
Abstract
Single-cell RNA sequencing (scRNA-seq) allows researchers to collect large catalogues detailing the transcriptomes of individual cells. Unsupervised clustering is of central importance for the analysis of these data, as it is used to identify putative cell types. However, there are many challenges involved. We discuss why clustering is a challenging problem from a computational point of view and what aspects of the data make it challenging. We also consider the difficulties related to the biological interpretation and annotation of the identified clusters.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others
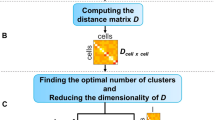

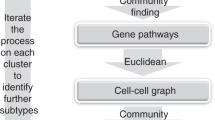
Change history
22 January 2019
During typesetting of this article, errors were inadvertently introduced to the hyperlinked URLs of some of the clustering tools in table 1 (Seurat, CIDR, pcaReduce and mpath), as well as to the numbering of the bold-text annotations in the reference list. The article has now been corrected online. The editors apologize for this error.
References
Tang, F. et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009).
10x Genomics. 10X Genomics single cell gene expression datasets. 10xgenomics https://support.10xgenomics.com/single-cell-gene-expression/datasets (2017).
Macosko, E. Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Guo, M., Wang, H., Potter, S. S., Whitsett, J. A. & Xu, Y. SINCERA: a pipeline for single-cell RNA-Seq profiling analysis. PLOS Comput. Biol. 11, e1004575 (2015).
Stegle, O., Teichmann, S. A. & Marioni, J. C. Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 16, 133–145 (2015).
Lun, A. T. L., McCarthy, D. J. & Marioni, J. C. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. [version 2; referees: 3 approved, 2 approved with reservations]. F1000Res 5, 2122 (2016).
Haque, A., Engel, J., Teichmann, S. A. & Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 9, 75 (2017).
Satija, R. SEURAT - R toolkit for single cell genomics: single cell integration in Seurat v3.0. satijalab.org https://satijalab.org/seurat/ (2015). References 4 and 9 are unsupervised clustering methods based on the Louvain method that have been shown to perform very well for large scRNA-seq data sets.
Kiselev, V. et al. Analysis of single cell RNA-seq data course. hemberg-lab.github https://hemberg-lab.github.io/scRNA.seq.course/ (2018).
Jain, A. K. Data clustering: 50 years beyond K-means. Pattern Recognit. Lett. 31, 651–666 (2010).
Quake, S. R., Wyss-Coray, T., Darmanis, S. & The Tabula Muris Consortium. Transcriptomic characterization of 20 organs and tissues from mouse at single cell resolution creates a Tabula Muris. Preprint at bioRxiv https://doi.org/10.1101/237446 (2017).
Zeisel, A. et al. Molecular architecture of the mouse nervous system. Preprint at bioRxiv https://doi.org/10.1101/294918 (2018).
Han, X. et al. Mapping the mouse cell atlas by Microwell-Seq. Cell 172, 1091–1107 (2018). References 12–14 are large collections of scRNA-seq data from mouse, and they give an indication of what a full atlas could look like.
Reid, A. J. et al. Single-cell RNA-seq reveals hidden transcriptional variation in malaria parasites. eLife 7, e33105 (2018).
Davie, K. et al. A single-cell transcriptome atlas of the aging Drosophila brain. Cell 174, 982–998 (2018).
Cusanovich, D. A. et al. The cis-regulatory dynamics of embryonic development at single-cell resolution. Nature 555, 538–542 (2018).
Rozenblatt-Rosen, O., Stubbington, M. J. T., Regev, A. & Teichmann, S. A. The Human Cell Atlas: from vision to reality. Nature 550, 451–453 (2017).
Bellman, R. Dynamic Programming (Courier Corporation, 2013).
Brennecke, P. et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods 10, 1093–1095 (2013).
Lloyd, S. Least squares quantization in PCM. IEEE Trans. Inform. Theory 28, 129–137 (1982).
Kiselev, V. Y. et al. SC3: consensus clustering of single-cell RNA-seq data. Nat. Methods 14, 483–486 (2017). SC3 is a user-friendly clustering method that works very well for smaller data sets.
Grün, D. et al. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 525, 251–255 (2015).
Wang, B., Zhu, J., Pierson, E., Ramazzotti, D. & Batzoglou, S. Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning. Nat. Methods 14, 414–416 (2017).
Lin, P., Troup, M. & Ho, J. W. K. CIDR: Ultrafast and accurate clustering through imputation for single-cell RNA-seq data. Genome Biol. 18, 59 (2017).
Zeisel, A. et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 (2015).
Žurauskiene˙, J. & Yau, C. pcaReduce: hierarchical clustering of single cell transcriptional profiles. BMC Bioinformatics 17, 140 (2016).
Tasic, B. et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci. 19, 335–346 (2016).
Blondel, V. D., Guillaume, J.-L., Lambiotte, R. & Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. 2008, 10008 (2008).
Xie, J., Kelley, S. & Szymanski, B. K. Overlapping community detection in networks. ACM Comput. Surv. 45, 1–35 (2013).
Lancichinetti, A. & Fortunato, S. Community detection algorithms: a comparative analysis. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 80, 056117 (2009).
Levine, J. H. et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 162, 184–197 (2015).
Mereu, E. et al. matchSCore: matching single-cell phenotypes across tools and experiments. Preprint at bioRxiv https://doi.org/10.1101/314831 (2018).
Freytag, S., Lonnstedt, I., Ng, M. & Bahlo, M. Cluster headache: comparing clustering tools for 10X single cell sequencing data. Preprint at bioRxiv https://doi.org/10.1101/203752 (2017).
Menon, V. Clustering single cells: a review of approaches on high-and low-depth single-cell RNA-seq data. Brief. Funct. Genom. 17, 240–245 (2018).
Fortunato, S. & Barthélemy, M. Resolution limit in community detection. Proc. Natl Acad. Sci. USA 104, 36–41 (2007).
Kleinberg & Jon. An impossibility theorem for clustering (2002).
Wolpert, D. H. & Macready, W. G. No free lunch theorems for optimization. IEEE Trans. Evol. Computat. 1, 67–82 (1997).
Saelens, W., Cannoodt, R., Todorov, H. & Saeys, Y. A comparison of single-cell trajectory inference methods: towards more accurate and robust tools. Preprint at bioRxiv https://doi.org/10.1101/276907 (2018).
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014).
Ji, Z. & Ji, H. TSCAN: pseudo-time reconstruction and evaluation in single-cell RNA-seq analysis. Nucleic Acids Res. 44, e117 (2016).
Deng, Q., Ramsköld, D., Reinius, B. & Sandberg, R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343, 193–196 (2014).
Peters, G., Crespo, F., Lingras, P. & Weber, R. Soft clustering – fuzzy and rough approaches and their extensions and derivatives. Int. J. Approx. Reason. 54, 307–322 (2013).
Wolf, F. A. et al. Graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Preprint at bioRxiv https://doi.org/10.1101/208819 (2017).
Chen, J., Schlitzer, A., Chakarov, S., Ginhoux, F. & Poidinger, M. Mpath maps multi-branching single-cell trajectories revealing progenitor cell progression during development. Nat. Commun. 7, 11988 (2016).
Andrews, T. S. & Hemberg, M. Dropout-based feature selection for scRNASeq. Preprint at bioRxiv https://doi.org/10.1101/065094 (2018).
van Dijk, D. et al. Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729 (2018).
Li, W. V. & Li, J. J. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat. Commun. 9, 997 (2018).
Jiang, L. et al. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 21, 1543–1551 (2011).
Grün, D., Kester, L. & van Oudenaarden, A. Validation of noise models for single-cell transcriptomics. Nat. Methods 11, 637–640 (2014).
Fan, J. et al. Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat. Methods 13, 241–244 (2016).
Kharchenko, P. V., Silberstein, L. & Scadden, D. T. Bayesian approach to single-cell differential expression analysis. Nat. Methods 11, 740–742 (2014).
Vallejos, C. A., Risso, D., Scialdone, A., Dudoit, S. & Marioni, J. C. Normalizing single-cell RNA sequencing data: challenges and opportunities. Nat. Methods 14, 565–571 (2017).
Severson, D. T., Owen, R. P., White, M. J., Lu, X. & Schuster-Böckler, B. BEARscc determines robustness of single-cell clusters using simulated technical replicates. Nat. Commun. 9, 1187 (2018).
Buttner, M., Miao, Z., Wolf, A., Teichmann, S. A. & Theis, F. J. Assessment of batch-correction methods for scRNA-seq data with a new test metric. Preprint at bioRxiv https://doi.org/10.1101/200345 (2017).
Gilad, Y. & Mizrahi-Man, O. A reanalysis of mouse ENCODE comparative gene expression data. [version 1; referees: 3 approved, 1 approved with reservations]. F1000Res 4, 121 (2015).
Tung, P.-Y. et al. Batch effects and the effective design of single-cell gene expression studies. Sci. Rep. 7, 39921 (2017).
Haghverdi, L., Lun, A. T. L., Morgan, M. D. & Marioni, J. C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421–427 (2018).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018). References 58 and 59 present the first two methods for correcting batch effects to merge samples.
Baran-Gale, J., Chandra, T. & Kirschner, K. Experimental design for single-cell RNA sequencing. Brief. Funct. Genom. 17, 233–239 (2018).
Gallego Romero, I., Pai, A. A., Tung, J. & Gilad, Y. RNA-seq: impact of RNA degradation on transcript quantification. BMC Biol. 12, 42 (2014).
Ferreira, P. G. et al. The effects of death and post-mortem cold ischemia on human tissue transcriptomes. Nat. Commun. 9, 490 (2018).
Wu, Y. E., Pan, L., Zuo, Y., Li, X. & Hong, W. Detecting activated cell populations using single-cell RNA-seq. Neuron 96, 313–329 (2017).
Petukhov, V. et al. dropEst: pipeline for accurate estimation of molecular counts in droplet-based single-cell RNA-seq experiments. Genome Biol. 19, 78 (2018).
Ilicic, T. et al. Classification of low quality cells from single-cell RNA-seq data. Genome Biol. 17, 29 (2016).
DePasquale, E. A. K. et al. DoubletDecon: cell-state aware removal of single-cell RNA-seq doublets. Preprint at bioRxiv https://doi.org/10.1101/364810 (2018).
Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Preprint at bioRxiv https://doi.org/10.1101/357368 (2018).
McGinnis, C. S., Murrow, L. M. & Gartner, Z. J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Preprint at bioRxiv https://doi.org/10.1101/352484 (2018).
Freytag, S., Tian, L., Lönnstedt, I., Ng, M. & Bahlo, M. Comparison of clustering tools in R for medium-sized 10x Genomics single-cell RNA-sequencing data. [version 1; referees: 1 approved, 2 approved with reservations]. F1000Res 7, 1297 (2018).
Buettner, F. et al. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 33, 155–160 (2015).
Scialdone, A. et al. Computational assignment of cell-cycle stage from single-cell transcriptome data. Methods 85, 54–61 (2015).
Tirosh, I. et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313 (2016).
Cole, M. B. et al. Performance assessment and selection of normalization procedures for single-cell RNA-seq. Preprint at bioRxiv https://doi.org/10.1101/235382 (2017).
Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Jiang, L., Chen, H., Pinello, L. & Yuan, G.-C. GiniClust: detecting rare cell types from single-cell gene expression data with Gini index. Genome Biol. 17, 144 (2016).
Villani, A.-C. et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, eaah4573 (2017). This study is a good example of how scRNA-seq was used to identify new cell types, which were subsequently confirmed by functional assays.
Campbell, J. N. et al. A molecular census of arcuate hypothalamus and median eminence cell types. Nat. Neurosci. 20, 484–496 (2017).
van der Maaten, L. & Hinton, G. Visualizing data using t-SNE. J. Machine Learn. Res. 9, 2579–2605 (2008).
McInnes, L. & Healy, J. UMAP: uniform manifold approximation and projection for dimension reduction. Preprint at a rXiv https://arxiv.org/abs/1802.03426 (2018).
Xu, C. & Su, Z. Identification of cell types from single-cell transcriptomes using a novel clustering method. Bioinformatics 31, 1974–1980 (2015).
Pollen, A. A. et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat. Biotechnol. 32, 1053–1058 (2014). This study shows that shallow sequencing can be sufficient to distinguish cell types.
Kolodziejczyk, A. A. et al. Single cell RNA-sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell 17, 471–485 (2015).
Fan, X. et al. Single-cell RNA-seq transcriptome analysis of linear and circular RNAs in mouse preimplantation embryos. Genome Biol. 16, 148 (2015).
Shah, S., Lubeck, E., Zhou, W. & Cai, L. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron 92, 342–357 (2016).
Wang, F. et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 14, 22–29 (2012).
Chen, K. H., Boettiger, A. N., Moffitt, J. R., Wang, S. & Zhuang, X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015).
Baron, M. et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 3, 346–360 (2016).
Muraro, M. J. et al. A single-cell transcriptome atlas of the human pancreas. Cell Syst. 3, 385–394 (2016).
Wang, Y. J. et al. Single-cell transcriptomics of the human endocrine pancreas. Diabetes 65, 3028–3038 (2016).
Segerstolpe, Å. et al. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. 24, 593–607 (2016).
Xin, Y. et al. RNA sequencing of single human islet cells reveals type 2 diabetes genes. Cell Metab. 24, 608–615 (2016).
Kiselev, V. Y., Yiu, A. & Hemberg, M. scmap: projection of single-cell RNA-seq data across data sets. Nat. Methods 15, 359–362 (2018).
Crow, M., Paul, A., Ballouz, S., Huang, Z. J. & Gillis, J. Characterizing the replicability of cell types defined by single cell RNA-sequencing data using MetaNeighbor. Nat. Commun. 9, 884 (2018). References 92 and 93 present methods for comparing clusters across data sets without merging.
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Sato, K., Tsuyuzaki, K., Shimizu, K. & Nikaido, I. CellFishing.jl: an ultrafast and scalable cell search method for single-cell RNA-sequencing. Preprint at bioRxiv https://doi.org/10.1101/374462 (2018).
Srivastava, D., Iyer, A., Kumar, V. & Sengupta, D. CellAtlasSearch: a scalable search engine for single cells. Nucleic Acids Res. 46, W141–W147 (2018).
Meehan, T. F. et al. Logical development of the cell ontology. BMC Bioinformatics 12, 6 (2011).
Aevermann, B. D. et al. Cell type discovery using single-cell transcriptomics: implications for ontological representation. Hum. Mol. Genet. 27, R40–R47 (2018).
Bakken, T. et al. Cell type discovery and representation in the era of high-content single cell phenotyping. BMC Bioinformatics 18, 559 (2017).
Saunders, A. et al. A single-cell atlas of cell types, states, and other transcriptional patterns from nine regions of the adult mouse brain. Preprint at bioRxiv https://doi.org/10.1101/299081 (2018).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Trapnell, C. Defining cell types and states with single-cell genomics. Genome Res. 25, 1491–1498 (2015).
Montoro, D. T. et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324 (2018).
Plasschaert, L. W. et al. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560, 377–381 (2018).
Pal, B. et al. Construction of developmental lineage relationships in the mouse mammary gland by single-cell RNA profiling. Nat. Commun. 8, 1627 (2017).
Hu, Y. et al. Single cell multi-omics technology: methodology and application. Front. Cell Dev. Biol. 6, 28 (2018).
Bock, C., Farlik, M. & Sheffield, N. C. Multi-omics of single cells: strategies and applications. Trends Biotechnol. 34, 605–608 (2016).
Macaulay, I. C., Ponting, C. P. & Voet, T. Single-cell multiomics: multiple measurements from single cells. Trends Genet. 33, 155–168 (2017).
Ostuni, R. et al. Latent enhancers activated by stimulation in differentiated cells. Cell 152, 157–171 (2013).
Gao, S. et al. Tracing the temporal-spatial transcriptome landscapes of the human fetal digestive tract using single-cell RNA-sequencing. Nat. Cell Biol. 20, 721–734 (2018).
Edsgärd, D., Johnsson, P. & Sandberg, R. Identification of spatial expression trends in single-cell gene expression data. Nat. Methods 15, 339–342 (2018).
Moncada, R. et al. Building a tumor atlas: integrating single-cell RNA-Seq data with spatial transcriptomics in pancreatic ductal adenocarcinoma. Preprint at bioRxiv https://doi.org/10.1101/254375 (2018).
Pandey, S., Shekhar, K., Regev, A. & Schier, A. F. Comprehensive identification and spatial mapping of habenular neuronal types using single-cell RNA-seq. Curr. Biol. 28, 1052–1065 (2018).
Angerer, P. et al. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241–1243 (2016).
Grün, D. et al. De novo prediction of stem cell identity using single-cell transcriptome data. Cell Stem Cell 19, 266–277 (2016).
Acknowledgements
The authors thank J. Elias for help with the figures. They also thank D. McCarthy for helpful discussions and J. Westoby for feedback on the manuscript.
Reviewer information
Nature Reviews Genetics thanks A. Ziesel and the other, anonymous reviewer(s) for their contribution to the peer review of this work.
Author information
Authors and Affiliations
Contributions
All authors contributed to all aspects of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related Links
BackSPIN: https://github.com/linnarsson-lab/BackSPIN
CIDR: https://github.com/VCCRI/CIDR
GiniClust: https://github.com/lanjiangboston/GiniClust
pcaReduce: https://github.com/JustinaZ/pcaReduce
mpath: https://github.com/JinmiaoChenLab/Mpath
PhenoGraph: https://github.com/jacoblevine/PhenoGraph
RaceID: https://github.com/dgrun/RaceID
RaceID2: https://github.com/dgrun/StemID
RaceID3: https://github.com/dgrun/RaceID3_StemID2
SC3: http://bioconductor.org/packages/release/bioc/html/SC3.html
scanpy: https://github.com/theislab/scanpy
Seurat (latest): https://satijalab.org/seurat/
SIMLR: https://bioconductor.org/packages/release/bioc/html/SIMLR.html
SINCERA: https://github.com/xu-lab/SINCERA
SNN-Cliq: http://bioinfo.uncc.edu/SNNCliq/
TSCAN: https://bioconductor.org/packages/release/bioc/html/TSCAN.html
Glossary
- Unsupervised clustering
-
The process of grouping objects based on similarity but without any ground truth or labelled training data.
- Feature selection
-
A collection of statistical approaches that identify and retain only variables that are most relevant to the underlying structure of the data set.
- Dimensionality reduction
-
A collection of statistical approaches that reduces the number of variables in a data set. It often refers specifically to methods that recombine the original variables into a new set of non-redundant variables. Dimensionality reduction can help in identifying important patterns and reducing the amount of computations needed.
- Greedy
-
An algorithm that, at each step, chooses the option that leads to the greatest reduction of the cost function. Greedy algorithms are often fast, but they may fail to find the optimal solution.
- Graphs
-
Each graph consists of a set of nodes connected to each other with a set of edges. In single-cell RNA sequencing, nodes are cells, and edges are determined according to cell–cell pairwise distances.
- Heuristic optimization
-
A method for solving a problem that is designed to sacrifice accuracy in favour of speed. These methods are often based on approximations and cannot be guaranteed to find the best solution.
- Bootstrapping
-
A statistical approach in which data sets are randomly sampled and reanalysed to assess the robustness of a result.
- Gaussian mixture model
-
A statistical model of one or more normal distributions. When fitted to data, each normal distribution can be interpreted as a distinct cluster of points.
- Cell ontology
-
A hierarchical organization of controlled vocabulary to describe properties of (and relationships between) different cell types.
Rights and permissions
About this article

Cite this article
Kiselev, V.Y., Andrews, T.S. & Hemberg, M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat Rev Genet 20, 273–282 (2019). https://doi.org/10.1038/s41576-018-0088-9
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41576-018-0088-9
This article is cited by
-
scSMD: a deep learning method for accurate clustering of single cells based on auto-encoder
BMC Bioinformatics (2025)
-
Multimodal learning for mapping genotype–phenotype dynamics
Nature Computational Science (2025)
-
k-Means-MIND: comparing seeds without repeated k-means runs
Neural Computing and Applications (2025)
-
p-clustval: a novel \(p\)-adic approach for enhanced clustering of high-dimensional single-cell RNASeq data
International Journal of Data Science and Analytics (2025)
-
iIMPACT: integrating image and molecular profiles for spatial transcriptomics analysis
Genome Biology (2024)
