledgements
The authors thank the developers of the methods discussed for the insightful feedback they provided. S.M.-D. was funded by the LiSyM-cancer network supported by the German Federal Ministry of Education and Research (BMBF; Funding number 031L0257B).
Author information
Authors and Affiliations
Contributions
All authors researched data for the article. P.B.-i-M., L.W., S.M.-D., R.O.R.F., R.A. and J.S.-R. contributed substantially to discussion of the content. P.B.-i-M., L.W., R.A. and J.S.-R. wrote the article. All authors reviewed and/or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
J.S.-R. reports funding from GSK, Pfizer and Sanofi, and consultant fees from Travere Therapeutics, Stadapharm and Astex Pharmaceutical. R.A. is an employee of Altos Labs. The other authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Genetics thanks X. Yang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Glossary
- Assay for transpose-accessible chromatin with sequencing
-
(ATAC-seq). A technique to identify accessible DNA regions using hyperactive Tn5 transposase.
- Betweenness centrality
-
A network centrality measure representing the number of appearances of a node in the shortest path of any other two nodes in the network.
- Chromatin
-
A higher-order filamentous structure of DNA–protein complex that can exist in a condensed or uncondensed state.
- Chromatin immunoprecipitation followed by sequencing
-
(ChIP-seq). A technique to analyse protein interactions with accessible DNA regions using chromatin immunoprecipitation followed by DNA sequencing.
- cis-Regulatory elements
-
(CREs). Non-coding DNA regions that regulate the transcription of nearby genes upon binding of transcription factors (TFs). These include promoters, enhancers and silencers.
- Cleavage under targets and tagmentation
-
(CUT&Tag). An antibody-based technique to analyse protein interactions with accessible DNA regions using transposase Tn5-mediated tagmentation followed by DNA sequencing.
- Closeness centrality
-
A network centrality measure describing the average distance (length of the shortest path) of a node to all other nodes.
- Degree centrality
-
A network centrality measure describing the number of edges (degree) of a node.
- DNA binding sites
-
DNA sequences where transcription factors can bind to drive gene regulation, usually represented as nucleotide patterns known as motifs.
- Eigenvector centrality
-
A network centrality measure describing the importance of a node in the network based on the centrality of its neighbours.
- Enhancers
-
Distal regulatory DNA regions where transcription regulatory proteins can bind and activate transcription.
- Expression quantitative trait loci
-
Genomic locations whose sequence variation is associated with changes in gene expression.
- Gene regulatory networks
-
(GRNs). Network representations of molecular interactions between transcriptional regulators and target genes.
- Genome-wide association studies
-
Analysis approach to identify frequently appearing single-nucleotide polymorphisms in the genome across a large cohort of individuals.
- Hi-C
-
A technique to study chromatin conformation in three dimensions to identify genomic sequences that might be distal to each other in linear distance but closer in the 3D space.
- Metacells
-
Groups of cells with a similar molecular profile that can be aggregated into a single omics profile to reduce sparsity of the data.
- Motif matcher algorithms
-
String matching algorithms to detect transcription factor binding sites in DNA sequences.
- Network centrality
-
A group of graph theory metrics that defines the relative importance of a node in a network.
- Peaks
-
Regions of accessible chromatin that form the read-out of epigenetic sequencing techniques.
- Promoter
-
A regulatory region in the genome located before the transcriptional start site of a gene.
- Silencers
-
Distal regulatory DNA regions where transcription regulatory proteins can bind and repress transcription.
- Single-nucleotide polymorphisms
-
(SNPs). DNA sequence variations caused by substitution of a single nucleotide in a specific position.
- Topologically associating domains
-
Self-interacting genomic regions with high interaction frequency of sequences within the domain and relative isolation from neighbouring regions, forming a 3D chromosome structure.
- Transcription factors
-
(TFs). Proteins that modify the rate of transcription by binding to specific DNA sequences.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Badia-i-Mompel, P., Wessels, L., Müller-Dott, S. et al. Gene regulatory network inference in the era of single-cell multi-omics. Nat Rev Genet 24, 739–754 (2023). https://doi.org/10.1038/s41576-023-00618-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41576-023-00618-5
This article is cited by
-
Considerations for building and using integrated single-cell atlases
Nature Methods (2025)
-
Integrated analysis of single-cell and bulk RNA sequencing data reveals a myeloid cell-related regulon predicting neoadjuvant immunotherapy response across cancers
Journal of Translational Medicine (2024)
-
scStateDynamics: deciphering the drug-responsive tumor cell state dynamics by modeling single-cell level expression changes
Genome Biology (2024)
-
Deciphering the topological landscape of glioma using a network theory framework
Scientific Reports (2024)
-
Decoding the principle of cell-fate determination for its reverse control
npj Systems Biology and Applications (2024)
