
Abstract
There is an increasing amount of evidence that biomolecular condensates are linked to neurodegenerative diseases associated with protein aggregation, such as Alzheimer’s disease and amyotrophic lateral sclerosis, although the mechanisms underlying this link remain elusive. In this Review, we summarize the possible connections between condensates and protein aggregation. We consider both liquid-to-solid transitions of phase-separated proteins and the partitioning of proteins into host condensates. We distinguish five key factors by which the physical and chemical environment of a condensate can influence protein aggregation, and we discuss their relevance in studies of protein aggregation in the presence of biomolecular condensates: increasing the local concentration of proteins, providing a distinct chemical microenvironment, introducing an interface wherein proteins can localize, changing the energy landscape of aggregation pathways, and the presence of chaperones in condensates. Analysing the role of biomolecular condensates in protein aggregation may be essential for a full understanding of amyloid formation and offers a new perspective that can help in developing new therapeutic strategies for the prevention and treatment of neurodegenerative diseases.

Similar content being viewed by others
Introduction
Amyloid protein aggregate formation is one of the hallmarks of neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) and Huntington’s disease (HD)1. Currently, there are around 50 known protein precursors related to amyloid disease, such as amyloid-β (AD-related), α-synuclein (αSyn, PD-related) and huntingtin (HD-related). Most, but not all, of the known proteins are intrinsically disordered proteins (IDPs), which can undergo cross-beta sheet stacking to form insoluble, thermodynamically stable fibrillar aggregates2,3. The formation of these fibrils ultimately results in microscopic inclusions visible in histological slides of patients (Fig. 1). Studies have suggested that the toxicity of amyloids is caused not only by fibrillar aggregates but also by intermediate oligomeric species2,4,5. Nonetheless, understanding the complete protein aggregation pathway is necessary for understanding the symptoms and potential origins of amyloidogenic disease1.

a, Specific brain regions are affected by the different neurodegenerative diseases, showing pathological aggregates in histological slides. Many of the proteins involved in these pathological aggregates have been shown to be capable of liquid–liquid phase separation in vitro and in vivo. The arrowheads indicate tau condensates. Scale bars, 10 μm. b, α-Synuclein liquid droplets as visualized by differential interference contrast (DIC) microscopy can undergo a time-dependent transformation into solid fibrillar aggregates. This is seen by an increase in thioflavin S (ThS) signal and eventually the formation of ThioS-positive fibrils, which corresponds to fibril formation when studied by transmission electron microscopy (TEM). c, The patient mutation G156E in fused in sarcoma (FUS) can cause liquid-to-solid transitions in reconstituted FUS condensates over the course of 8 h, whereas wild-type FUS remains liquid. Part a reprinted with permission from ref. 126, Elsevier. Part b reprinted from ref. 50, Springer Nature Limited. Part c reprinted with permission from ref. 39, Elsevier.
Although protein deposits in the brain have been known for more than 100 years (ref. 1), their formation pathways and factors that act upon those pathways are still not fully understood. Models of general protein aggregation exist and have reasonable predictive powers for in vitro models6. However, a cell is a complex, inhomogeneous and crowded environment wherein many other factors need to be taken into account, such as the potential phase separation of aggregation-prone proteins, the presence of other biomolecular condensates and their interfaces, and chaperones — both inside and outside condensates. The relation between biomolecular condensates and protein aggregation has been gaining attention, in part owing to the understanding that IDPs are often involved in the formation of both condensates and fibrillar aggregates7,8,9.
Biomolecular condensates are compartments formed by the liquid–liquid phase separation (LLPS) of various biomolecules such as proteins, nucleic acids, lipids and small molecules10,11. LLPS has been extensively studied in colloid science and chemistry, wherein the term coacervation was coined. Coacervation is a process involving the condensation of polymeric molecules into a dense, liquid phase, analogous to the formation of biomolecular condensates. It was first observed in 1929 in solutions containing oppositely charged biopolymers or isoelectric proteins mixed with (poly)phenols12. In the past decade, LLPS has gained renewed attention in the context of cellular biomolecular condensates. Such condensates are hypothesized to act as storage sites, organizational hubs and reaction crucibles within the cell, by concentrating or excluding biomolecules locally, altering transition state energies, and buffering concentrations10,13,14,15. Furthermore, several experimental studies have found a link between LLPS and protein aggregation16,17. Stress granules are biomolecular condensates that are known to co-localize with cytoplasmic aggregates formed by the TAR DNA-binding protein 43 (TDP-43), and they are hypothesized to function as a nucleation site for TDP-43 aggregate formation18. They have also been shown to interact with many other proteins related to neurodegenerative diseases, such as fused in sarcoma (FUS), ataxin 2 (ATXN2) and heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1)16. Many of these proteins undergo LLPS in vitro and in vivo (Fig. 1a), which can precede liquid-to-solid transition (LST) and aggregate formation (Fig. 1b,c).
Despite the evidence linking condensates with aggregation, a comprehensive framework of mechanisms by which LLPS compartments influence amyloid aggregation has yet to be established and systematically investigated. In this Review, we will explore the two main pathways through which protein aggregation can be affected by (the presence of) condensates: (1) LST following the condensation of aggregation-prone protein and (2) the interaction of aggregation-prone proteins with a host biomolecular condensate composed predominantly of different biomolecules (Fig. 2a).

a, Protein aggregation within LLPS compartments can occur in condensates which consist of the aggregation-prone protein (idemic condensate aggregation) or by partitioning into a host condensate as a guest. b, Five main factors are identified as key influencers of protein aggregation in condensates. (1) Condensates can concentrate guest proteins based on their partition coefficient, KP, leading to faster nucleation. The monomer concentration is replenished with external proteins, causing a positive feedback loop with enhanced aggregation. (2) The inside of the condensates is viscous, potentially slowing down aggregation for protein aggregates with fast elongation. (3) Chaperones can co-localize inside condensates and use the principles of enhanced local concentration to prevent aggregation within condensates. However, if they do not partition, this could be a pathway to unchecked misfolding and eventually aggregation. (4) When proteins localize to the interface of condensates, they can undergo heterogeneous nucleation and enhance aggregation, or potentially stabilize their conformation through specific limited orientations. (5) The chemical environment within condensates can alter the compaction, conformation, or dynamics of changing conformations of proteins, leading to an altered aggregation landscape.
To differentiate between the transition of a condensate formed by amyloidogenic proteins and a host–guest situation, we introduce the term ‘idemic’ condensate aggregation to refer to the gradual transition of a condensate consisting mainly of an amyloidogenic protein into an aggregated state. For both pathways, there are several factors originating from the presence of a condensate that modulate the aggregation process compared to protein aggregation taking place in solution. We will discuss five key factors of the condensate microenvironment that have attracted attention in the context of protein aggregation and stability (Fig. 2b): (1) the local concentration, (2) reduced diffusivity inside the viscoelastic and crowded condensate networks, (3) interfacial localization, (4) local interactions that may alter the conformation and intrinsic stability of proteins, and (5) co-localization of chaperones in some condensates.
Mechanisms and kinetics of protein aggregation in vitro
To understand how amyloid protein aggregation can be influenced by the presence of biomolecular condensates, we start by briefly discussing a model of protein aggregation that was found to describe the aggregation process quantitatively in vitro and in model organisms6,19,20,21,22 (Fig. 3a), as this will lay the foundation for later discussions of the effect of condensates. To help explain the trends associated with the five factors introduced above (Fig. 2b), we show how typical protein aggregation curves, based on a work by Lipiński et al.23, vary with these factors. We specifically consider amyloid formation, a form of protein aggregation in which fibrils are stabilized by the cross β-sheet structure with a continuous array of hydrogen bonds24.

a, The kinetic processes involved in protein aggregation. Monomers form reversible oligomers or fibrils by primary nucleation (kn), which grow by elongation (k+) through addition of monomers at both ends and shrink by dissociation (k−). Fibrils can fragment (kb), producing additional growth ends. New fibrils can also form by secondary nucleation (k2) on the surface of existing fibrils. b, Single-phase aggregation with varying nucleation rate constants, kn. Higher nucleation rates lead to lower tlag times, with similar vmax. c, Single-phase aggregation with varying secondary nucleation rate constants, k2. Both the tlag and vmax are affected by slower secondary nucleation. d, Varying elongation rates for aggregation modelled in a single phase. Parameters used for simulations unless shown otherwise: kn = 1 × 10−7 μM h−1, k+ = 200 μM h−1 and k2 = 0.1 μM h−1.
The first step to amyloid formation is primary nucleation, in which two or more monomers (M) form a fibril. Such fibrils are in principle thermodynamically reversible25, although on practical timescales and under physiological conditions they can be considered irreversible for simplicity26. Monomers can also form reversible oligomers, but for simplicity we only consider the direct transition from monomers to fibrils. Nucleation can occur when the monomers interact in solution (homogeneous nucleation), or when the monomers interact at an interface, such as an air–water interface (heterogeneous nucleation). Fibrils can be elongated on either end by the addition of monomers (the rate of this process is dependent on the total concentration of fibrils [Fib]). Fibrils have the capacity to nucleate new fibrils, through surface-catalysed secondary nucleation, in which two or more monomers create a new fibril nucleus on the surface of an existing fibril (the rate of this process is dependent on both the monomer concentration and the total mass of aggregates [Agg]). The final mechanism that contributes to fibril formation is fragmentation, in which a fibril breaks into two parts. Amyloid fibril formation has the characteristics of an autocatalytic reaction, with a sigmoidal increase in aggregate mass because of the fast elongation of aggregates and secondary nucleation and fragmentation leading to more aggregates. The lag phase, the length of which is determined mostly by the nucleation rate (Fig. 3b), is followed by the rapid growth phase, when the elongation and secondary nucleation processes dominate (Fig. 3c,d).
The change in fibril concentration as a function of time (\(\frac{{\rm{d}}[{\rm{Fib}}]}{{\rm{d}}t}\)) is described by equation (1) and includes primary and secondary nucleation and fragmentation (Fig. 3a). The primary nucleation rate constant is given by kn, [M] is the concentration of monomers, and nc describes the reaction order of primary nucleation. The last part of the equation describes the secondary nucleation rate, which is a function of the secondary nucleation rate constant k2, [M] with reaction order n2, as well as the total mass concentration of fibrils [Agg], owing to secondary nucleation being dependent on the total amount of fibril surface, not the concentration of fibrils.
The change in fibril mass, \(\frac{{\rm{d}}[{\rm{Agg}}]}{{\rm{d}}t}\), is equal to the elongation rate (equation (2)) if we assume that mass changes related to the nucleation reactions can be neglected, which is justified by the fact that the elongation reaction is typically several orders of magnitude faster than nucleation. The elongation rate is given by k+, and the factor of 2 accounts for the fact that elongation can occur at both ends of a fibril.
The total aggregate mass concentration is usually monitored with fluorescent molecules which show enhanced fluorescence when binding to amyloid fibrils, for example, thioflavin T (ThT)27. Other techniques, such as nuclear magnetic resonance (NMR) and circular dichroism spectroscopy, can also be used28,29.
If the aggregation mechanism is known, fitting the kinetic model can provide information about the kinetic rate constants of reactions in the aggregation cycle. Although knowledge of the total aggregate mass is often sufficient, other methods such as electron paramagnetic resonance (EPR)30, Förster resonance energy transfer (FRET)31,32, single-molecule spectroscopy33, fluorescence lifetime imaging (FLIM)34 and mass photometry35 can be applied to identify oligomeric species and characterize the details of their molecular structure. The typical timescale of amyloid fibril formation can range from hours to days, depending not only on the aggregating protein but also on environmental factors, such as pH, salt concentration, crowding, and the presence of membranes and condensates. In this Review, we simulate and discuss how condensates (such as a two-phase system) can influence protein aggregation, assuming that the monomeric, non-aggregated protein can transfer between the two phases.
Liquid-to-fibril transition of biological condensates
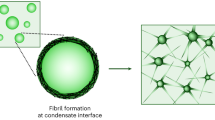
Proteins with the ability to form fibrillar aggregates often undergo LLPS on their own, and this condensed liquid phase can be a (metastable) intermediate on the way to aggregates (Fig. 1b,c). We call this situation idemic condensate aggregation, to distinguish it from the amyloid formation by proteins that are present as ‘guests’ in host condensates, and from the LST of condensates that results in a non-fibrillar, gel-like state16,36,37. It is commonly agreed that the formation of fibrils can be initiated in the dilute phase, as a combination of primary and secondary nucleation. Fibril formation in idemic condensates can occur through alternative pathways, and in addition, the condensed liquid phase can facilitate fibril formation by increasing the local concentration of proteins, providing an interface on which nucleation is enhanced38.
The two routes — nucleation in the dilute phase and liquid-to-solid transition — are not mutually exclusive, and their importance may differ for different proteins and in different contexts (for example, in vitro compared to in living cells)38. The ability to undergo LLPS has been shown either in vivo or in vitro for proteins such as FUS36,39, hnRNPA1 (refs. 16,37) and TDP-43 (ref. 16). Neurodegenerative disease-related mutations within the low-complexity domains were also found to disrupt LLPS and lead to aberrant, solidified compartments36,39,40,41. However, it should be noted that not all LSTs of condensates result in the formation of template-competent amyloid fibrils, as observed in vitro for TDP-43 and parkin-interacting substrate condensates42,43,44. In such cases, LST may suppress amyloid fibril formation by kinetically trapping protein monomers in a solidified condensate. Moreover, even in the case of an LST involving the formation of template-competent amyloid fibrils, model systems based on in vitro LLPS and a further LST of pure amyloidogenic proteins, is only a great simplification of the processes occurring in living cells, whose complex composition practically excludes the possible formation of single-component condensates. Nevertheless, studying these processes in simple systems can still provide information about the general LST mechanisms and its role in altering the rate of amyloid formation.
For example, idemic condensate aggregation was shown in a work by Linsenmeier et al., in which amyloid formation by the low-complexity domain of hnRNPA1 was promoted at the interface of the condensates45. They explain that the interface of idemic condensates can promote nucleation of fibrils by locally increasing the concentration of monomers and stabilizing their aggregation-prone conformation. For this particular system, amyloid formation only occurred when condensates were formed. The LST mechanism of hnRNPA1 seems to be different from αSyn and tau, suggesting that it is protein specific.
Studies of tau protein, which has a role in microtubule assembly and stabilization but can also form pathological aggregates, have shown that it has a propensity to undergo LLPS46,47. A more detailed investigation of the LST process occurring in tau condensates revealed that the environment of the liquid droplets promotes a more extended protein conformation, facilitating intermolecular interactions, and further clustering and aggregation48. An elegant study by Boyko et al. shows that the aggregation kinetics of phase-separating proteins should not be affected by the overall concentration of tau if it falls within the two-phase region of the phase diagram49. This is because the concentration of tau in the droplet, which determines the nucleation and elongation rates, remains constant. They also show it can be regulated by the addition of non-aggregating mutants that co-localize in the dense-liquid phase49. This case of ‘diluting’ condensed tau, even though droplets are formed by mutants of the same protein, is effectively similar to host–guest systems wherein the aggregation of protein is only one of the components (see section ‘Host–guest aggregation’).
A work by Ray et al. shows that PD-related protein αSyn undergoes LLPS in the presence of polyethylene glycol (PEG) as a crowding agent50 and that the liquid condensates can serve as nucleation spots for amyloid aggregation51. Although most evidence now indicates that nucleation probably occurs at the interface of idemic condensates, FLIM measurements on idemic αSyn condensates in PEG-based solutions by Ray et al. suggest that the LST of αSyn is initiated primarily in the centre of the droplets under these conditions, thereby creating a solid-like core. Upon fusion and compositional ripening, monomers are transported from homogeneous liquid droplets that have not yet formed a solid-like core to droplets containing a solid-like core. Interestingly, this transport seems to be arrested in condensates that underwent a complete liquid-to-solid transition and formed solid particles51.
An open question concerning the connection between LLPS of pure amyloidogenic proteins in vitro and pathological LST (or aggregation in general) is whether phase separation of these proteins in vivo occurs similarly to in vitro studies. Condensation of pure proteins typically requires concentrations to be higher than physiological concentrations (either of the protein itself or of additives such as PEG)50,52. Similar to in vitro assays, in vivo studies are usually performed in systems wherein proteins are overexpressed to reach very high concentrations53. However, it is possible that even at lower concentrations of protein, small clusters locally enriched in the aggregating proteins with liquid-like properties can exist. These can emerge owing to the heterogeneous cell environment with locally increased crowding, or at biological interfaces (such as lipid–membrane or liquid–liquid interfaces)35.
Host–guest aggregation
Proteins that can form fibrillar aggregates may also partition into condensates formed by other components and undergo aggregation inside or at the surface of these condensates (‘host–guest aggregation’, Fig. 2a). This pathway of aggregation does not necessarily require high overall concentrations of the aggregating protein or even the intrinsic capability of the protein to undergo LLPS. As a result, we suspect that it may be a rather general pathway by which LLPS affects protein aggregation, and a large group of proteins may be involved.
Both idemic condensate aggregation and host–guest aggregation are influenced by various factors that affect protein aggregation. In principle, the factors affecting protein aggregation are the same because they are independent of the aggregation-prone protein being the main constituent of the LLPS condensate (idemic) or a small fraction of the material (host–guest). We will cover each identified factor, namely partitioning, viscosity and crowding, interfacial accumulation, energy modulation, and the presence of chaperones, discuss the literature in which these factors have been identified, and, where applicable, estimate their relative importance for both pathways based on the model of amyloid protein aggregation in two-phase systems23,26.
For example, aggregation of αSyn is affected by the presence of complex coacervates in vitro23. Depending on the type of coacervate, protein could partition into the condensed phase or accumulate at the interface, affecting the aggregation kinetics in different ways. Although interfacial accumulation in general accelerated aggregation (similarly to the idemic systems), accumulation of protein inside coacervates could cause stabilization of monomers and slower aggregation. Sequestration of monomers upon partitioning has also been shown for the 1–42 fragment of amyloid beta (Aβ-42), with host condensates formed by different low-complexity domains54.
So far, host–guest interactions have not been studied very well in vivo. However, a work by Choi et al. in cells shows that the interfaces of host biomolecular condensates can accelerate coarsening of small aggregates and promote the generation of larger aggregates55. They use a light-activated condensate system derived from p62 protein/sequestosome 1 and studied its influence on polyQ aggregation. As the authors hypothesize, the coarsening of small aggregates into larger ones may actually have a protective role in cells, by reducing the accessible surface area of aggregates to interfere with healthy, functional proteins.
Factors affecting protein aggregation
Protein aggregation of partitioned guest protein and idemic condensate aggregation can be affected by major factors that originate owing to the presence of a condensate: (1) local concentration, (2) reduced diffusivity inside the condensate, (3) interfacial localization, (4) interactions with the condensate matrix altering the protein conformation and stability, and (5) the presence of chaperones (Fig. 2b). These factors are, in theory, relevant for both idemic and host–guest condensate aggregation, but their relative importance may be different in the two situations. We note that additional factors such as flow-induced shear can also influence protein aggregation nucleation in condensates. However, we will limit our discussion in this Review to amyloid formation in the presence of condensates without shear and in or close to equilibrium56. We will discuss the effect of each of these factors using examples from the literature and, where possible, theoretical trends to show the expected relevance of the effect.
Enhanced local concentration of aggregating proteins
Condensates can spontaneously take up molecules from their surroundings. These partitioning molecules can be seen as client molecules in the host condensate environment and are assumed to be able to freely move in and out of the condensate. The degree of client molecules in condensates is governed by the free energy of transport of the solute in the different phases \((\Delta {G}^{{\rm{t}}{\rm{r}}}=-\,RT{\rm{l}}{\rm{n}}K)\) (ref. 57). Partitioning can affect aggregation by locally enhancing the concentration of the protein58 and by affecting the protein folding as discussed in the section ‘The protein aggregation pathway and energy landscape’. Partitioning of amyloidogenic proteins into condensates has been shown, namely Aβ-42 into condensates derived from DEAD-box proteins LAF-1, Dbp1 and Ddx454, and αSyn and insulin into several model coacervates23,59. There are many studies involving the partitioning of non-amyloid proteins into condensates, such as F-actin60 and sequestration of proteins into stress granules61, and the accumulation of misfolded luciferin into the nucleolus62. Comparably, in idemic condensate aggregation, local concentrations are also enhanced. In both pathways, an enhanced local concentration can lead to faster aggregation compared to having the same total number of protein monomers in a single, dilute phase.
From a thermodynamic point of view, reaction rates, including aggregation, are determined by the activity of the reacting species rather than the concentration. Bauermann et al. analysed how phase equilibrium of one of the reacting species between a dilute and a condensed phase would influence the kinetics of mass action. They showed that theoretically, in such cases — which may resemble idemic condensate aggregation — the activity of these species is equal in both phases, despite higher local concentration in the condensed phase63. In other words, a high local concentration of reactants owing to phase separation is coupled with a low activity coefficient. Differences in reaction rates, such as aggregation, would in that case always be caused by differences in the rate coefficient (which can include other factors shown in Fig. 2b). However, local concentrations have a role in reactions of guest proteins in host condensates26 — in line with the principles of chemical kinetics during micellar catalysis or emulsion polymerizations. Moreover, condensates in vivo are far from simple, single-component liquids; instead, they are complex fluids with a multicomponent nature and sometimes hierarchical organization. Whether a phase equilibrium is maintained in condensates partaking in idemic protein aggregation remains to be seen. Quantification of local concentrations and amyloid formation rates will, therefore, be key to establishing the role of local protein concentrations in both idemic and host–guest protein aggregation.
To better understand how a locally enhanced concentration can lead to faster aggregation in a host–guest system, Weber et al. showed that the accumulation or concentration of monomeric aggregating proteins in a host compartment, such as a condensate, can lead to increased aggregation rates23,26. In this theoretical study, protein aggregation within two phases was modeled with different partition coefficients (KP), which represent the ratio of the concentration of protein in a condensate phase to the concentration in the dilute phase. It was found that even weak partitioning of monomers can lead to differences on the scale of orders of magnitude in the final aggregate concentration between the two phases. This difference is driven by a positive feedback loop. When nucleation occurs in a phase (higher tendency to take place in the phase with a higher concentration), the monomeric concentration decreases and is replenished by monomers from the other phase. Through this effect, the elongation of aggregates is attenuated, and the autocatalytic aspect of aggregation is enhanced (Fig. 4a), nearly reaching aggregation rates of a single highly concentrated phase. In Weber’s model, they find that compartment volume and partitioning are determining factors for the total number of aggregates, in addition to reaction orders nc and n2. Interestingly, phase separation may also be exploited to enhance the potency of aggregation inhibitors through the same local concentration enhancement64. The enhanced aggregation owing to partitioning has also been observed in vitro, using the model coacervate system of [RRASL]3/polyU that hosts αSyn at an increased concentration23. In vitro studies such as these allow better control over conditions, and the aggregation process and the resulting fibrils can be measured in greater detail. For example, real-time measurements of the dilute phase concentration of IDPs could be used as a proxy for the aggregation process65. Altogether, this shows that the enhanced local concentration of amyloidogenic protein in condensates can lead to highly increased aggregation.

a, Increasing the partitioning coefficient of protein to the compartment speeds up aggregation. b, Increasing viscosity slows down aggregation. The numbers shown are relative to the viscosity of water, where η = 105 is the limit of typical condensates and η = 106 is representative of aged condensates. Simulations done at KP = 10. c, Changes in primary nucleation and elongation show how changes in energy modulation can affect aggregation. For panels a,b and f, kn = 1 × 10−7 μM h−1, k+ = 200 μM h−1, k2 = 0.1 μM h−1, KP = 10 and Vdilute/Vcondense = 100. For panel c, kn = 1 × 10−5 μM h−1, k+ = 1 μM h−1, k2 = 1 × 10−4 μM h−1, KP = 10 and Vdilute/Vcondense = 100. d, Full-length α-synuclein (FL-αSyn) can partition into [RRASL]3/polyU coacervates and localizes at the interface of pLys/pGlu coacervates. e, Condensates of hnRNPA1-B low-complexity domain (LCD) form a protein-rich interface, as evidenced by an increase in fluorescence of atto647-labelled hnRNPA1-B LCD. ThT fluorescence is evenly distributed throughout the condensate, indicating an absence of fibrils. f, Condensate solidification over time owing to an increase in aggregates present. Two scaling methods are shown, one in which the viscosity scales with [Agg]1 L3.4 (dashed back curve) and one which scales with [Agg]3.4 L3.4 (green curve), where L is the average length of the aggregates. Compared to a constant viscosity equal to water (grey line), higher scaling shows a considerably slowed down aggregation as more and more of the condensate is solidified. g, Ways that crowding agents can have differing effects on aggregation depending on the size of the crowder and aggregates: if there is an increase in volume (yellow rectangle) upon aggregation, aggregation is slowed down, and the opposite occurs if the volume is decreased upon aggregation. Part d is adapted from ref. 23 under a Creative Commons licence CC BY 4.0. Part e is adapted from ref. 45, Springer Nature Limited.
Viscosity and crowding of condensates
Another aspect of the distinct condensate environment is the reduced diffusivity of proteins owing to a higher local viscosity (η) compared to the dilute phase. Such higher viscosities appear to affect mostly translational diffusion of proteins in condensates, as shown by fluorescence recovery after photobleaching (FRAP) measurements66. Interestingly, IDPs have been found to undergo rapid conformational rearrangements and rotational diffusion, despite high condensate viscosities67. Nevertheless, translational diffusion impacts amyloid formation particularly strongly because it involves considerable mass transport and is bound by diffusion limitations. Simulations68 and in vitro work on αSyn69 show that high viscosity can indeed alter aggregation rates. The viscosity of membraneless organelles has been reported to range from 0.1 to 1,000 Pa s (refs. 67,70,71). On the basis of diffusion coefficients measured with FRAP for homotypic αSyn and FUS LC condensates, we estimate their nanoviscosity (viscosity as perceived by nanoscale probes) to be 0.1–0.5 Pa s (based on the Stokes–Einstein equation assuming a ~4-nm hydrodynamic radius)50,72,73. It should be noted that FRAP is a technique which requires multiple assumptions to quantify viscosity74,75, and because of relatively slow data accumulation, fast diffusion cannot be reliably measured. Alternative methods for determining viscosity such as micropipette-based techniques71, optical tweezers76,77, or fluorescence correlation spectroscopy, should be considered to provide more accurate estimates of viscosity.
It should also be noted that the measured viscosity of mesh-like solvated structures such as condensates is length-scale dependent. Using smaller-sized dyes as reporters for the diffusion coefficient and calculating the viscosity based on these values yields a nanoviscosity — which can be several orders of magnitude lower than the (macro)viscosity — experienced by probes that are much larger than the largest mesh inside a condensate78,79. To find the average mesh size for LAF-1 (a DDX3 RNA helicase present in P granules) RNA droplets in vitro, Wei et al. measured the experienced viscosity for probes of different sizes and found a difference of two orders of magnitude between the smaller and larger probes, giving an estimate mesh size between 3 and 6 nm (ref. 70). It can be argued that the nanoviscosity of probes with similar sizes to monomeric proteins is more relevant to aggregation than the macroviscosity because it says more about the diffusion of proteins. The macroviscosity only becomes relevant once the fibrils are larger than the mesh size.
Nevertheless, the viscosity of the condensate is typically several orders of magnitude higher than the surrounding dilute phase (at least in vitro), and this difference in viscosity can drastically alter the rate of aggregation. The effect of viscosity on protein aggregation was studied by measuring αSyn aggregation at increasing glycerol concentrations80. However, glycerol had a non-trivial effect on protein aggregation — a 40% glycerol solution greatly enhanced the protein aggregation of αSyn, whereas a 60% glycerol solution completely halted aggregation. The acceleration was owing to the stabilizing effect of glycerol on the partially folded intermediate monomer81, whereas at a high enough concentration, the decrease in diffusion slows down aggregation and eventually halts aggregation. This highlights that although altering viscosity with chemicals usually has multiple effects, it can also slow down aggregation.
To estimate the effect of viscosity on protein aggregation, we modelled the effect of increased experienced viscosity within compartments by rewriting the reaction rate constants as a combination of the inherent rate constant (kr, reaction rate in the limit of infinitely fast diffusion) and the diffusion limited rate constant (kD, reaction rate in the limit of infinitely high inherent rate constant, equation (3) (ref. 82)). The latter is proportional to the diffusion rate and inversely proportional to the viscosity of the reaction medium (Fig. 4b). This means that faster, up to diffusion-limited reactions, such as elongation, are affected more by the reduced diffusion than by slow reactions.
Typical viscosities of coacervates and condensates (below 105 times the viscosity of water) result in a marginal slowing down of aggregation. When the viscosity is increased by another order of magnitude, aggregation slows down considerably. The viscosity inside the compartments notably affects the overall reaction more when there is a high partitioning of the monomers into the compartment. For the trends, a partition coefficient of 10 was chosen. The effect of viscosity is only present if the elongation reaction is diffusion-limited (\({k}_{{\rm{r}}}\gg {k}_{{\rm{D}}}\)) and the nucleation rate is not high. We expect that the effect of viscosity inside condensates on protein aggregation is not a large factor in protein aggregation.
Aggregation of proteins in liquid condensates may cause solidification over time, effectively altering the experienced viscosity83. We simulate how an increasing viscosity as a function of the concentration of aggregates within condensates may influence aggregation (Fig. 4f) with different scaling factors based on the entanglement concentration84. The viscosity increases as \(\eta \sim {[{\rm{Agg}}]}^{1}{L}^{3.4}\) (where L is the average length of the aggregates given by \(\frac{[{\rm{Agg}}]}{[{\rm{Fib}}]}\)) up to a certain (entanglement) concentration, and then increase with \(\eta \sim {[{\rm{Agg}}]}^{3.4}{L}^{3.4}\). Because the entanglement concentration depends on the nature of the polymer (fibril), we show how the viscosity scales in both limiting cases. The lower scaling of viscosity leads to aggregation that is equal to having a constant viscosity of water, for this particular monomer concentration and set of aggregation rate constants. However, if the viscosity within condensates increases with the higher scaling factor at any point (Fig. 4f, area in green), the aggregation rate slows down substantially. Compared to having more viscous condensates, solidification as a result of aggregation is a more general way in which viscosity affects protein aggregation in condensates. We expect that condensate hardening can be an important factor in prolonging protein aggregation, especially in the case of idemic condensate aggregation, wherein the majority of the condensate aggregates over time. It is also worth noting that the LST of condensates can lead to non-homogeneous distributions of condensate material, further complicating the kinetics of the process. However, a more detailed consideration of the spatial distribution of aggregates on the kinetics is beyond the scope of this Review85.
In the case of host–guest aggregation, the biological function of the host biomolecular condensate may be lost as it solidifies, and cellular physiology may be disturbed. The hardening of biomolecular condensates may have consequences for the progression of neurodegenerative disease, but the potential impact remains unstudied. For example, stress granules, which form in response to cellular stress and sequester proteins, may harden upon aggregation and lose their protective function. If protein aggregation is found to occur within or at the interface of cellular condensates in vivo, the loss of the biological function of condensates upon hardening could be a new direction for studies.
Alongside the altered viscosity inside condensates, an additional crowding effect within condensates must also be considered. To examine the effect of crowding on protein aggregation, the aggregation of αSyn within similarly viscous solutions of glycerol and PEG 3350 was compared. It was found that fibrillization was considerably faster with 15% PEG 3350 than with 40% glycerol (which have similar viscosities), resulting in clustered fibrils80. Similar effects have been found using insulin and αSyn in combination with various concentrations of dextran 100 and hydroxypropyl cellulose 370 (HPC 370), showing that dextran enhances protein aggregation owing to crowding, whereas HPC slows it down86. Computational studies have shown that chemical crowders can also both accelerate or suppress aggregation87 (Fig. 4g). The acceleration can be understood in terms of excluded volume, wherein proteins take up less excluded volume once aggregated and, thus, increase the entropy of the crowders, making aggregation more probable. However, when the excluded volume of aggregated proteins is larger than the excluded volume of the crowders joining up, aggregation slows down or is suppressed. We can, thus, conclude that the crowding effect within condensates is dependent on the constituents of the compartment and the excluded volume of the specific protein undergoing aggregation.
Interfacial effects
Aside from compartmentalization, LLPS droplets also provide interfaces wherein aggregating proteins may accumulate and undergo an alternative aggregation process or be sequestered88 (Fig. 2b). It is known that proteins such as αSyn89,90 (Fig. 4d) and Aβ (ref. 91) localize to interfaces such as lipid membranes, water–air interfaces and the interfaces of condensates23. Interfaces can generally alter aggregation in two ways: by increasing the local concentration or by altering the conformation of the interface-bound protein to stabilize or promote aggregation92,93. Altered aggregation at the interface is described as heterogeneous nucleation, as opposed to homogeneous nucleation in solution23,45,88,94. In the case of no saturation (not all available binding sites at the interface are occupied), an increase of proteins at the interface of condensates can lead to increased aggregation (Fig. 5a). In vitro results show that interfacial localization can greatly enhance aggregation, as shown with αSyn primary nucleation being three orders of magnitude larger when localized to small unilamellar vesicles, but aggregation also decreased when all αSyn is bound with no monomeric proteins in solution89.

a, Different numbers of interfacial binding sites (I0) can speed up aggregation within non-saturated interfaces. b, Model coacervates RP3/polyU and pLys/pGlu show enhanced intramolecular Förster resonance energy transfer (FRET) efficiency of full-length α-synuclein (FL-αSyn) using Alexa Fluor 546 and Alexa Fluor 647 at label positions close to the region responsible for β-sheet formation. c, Transmission electron microscopy images of FL-αSyn aggregates formed in the presence of RP3/polyU coacervates. Spherical aggregates are seen with fibrils pointing away from the surface. d, At 0 h, labelled hnRNPA1-B low-complexity domain (LCD) is accumulated at the interface of homotypic condensates whereas the ThT signal is equally distributed throughout the image. After 48 h, B-LCD shows aggregate-like structures originating from the condensates. ThT fluorescence is increased at the interface of the condensates indicating a beta-sheet structure and fibrils have grown out of the condensates. e, Top panel: p62 condensate accumulates PolyQ aggregates at their interface. Bottom panel: Radial plots of the normalized intensity show accumulation of polyQ at the interface, as indicated by an increase in relative intensity at the condensate boundary (r/Rcondensate boundary = 1). Part b is adapted from ref. 23 under a Creative Commons licence CC BY 4.0. Part c is adapted from ref. 23 under a Creative Commons licence CC BY 4.0. Part d is adapted from ref. 45, Springer Nature Limited. Part e is reprinted from ref. 55 under a Creative Commons licence CC BY 4.0.
The coating of condensates by amyloidogenic proteins may be a common phenomenon. Interfacial localization and aggregation occurring at the interface of charged coacervates have already been observed for αSyn23 (Fig. 5b,c). We hypothesize that owing to the amphiphilic properties of some amyloidogenic proteins combined with the surface charge of coacervates, they are driven to the interface by electrostatic effects. Furthermore, Linsenmeier et al. show that idemic condensates consisting of splicing variants of hnRNPA1, a protein involved in ALS, catalyse hnRNPA1 fibril formation on their surface, and that aggregation could be reduced by introducing protein-based surfactant molecules45 (Fig. 5d). In another case, MEG-3 protein clusters were found to localize at the interface of PGL droplets and act as Pickering agents to slow coarsening95. Similar findings have been made for stress granules wherein protein–RNA assemblies adsorb to their interface96. The surface-to-volume ratio of cellular condensates may also lead to more pronounced interfacial effects because cellular condensates are relatively small compared to condensates used in in vitro studies. Pickering effects and condensate interfacial charge could help stabilize smaller droplets with relatively large surface area95,97. Together, these results suggest that interfacial localization of proteins or protein clusters may have an important role in vivo and that it is also potentially relevant for aggregation.
However, two alternative possibilities in vivo are that non-aggregation-prone biomolecules occupy the interface of condensates more strongly than amyloidogenic proteins in healthy states, preventing aggregation, and that condensates in vivo are insufficiently surface charged to concentrate proteins at their interface. Although interfacial effects based on surface charge could be less pronounced in vivo, interfacial localization based on hydrophobicity may be sufficient to drive proteins to the interface for certain proteins and condensates. Although the evidence for in vivo interfacial accumulation is limited, a work by Choi et al. shows that artificial light-induced condensation can give rise to condensates that interfacially accumulate aggregation-prone proteins55 (Fig. 5e). Interestingly, they also studied the effect of condensate formation and dissolution cycles on protein aggregate formation and observed an increase in the average aggregate size after successive cycles. This may be particularly relevant for in vivo environments wherein most condensates are transient. Combined, these results suggest that the interface of condensates could have an important role in the process of fibrillar aggregate formation.
The protein aggregation pathway and energy landscape
Besides altering the spatial distribution of proteins and the physicochemical properties of the environment in which aggregation takes place, the presence of biomolecular condensates can also alter the energy landscape and protein aggregation pathway. Protein aggregation and amyloid formation are complex processes that involve hundreds of intramolecular and intermolecular interactions, consolidated in an energy landscape with a multitude of local minima and a complex overall shape. In such energy landscapes, the amyloid state is typically depicted as a deep global minimum with steep gradients, reflecting the practical irreversibility of most amyloid structures98. Theoretically, the energy landscape contains all the information about the pathways and kinetics of protein aggregation. However, in practice, the inherent complexity of protein aggregation has hampered efforts to quantify the underlying energy landscape. Nevertheless, viewing the aggregation process from the underlying energy landscape perspective can be useful for understanding how biomolecular condensates could impact protein aggregation63.
Nucleation and elongation rates are key in the overall speed of aggregation (Fig. 4c) and especially nucleation rates are linked to the conformation of a protein99. For instance, simulations with αSyn have shown that it can transition between several conformationally distinct aggregation-prone and aggregation-resistant metastable states. In a crowded environment, compaction of all metastable conformations was observed, strengthening the intramolecular interactions in the monomeric protein and possibly stabilizing it from aggregation100. A similar conformational effect could occur in condensates: the conformation of a monomeric protein may be altered when it is a part of a condensate or located at the surface of a condensate, as a result of intermolecular interactions between the (client) protein and other condensate components, and the conformational entropy at the condensate interface101. Lattice-based Monte Carlo simulations have shown that IDPs with a sticker-and-spacer architecture exhibit a more extended conformation inside condensates than in the dilute phase, contrary to the effect of crowding101 (Fig. 6a). Intramolecular FRET experiments of tau also suggest an expanded conformation, as shown by a decrease in single-molecule FRET efficiency between spaced-out FRET pairs on tau in LLPS conditions compared to in solution48 (Fig. 6b). By contrast, NMR experiments on FUS LC showed no evidence for structural differences between the protein in the dilute and condensed phase73. Interestingly, simulations predict that the conformation of proteins at the interface is the most extended, which could result in an enhanced aggregation rate specifically at the condensate interface23,101 (Fig. 6c). These extended conformations at the interface may help give rise to heterogeneous nucleation23,45,88,94. It is important to note that these simulations concerned condensate-forming IDPs (giving rise to idemic protein aggregation) — the conformation of client IDPs has not been studied in detail yet.

a, Simulated radius of gyration (Rg) of wild-type A1-low-complexity domain (LCD) within the dilute and dense phases against the width of the phase separation (ω) defined as \({\log }_{10}(\frac{{c}_{{\rm{dilute}}}}{{c}_{{\rm{dense}}}})\). Conformations inside the dense phase are more expanded. b, Intramolecular Förster resonance energy transfer (FRET) of tau in non-liquid–liquid phase separation (LLPS) conditions and in LLPS conditions shows that upon phase separation the FRET efficiency decreases, indicating an expanded conformation. The blue-shaded region indicates the inactive acceptor region. c, Left panel: a schematic view of how polymer chains at the radial shell of interest (area between dashed lines) can be oriented with respect to the condensate. The number of residues (red) can be counted and divided by the number of chains with a residue in this area (blue) to calculate how perpendicular the chains are to the condensate. A higher number indicates more perpendicular conformations. Right panel: simulated data of wild-type hnRNPA1-LCD showing the number of distinct chains per residue plotted against the distance from the centre of the condensate. At the interface (blue-shaded region), there is a higher number of chains per residue, indicating a higher perpendicularity than the inside of the condensate. d, Aggregation of 2 μM amyloid-β 1-42 (Aβ-42) solution, expressed by the fibril mass fraction, without (black) and with (blue) 10 μM LAF-1-AK-LAF-1 condensates, which are able to sequester the Aβ-42. e, Misfolded proteins can partition into otherwise healthy host condensates such as stress granules and aggregate within them, causing solidification of condensates127. Chaperones can disassemble condensates in the early stages and degrade the aggresome in case of aggregation. f, Fluorescence recovery after photobleaching (FRAP) of FUS-EGFP condensates with (+) and without (−) Hsp70 chaperones show that the addition of Hsp70 helps prevent LST. g, In the left panel, FRAP of luciferase-GFP within nucleoli, both without (−) and with (+) heat shock (HS), and with the inclusion of 2-phenylbenzothiazole (PBT), a luciferase stabilizer, is shown. PBT proves effective in preventing nucleolar aggregation. In the right panel, the introduction of VER-155008, an Hsp70 inhibitor, hinders the function of Hsp70 in nucleoli, impeding the prevention of solidification. Rec refers to the state after the recovery from the heat shock. FUS, fused in sarcoma; PEG, polyethylene glycol. Part a is reprinted from ref. 101 under a Creative Commons licence CC BY 4.0. Part b is adapted from ref. 48 under a Creative Commons licence CC BY 4.0. Part c is adapted from ref. 101 under a Creative Commons licence CC BY 4.0. Part d is reprinted with permission from ref. 54, RSC. Part f is reprinted with permission from ref. 118, Elsevier. Part g is reprinted with permission from ref. 62, AAAS.
An altered conformation affects the intrinsic stability of a protein and impacts the probability of the protein sampling a range of conformations that lead to aggregation. For example, for αSyn, it has been shown that intramolecular interactions between the N-terminal and C-terminal region of the protein contribute to its stability102 and that sampling of locally extended β-strand-like conformations may contribute to aggregate growth and secondary nucleation103. Insights into protein conformation inside condensates is, therefore, relevant for understanding not only condensate structure and function but also the propensity of aggregation of client and scaffold proteins, although the precise effects depend on the molecular details of the aggregation process, as illustrated by αSyn. Molecular dynamics simulations and NMR experiments have shown that IDPs inside condensates exhibit slower backbone, segmental and collective dynamics than IDPs in both a dilute phase and crowded media with similar viscosities, suggesting that intermolecular interactions between IDPs are responsible for the decreased dynamics104. These results suggest that protein aggregation may be slowed down inside condensates, as a result of the restricted fluctuations. An example of this was found by Küffner et al., who show that Aβ-42 is sequestered within LAF-1-AK-LAF condensates54 (Fig. 6d).
Finally, the energy landscape is also affected by the solvation of proteins. Aggregation-prone globular proteins can be stabilized by co-solvents such as glycerol, driven by the interactions of glycerol with hydrophobic surface regions of the native proteins, inhibiting protein unfolding and stabilizing any partially unfolded species81. The environment inside condensates is characterized by a lower polarity than the surrounding solution. A more hydrophobic environment inside condensates67,105,106,107 will probably lead to decompaction of the hydrophobic pockets of proteins and a hydration-driven compaction of hydrophilic regions. How this affects disordered proteins, such as αSyn, is difficult to predict. The potentially expanded conformation of proteins inside condensates is expected to relate to an increase in aggregation. However, owing to a competition in interactions with condensate material or other guest molecules, it is expected that an expanded conformation may still lead to decreased aggregation54 depending on interaction strength with the condensate material.
Together, insights into structural characteristic and conformational dynamics of proteins (both host and client) inside condensates may hold another key to understanding the role of condensates in protein aggregation. Protein conformations inside condensates and at the surface of condensates are not easily determined experimentally but can be probed by rigorous FRET108,109, EPR110, or NMR111,112 studies. How changes to protein conformation affect their aggregation further depends on the details of the aggregation pathway. Characteristics of the energy landscape may be estimated by measuring the kinetics of aggregation in a spatially resolved manner and extracting aggregation rate constants23.
Chaperone activity and condensates
Chaperones have a crucial role in cellular protein quality control and have attracted considerable attention in the study of cellular aging and protein aggregation24. Chaperones are proteins that assist in the proper folding, assembly and stabilization of other proteins, preventing misfolding and aggregation113. Small heat shock proteins (sHsps) were shown to inhibit amyloid fibril formation under physiological conditions in vitro in 2001, using apolipoprotein C-II114. Since then, interactions between sHsps and many amyloidogenic proteins have been found115. For instance, Hsp70 is capable of dissolving preformed αSyn fibrils in vitro, and at least five other sHsps have been shown to interact and inhibit aggregation116,117. Mechanistically, chaperones can target specific steps in the aggregation pathway. Hsp70 can target primary or secondary nucleation processes in Ure2 aggregation, resulting in a delayed lag phase118, αB-crystallin can reduce the elongation of Aβ fibrils119, and Hsp110 with Hsp70 and Hsp40 have been shown to disaggregate fibrils of Aβ, tau, αSyn and amylin120. Soluble oligomers can also be targeted by Hsp90, which has also been implicated in PD121. A work by Zhang et al. has shown that TRIM11 is capable of various aggregation-related functions for tau, such as maintaining solubility, preventing seeding of aggregates, disaggregating aggregates, and even preventing disease phenotype progression in mice model systems122. Collectively, chaperones are important in regulating amyloid formation by targeting intermediate species, maintaining solubility of monomers, and clearing out formed aggregates.
Chaperones have also been associated with stabilizing phase separations of amyloid-forming proteins118,123,124, highlighting the importance of understanding their interactions with LLPS (Fig. 6e). Many chaperones are involved in inhibiting amyloid formation (such as Hsp27, Hsp40 and Hsp70), and others have been shown to be able to localize within stress granules and inhibit the LST of FUS (Fig. 6f). This is probably because the motifs targeted by sHsps to prevent aggregation are also involved in LLPS115. Hsp70 has also been shown to modulate the liquid properties of stress granules, demonstrating that the role of chaperones in regulating protein aggregation may not be limited to direct intervention in the protein aggregation pathway.
Similar to how protein aggregation inhibition by small molecules can be enhanced by partitioning into condensates together with amyloidogenic proteins, chaperones may exploit the same mechanisms64. Because condensates may provide an environment that promotes amyloid formation, the presence of sHsps with potentially locally increased concentrations may be necessary to prevent rampant aggregation. Frottin et al. demonstrated that Hsp70 accumulates in the nucleolus during cellular stress, alongside misfolded proteins, to refold the proteins62. Afterwards, the proteins are released from the nucleolus in their refolded form (Fig. 6g). Additionally, the activity of chaperone complexes involved in disaggregation, such as the aforementioned Hsp104 with Hsp70 and Hsp40 or the synergistic action between DNAJB1 and Hsp110, may be enhanced by localizing inside condensates117,120. There is also evidence that some chaperones specifically target energetically frustrated sites of partially misfolded proteins, highlighting the importance of protein conformation dynamics within condensates when potentially interacting with chaperones125. This protein aggregation inhibition can be achieved through direct interactions with the monomeric protein, misfolded intermediates, disassembly of aggregates, or by modulating the condensate properties themselves.
Conclusions and perspectives
The mechanism of protein aggregation within the cellular context has been a longstanding question in understanding neurodegenerative disease. The complex cellular environment, including the presence of liquid–liquid phase-separated condensates, is attracting increasing interest and its role in protein aggregation is only now beginning to be investigated. Currently, how important biomolecular condensate-mediated aggregation pathways are for protein aggregation-related disease states is unclear. Although in vitro studies can be useful for understanding the process in a controlled environment, not all in vitro conditions are relevant for or representative of (pre-)disease states in cells. This underscores the need for both in vitro and in vivo studies. In this Review, we discussed the possible ways in which LLPS could in principle alter protein aggregation. Future experiments with IDPs in conditions that mimic the cellular environment as closely as possible are key to establishing the actual role of condensates in understanding physiological protein aggregation. Nevertheless, from a physicochemical point of view, it seems plausible that condensates can alter the process of protein aggregation.
Both idemic condensate aggregation, in which the aggregation-prone protein itself phase separates and undergoes a transition from liquid to solid, fibrillar state, and host–guest condensate aggregation, in which an aggregation-prone protein partitions into a condensate, are relevant for understanding protein aggregation. We propose that five key factors related to the presence of a condensate compartment affect the aggregation kinetics in both situations (Fig. 1).
We expect that condensates in vivo influence amyloid protein aggregation by both partitioning and interface effects, depending on the properties of the protein (for example, hydrophobicity may promote partitioning into the condensates, whereas amphiphilicity promotes accumulation at the interface) and on the properties of the condensates (such as the presence of domains that can bind to specific sites of the aggregating protein). We note that the properties that drive partitioning of proteins will also be important in the protein conformation when interacting with the condensate material and that these effects are not understood separately. In terms of importance, we expect that surface effects from the condensates will be highly relevant for future studies and in determining the overall effect of condensates on protein aggregation. Viscosity and crowding effects by themselves may not be as important, but they can contribute to the altered kinetics and thermodynamics of aggregation. We consider the partition coefficient to be another key factor as it is probably related to the interaction strength of the protein to the condensate material (and, thus, potentially its conformation inside the condensates) and is necessary for viscosity and crowding effects to take place. Finally, chaperone function and activity inside condensates are currently understudied factors with considerable implications for our understanding of protein stability and aggregation in the complex and compartmentalized cellular environment.
Overall, the interactions between the condensates and the aggregating proteins offer a possible way by which cells can actually regulate protein aggregation. Sequestering monomers to inhibit fibrilization or speeding up the transition of toxic oligomers into aggregates could achieve this goal. However, LLPS can also be a pathway for the progression of disease — mutations or disturbances within phase separation-regulating pathways may potentially promote neurodegenerative disease. A full understanding of the interplay between factors involving phase separation and biological protein quality control can help develop new treatments and strategies to prevent pathological aggregation.
References
Ross, C. A. & Poirier, M. A. Protein aggregation and neurodegenerative disease. Nat. Med. 10, S10–S17 (2004).
Willbold, D., Strodel, B., Schröder, G. F., Hoyer, W. & Heise, H. Amyloid-type protein aggregation and prion-like properties of amyloids. Chem. Rev. 121, 8285–8307 (2021).
Dobson, C. M. The amyloid phenomenon and its links with human disease. Cold Spring Harb. Perspect. Biol. 9, a023648 (2017).
Emin, D. et al. Small soluble α-synuclein aggregates are the toxic species in Parkinson’s disease. Nat. Commun. 13, 5512 (2022).
Cascella, R. et al. Probing the origin of the toxicity of oligomeric aggregates of α-synuclein with antibodies. ACS Chem. Biol. 14, 1352–1362 (2019).
Meisl, G. et al. Uncovering the universality of self-replication in protein aggregation and its link to disease. Sci. Adv. 8, 6831 (2022).
Alberti, S. & Hyman, A. A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 22, 196–213 (2021).
Alberti, S. & Hyman, A. A. Are aberrant phase transitions a driver of cellular aging? BioEssays 38, 959–968 (2016).
Vazquez, D. S., Toledo, P. L., Gianotti, A. R. & Ermácora, M. R. Protein conformation and biomolecular condensates. Curr. Res. Struct. Biol. 4, 285–307 (2022).
Nakashima, K. K., Vibhute, M. A. & Spruijt, E. Biomolecular chemistry in liquid phase separated compartments. Front. Mol. Biosci. 6, 21 (2019).
Bhattacharya, A. et al. Lipid sponge droplets as programmable synthetic organelles. Proc. Natl Acad. Sci. USA 117, 18206–18215 (2020).
de Jong, B. Coacervation. Proc. R. Acad. Amst. 32, 849–856 (1929).
Banani, S. F., Lee, H. O., Hyman, A. A. & Rosen, M. K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298 (2017).
Peeples, W. & Rosen, M. K. Mechanistic dissection of increased enzymatic rate in a phase-separated compartment. Nat. Chem. Biol. 17, 693–702 (2021).
Zhang, Y., Narlikar, G. J. & Kutateladze, T. G. Enzymatic reactions inside biological condensates. J. Mol. Biol. 433, 166624 (2021).
Molliex, A. et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133 (2015).
Nedelsky, N. B. & Taylor, J. P. Pathological phase transitions in ALS-FTD impair dynamic RNA–protein granules. RNA 28, 97–113 (2022).
Dewey, C. M. et al. TDP-43 aggregation in neurodegeneration: are stress granules the key? Brain Res. 1462, 16–25 (2012).
Törnquist, M. et al. Secondary nucleation in amyloid formation. Chem. Commun. 54, 8667–8684 (2018).
Michaels, T. C. T. et al. Chemical kinetics for bridging molecular mechanisms and macroscopic measurements of amyloid fibril formation. Annu. Rev. Phys. Chem. 69, 273–298 (2018).
Sinnige, T. et al. Kinetic analysis reveals that independent nucleation events determine the progression of polyglutamine aggregation in C. elegans. Proc. Natl Acad. Sci. USA 118, e2021888118 (2021).
Ignatova, Z. & Gierasch, L. M. Monitoring protein stability and aggregation in vivo by real-time fluorescent labeling. Proc. Natl Acad. Sci. USA 101, 523–528 (2004).
Lipiński, W. P. et al. Biomolecular condensates can both accelerate and suppress aggregation of α-synuclein. Sci. Adv. 8, eabq6495 (2022).
Knowles, T. P. J., Vendruscolo, M. & Dobson, C. M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396 (2014).
Farzadfard, A. et al. Thermodynamic characterization of amyloid polymorphism by microfluidic transient incomplete separation. Chem. Sci. 15, 2528–2544 (2024).
Weber, C., Michaels, T. & Mahadevan, L. Spatial control of irreversible protein aggregation. eLife 8, 42315 (2019).
Khurana, R. et al. Mechanism of thioflavin T binding to amyloid fibrils. J. Struct. Biol. 151, 229–238 (2005).
Wetzel, R. Amyloids, prions & other aggregates. Methods Enzymol. 309, 3–820 (1999).
Hellstrand, E., Boland, B., Walsh, D. M. & Linse, S. Amyloid β-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process. ACS Chem. Neurosci. 1, 13–18 (2010).
Zurlo, E. et al. In situ kinetic measurements of α-synuclein aggregation reveal large population of short-lived oligomers. PLoS ONE 16, e0245548 (2021).
Fakhree, M. A. A., Nolten, I. S., Blum, C. & Claessens, M. M. A. E. Different conformational subensembles of the intrinsically disordered protein α-synuclein in cells. J. Phys. Chem. Lett. 9, 1249–1253 (2018).
Veldhuis, G., Segers-Nolten, I., Ferlemann, E. & Subramaniam, V. Single-molecule FRET reveals structural heterogeneity of SDS-bound α-synuclein. ChemBioChem 10, 436–439 (2009).
Iljina, M. et al. Quantitative analysis of co-oligomer formation by amyloid-beta peptide isoforms. Sci. Rep. 6, 28658 (2016).
Tittelmeier, J., Druffel-Augustin, S., Alik, A., Melki, R. & Nussbaum-Krammer, C. Dissecting aggregation and seeding dynamics of α-Syn polymorphs using the phasor approach to FLIM. Commun. Biol. 5, 1345 (2022).
Ray, S. et al. Mass photometric detection and quantification of nanoscale α-synuclein phase separation. Nat. Chem. 15, 1306–1316 (2023).
Murakami, T. et al. ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88, 678–690 (2015).
Lin, Y., Protter, D. S. W., Rosen, M. K. & Parker, R. Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell 60, 208–219 (2015).
Mathieu, C., Pappu, R. V. & Taylor, J. P. Beyond aggregation: pathological phase transitions in neurodegenerative disease. Science 370, 55–60 (2020).
Patel, A. et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077 (2015).
Kim, H. J. et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473 (2013).
Conicella, A. E., Zerze, G. H., Mittal, J. & Fawzi, N. L. ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 24, 1537–1549 (2016).
Kang, H. et al. PARIS undergoes liquid–liquid phase separation and poly(ADP‐ribose)‐mediated solidification. EMBO Rep. 24, e56166 (2023).
Gruijs da Silva, L. A. et al. Disease‐linked TDP‐43 hyperphosphorylation suppresses TDP‐43 condensation and aggregation. EMBO J. 41, e108443 (2022).
Tomaszewski, A. et al. Solid-to-liquid phase transition in the dissolution of cytosolic misfolded-protein aggregates. iScience 26, 108334 (2023).
Linsenmeier, M. et al. The interface of condensates of the hnRNPA1 low-complexity domain promotes formation of amyloid fibrils. Nat. Chem. 15, 1340–1349 (2023).
Wegmann, S. et al. Tau protein liquid–liquid phase separation can initiate tau aggregation. EMBO J. 37, e98049 (2018).
Boyko, S. et al. Liquid-liquid phase separation of tau protein: the crucial role of electrostatic interactions. J. Biol. Chem. 294, 11054–11059 (2019).
Wen, J. et al. Conformational expansion of tau in condensates promotes irreversible aggregation. J. Am. Chem. Soc. 143, 13056–13064 (2021).
Boyko, S., Surewicz, K. & Surewicz, W. K. Regulatory mechanisms of tau protein fibrillation under the conditions of liquid–liquid phase separation. Proc. Natl Acad. Sci. USA 117, 31882–31890 (2020).
Ray, S. et al. Synuclein aggregation nucleates through liquid–liquid phase separation. Nat. Chem. 12, 705–716 (2020).
Ray, S. et al. Spatiotemporal solidification of α-synuclein inside the liquid droplets. Preprint at https://doi.org/10.1101/2021.10.20.465113 (2021).
Sawner, A. S. et al. Modulating α-synuclein liquid-liquid phase separation. Biochem 60, 3676–3696 (2021).
Hardenberg, M. C. et al. Observation of an α-synuclein liquid droplet state and its maturation into Lewy body-like assemblies. J. Mol. Cell Biol. 13, 282–294 (2021).
Küffner, A. M. et al. Sequestration within biomolecular condensates inhibits Aβ-42 amyloid formation. Chem. Sci. 12, 4373–4382 (2021).
Choi, C. H., Lee, D. S. W., Sanders, D. W. & Brangwynne, C. P. Condensate interfaces can accelerate protein aggregation. Biophys. J. 123, 1404–1413 (2023).
Shen, Y. et al. Biomolecular condensates undergo a generic shear-mediated liquid-to-solid transition. Nat. Nanotechnol. 15, 841–847 (2020).
Riback, J. A. et al. Composition-dependent thermodynamics of intracellular phase separation. Nature 581, 209–214 (2020).
Elbaum-Garfinkle, S. et al. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl Acad. Sci. USA 112, 7189–7194 (2015).
Onuchic, P. L., Milin, A. N., Alshareedah, I., Deniz, A. A. & Banerjee, P. R. Divalent cations can control a switch-like behavior in heterotypic and homotypic RNA coacervates. Sci. Rep. 9, 12161 (2019).
McCall, P. M. et al. Partitioning and enhanced self-assembly of actin in polypeptide coacervates. Biophys. J. 114, 1636–1645 (2018).
Samanta, N. et al. Sequestration of proteins in stress granules relies on the in-cell but not the in vitro folding stability. J. Am. Chem. Soc. 143, 19909–19918 (2021).
Frottin, F. et al. The nucleolus functions as a phase-separated protein quality control compartment. Science 365, 342–347 (2019).
Bauermann, J., Laha, S., McCall, P. M., Jülicher, F. & Weber, C. A. Chemical kinetics and mass action in coexisting phases. J. Am. Chem. Soc. 144, 19294–19304 (2022).
Michaels, T. C. T., Mahadevan, L. & Weber, C. A. Enhanced potency of aggregation inhibitors mediated by liquid condensates. Phys. Rev. Res. 4, 043173 (2022).
Stender, E. G. P. et al. Capillary flow experiments for thermodynamic and kinetic characterization of protein liquid-liquid phase separation. Nat. Commun. 12, 7289 (2021).
Taylor, N. O., Wei, M. T., Stone, H. A. & Brangwynne, C. P. Quantifying dynamics in phase-separated condensates using fluorescence recovery after photobleaching. Biophys. J. 117, 1285–1300 (2019).
Yewdall, N. A., André, A. A. M., Lu, T. & Spruijt, E. Coacervates as models of membraneless organelles. Curr. Opin. Colloid Interface Sci. 52, 101416 (2021).
Pönisch, W., Michaels, T. C. T. & Weber, C. A. Aggregation controlled by condensate rheology. Biophys. J. 122, 197–214 (2023).
Ahmad, B., Chen, Y. & Lapidus, L. J. Aggregation of α-synuclein is kinetically controlled by intramolecular diffusion. Proc. Natl Acad. Sci. USA 109, 2336–2341 (2012).
Wei, M. T. et al. Phase behaviour of disordered proteins underlying low density and high permeability of liquid organelles. Nat. Chem. 9, 1118–1125 (2017).
Wang, H., Kelley, F. M., Milovanovic, D., Schuster, B. S. & Shi, Z. Surface tension and viscosity of protein condensates quantified by micropipette aspiration. Biophys. Rep. 1, 100011 (2021).
Li, J., Uversky, V. N. & Fink, A. L. Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochem 40, 11604–11613 (2001).
Murthy, A. C. et al. Molecular interactions underlying liquid–liquid phase separation of the FUS low-complexity domain. Nat. Struct. Mol. Biol. 26, 637–648 (2019).
Smisdom, N. et al. Fluorescence recovery after photobleaching on the confocal laser-scanning microscope: generalized model without restriction on the size of the photobleached disk. J. Biomed. Opt. 16, 046021 (2011).
Axelrod, D., Koppel, D. E., Schlessinger, J., Elson, E. & Webb, W. W. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 16, 1055–1069 (1976).
Jawerth, L. M. et al. Salt-dependent rheology and surface tension of protein condensates using optical traps. Phys. Rev. Lett. 121, 258101 (2018).
Zhou, H. X. Determination of condensate material properties from droplet deformation. J. Phys. Chem. B 124, 8372–8379 (2020).
Kalwarczyk, T. et al. Motion of nanoprobes in complex liquids within the framework of the length-scale dependent viscosity model. Adv. Colloid Interface Sci. 223, 55–63 (2015).
Bubak, G. et al. Quantifying nanoscale viscosity and structures of living cells nucleus from mobility measurements. J. Phys. Chem. Lett. 12, 294–301 (2021).
Munishkina, L. A., Cooper, E. M., Uversky, V. N. & Fink, A. L. The effect of macromolecular crowding on protein aggregation and amyloid fibril formation. J. Mol. Recognit. 17, 456–464 (2004).
Vagenende, V., Yap, M. G. S. & Trout, B. L. Mechanisms of protein stabilization and prevention of protein aggregation by glycerol. Biochem 48, 11084–11096 (2009).
Roussel, M. R. Foundations of Chemical Kinetics (IOP Publishing, 2023).
Abyzov, A., Blackledge, M. & Zweckstetter, M. Conformational dynamics of intrinsically disordered proteins regulate biomolecular condensate chemistry. Chem. Rev. 122, 6719–6748 (2022).
Rubinstein, M. & Colby, R. H. Polymer Physics (Oxford Univ. Press, 2003).
Garaizar, A. et al. Aging can transform single-component protein condensates into multiphase architectures. Proc. Natl Acad. Sci. USA 119, e2119800119 (2022).
Breydo, L. et al. The crowd you’re in with: effects of different types of crowding agents on protein aggregation. Biochim. Biophys. Acta Proteins Proteom. 1844, 346–357 (2014).
Schreck, J. S., Bridstrup, J. & Yuan, J. M. Investigating the effects of molecular crowding on the kinetics of protein aggregation. J. Phys. Chem. B 124, 9829–9839 (2020).
Grigolato, F. & Arosio, P. The role of surfaces on amyloid formation. Biophys. Chem. 270, 106533 (2021).
Galvagnion, C. et al. Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol. 11, 229–234 (2015).
Marie, G. et al. Acceleration of α-synuclein aggregation by exosomes. J. Biol. Chem. 290, 2969–2982 (2015).
Morinaga, A. et al. Critical role of interfaces and agitation on the nucleation of Aβ amyloid fibrils at low concentrations of Aβ monomers. Biochim. Biophys. Acta Proteins Proteom. 1804, 986–995 (2010).
Gray, J. J. The interaction of proteins with solid surfaces. Curr. Opin. Struct. Biol. 14, 110–115 (2004).
Zapadka, K. L., Becher, F. J., Gomes dos Santos, A. L. & Jackson, S. E. Factors affecting the physical stability (aggregation) of peptide therapeutics. Interface Focus 7, 20170030 (2017).
Camino, J. D., Gracia, P. & Cremades, N. The role of water in the primary nucleation of protein amyloid aggregation. Biophys. Chem. 269, 106520 (2021).
Folkmann, A. W., Putnam, A., Lee, C. F. & Seydoux, G. Regulation of biomolecular condensates by interfacial protein clusters. Science 373, 1218–1224 (2021).
Garcia-Jove Navarro, M. et al. RNA is a critical element for the sizing and the composition of phase-separated RNA–protein condensates. Nat. Commun. 10, 3230 (2019).
Welsh, T. J. et al. Surface electrostatics govern the emulsion stability of biomolecular condensates. Nano Lett. 22, 612–621 (2022).
Vabulas, R. M., Raychaudhuri, S., Hayer-Hartl, M. & Hartl, F. U. Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb. Perspect. Biol. 2, a004390 (2010).
Chirita, C. N., Congdon, E. E., Yin, H. & Kuret, J. Triggers of full-length tau aggregation: a role for partially folded intermediates. Biochemistry 44, 5862–5872 (2005).
Menon, S. & Mondal, J. Conformational plasticity in α-synuclein and how crowded environment modulates it. J. Phys. Chem. B 127, 4032–4049 (2023).
Farag, M. et al. Condensates formed by prion-like low-complexity domains have small-world network structures and interfaces defined by expanded conformations. Nat. Commun. 13, 7722 (2022).
Ohgita, T. et al. Intramolecular interaction kinetically regulates fibril formation by human and mouse α-synuclein. Sci. Rep. 13, 10885 (2023).
Kumari, P. et al. Structural insights into α-synuclein monomer–fibril interactions. Proc. Natl Acad. Sci. USA 118, e2012171118 (2021).
Guseva, S. et al. Liquid-liquid phase separation modifies the dynamic properties of intrinsically disordered proteins. J. Am. Chem. Soc. 145, 10548–10563 (2023).
Zhao, M. et al. Partitioning of small molecules in hydrogen-bonding complex coacervates of poly(acrylic acid) and poly(ethylene glycol) or pluronic block copolymer. Macromolecules 50, 3818–3830 (2017).
Huang, S. et al. Effect of small molecules on the phase behavior and coacervation of aqueous solutions of poly(diallyldimethylammonium chloride) and poly(sodium 4-styrene sulfonate). J. Colloid Interface Sci. 518, 216–224 (2018).
Lipiński, W. P. et al. Fibrils e merging from droplets: molecular guiding principles behind phase transitions of a short peptide-based condensate studied by solid-state NMR. Chem. Eur. J. 29, e202301159 (2023).
Leblanc, S. J., Kulkarni, P. & Weninger, K. R. Single molecule FRET: a powerful tool to study intrinsically disordered proteins. Biomolecules 8, 140 (2018).
Holmstrom, E. D. et al. Accurate transfer efficiencies, distance distributions, and ensembles of unfolded and intrinsically disordered proteins from single-molecule FRET. Methods Enzymol. 611, 287–325 (2018).
Bordignon, E. & Polyhach, Y. EPR techniques to probe insertion and conformation of spin-labeled proteins in lipid bilayers. Meth. Mol. Biol. 974, 329–355 (2013).
Maltseva, D. et al. Fibril formation and ordering of disordered FUS LC driven by hydrophobic interactions. Nat. Chem. 15, 1146–1154 (2023).
Dyson, H. J. & Wright, P. E. Insights into the structure and dynamics of unfolded proteins from nuclear magnetic resonance. Adv. Protein Chem. 62, 311–340 (2002).
Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 14, 630–642 (2013).
Hatters, D. M., Lindner, R. A., Carver, J. A. & Howlett, G. J. The molecular chaperone, α-crystallin, inhibits amyloid formation by apolipoprotein C-II. J. Biol. Chem. 276, 33755–33761 (2001).
Webster, J. M., Darling, A. L., Uversky, V. N. & Blair, L. J. Small heat shock proteins, big impact on protein aggregation in neurodegenerative disease. Front. Pharmacol. 10, 1047 (2019).
Bruinsma, I. B. et al. Inhibition of α-synuclein aggregation by small heat shock proteins. Proteins 79, 2956–2967 (2011).
Wentink, A. S. et al. Molecular dissection of amyloid disaggregation by human HSP70. Nature 587, 483–488 (2020).
Li, Y. et al. Hsp70 exhibits a liquid-liquid phase separation ability and chaperones condensed FUS against amyloid aggregation. iScience 25, 104356 (2022).
Shammas, S. L. et al. Binding of the molecular chaperone αb-crystallin to Aβ amyloid fibrils inhibits fibril elongation. Biophys. J. 101, 1681–1689 (2011).
Shorter, J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS ONE 6, e26319 (2011).
Daturpalli, S., Waudby, C. A., Meehan, S. & Jackson, S. E. Hsp90 inhibits α-synuclein aggregation by interacting with soluble oligomers. J. Mol. Biol. 425, 4614–4628 (2013).
Zhang, Z. Y. et al. TRIM11 protects against tauopathies and is down-regulated in Alzheimer’s disease. Science 381, eadd6696 (2023).
Liu, Z. et al. Hsp27 chaperones FUS phase separation under the modulation of stress-induced phosphorylation. Nat. Struct. Mol. Biol. 27, 363–372 (2020).
Gu, J. et al. Hsp40 proteins phase separate to chaperone the assembly and maintenance of membraneless organelles. Proc. Natl Acad. Sci. USA 117, 31123–31133 (2020).
Hiller, S. Chaperone-bound clients: the importance of being dynamic. Trends Biochem. Sci. 44, 517–527 (2019).
Zbinden, A., Pérez-Berlanga, M., De Rossi, P. & Polymenidou, M. Phase separation and neurodegenerative diseases: a disturbance in the force. Dev. Cell 55, 45–68 (2020).
Mateju, D. et al. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 36, 1669–1687 (2017).
Acknowledgements
This work was supported financially by a Vidi grant from the Netherlands Organization for Scientific Research (NWO).
Author information
Authors and Affiliations
Contributions
All authors contributed to the writing and editing of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Chemistry thanks Alexander Buell, Sara Linse and Soumik Ray, and the other anonymous referee(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Visser, B.S., Lipiński, W.P. & Spruijt, E. The role of biomolecular condensates in protein aggregation. Nat Rev Chem 8, 686–700 (2024). https://doi.org/10.1038/s41570-024-00635-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41570-024-00635-w
