
Abstract
Given the global burden of heart failure, strategies to understand the underlying cause or to provide prognostic information are critical to reducing the morbidity and mortality associated with this highly prevalent disease. Cardiomyopathies often have a genetic cause, and the field of heart failure genetics is progressing rapidly. Through a deliberate investigation, evaluation for a familial component of cardiomyopathy can lead to increased identification of pathogenic genetic variants. Much research has also been focused on identifying markers of risk in patients with cardiomyopathy with the use of genetic testing. Advances in our understanding of genetic variants have been slightly offset by an increased recognition of the heterogeneity of disease expression. Greater breadth of genetic testing can increase the likelihood of identifying a variant of uncertain significance, which is resolved only rarely by cellular functional validation and segregation analysis. To increase the use of genetics in heart failure clinics, increased availability of genetic counsellors and other providers with experience in genetics is necessary. Ultimately, through ongoing research and increased clinical experience in cardiomyopathy genetics, an improved understanding of the disease processes will facilitate better clinical decision-making about the therapies offered, exemplifying the implementation of precision medicine.
Key points
The frontier of genetics is rapidly advancing and, increasingly, genetic testing in heart failure clinics is associated with benefit for family screening and individual prognostication.
A focused evaluation of the clinical characteristics and inheritance pattern of heart failure or sudden cardiac death, in addition to consultation with a genetic counsellor when appropriate, can facilitate a successful genetic evaluation.
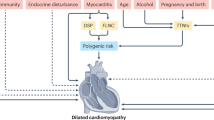
Genetic testing is recommended in all patients with familial dilated cardiomyopathy (DCM) to facilitate screening, whereas guideline recommendations for testing in patients with sporadic DCM differ, but specific clinical features might increase the yield of testing.
Genetic testing in DCM is currently associated with the identification of a culprit variant in approximately 15–25% of patients with sporadic DCM and approximately 20–40% of patients with familial DCM.
The identification of a pathogenic variant might have important predictive and therapeutic implications; however, expression of the phenotype might depend on environmental triggers.
All first-degree relatives of patients with familial DCM should undergo clinical screening; guidelines for screening of first-degree relatives of patients with sporadic DCM differ.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others


References
Mozaffarian, D. et al. Heart disease and stroke statistics — 2016 update a report from the American Heart Association. Circulation 133, e38–e360 (2016).
Bui, A. L., Horwich, T. B. & Fonarow, G. C. Epidemiology and risk profile of heart failure. Nat. Rev. Cardiol. 8, 30–41 (2011).
Ponikowski, P. et al. Heart failure: preventing disease and death worldwide. ESC Heart Fail. 1, 4–25 (2014).
Gerber, Y. et al. A contemporary appraisal of the heart failure epidemic in Olmsted County, Minnesota, 2000 to 2010. JAMA Intern. Med. 175, 996–1004 (2015).
Heidenreich, P. A. et al. Forecasting the impact of heart failure in the United States a policy statement from the American Heart Association. Circ. Heart Fail. 6, 606–619 (2013).
Redfield, M. M. et al. Burden of systolic and diastolic ventricular dysfunction in the community. JAMA 289, 194 (2003).
Hershberger, R. E., Hedges, D. J. & Morales, A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 10, 531–547 (2013).
Baldasseroni, S. et al. Left bundle-branch block is associated with increased 1-year sudden and total mortality rate in 5517 outpatients with congestive heart failure: a report from the Italian Network on Congestive Heart Failure. Am. Heart J. 143, 398–405 (2002).
Cahill, T. J., Ashrafian, H. & Watkins, H. Genetic cardiomyopathies causing heart failure. Circ. Res. 113, 660–675 (2013).
Burkett, E. L. & Hershberger, R. E. Clinical and genetic issues in familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 57, 1641–1649 (2011).
Jefferies, J. L. & Towbin, J. A. Dilated cardiomyopathy. Lancet 375, 752–762 (2010).
Michels, V. et al. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N. Engl. J. Med. 326, 77–82 (1992).
Grünig, E. et al. Frequency and phenotypes of familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 31, 186–194 (1998).
McNally, E. M. & Mestroni, L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ. Res. 121, 731–748 (2017).
Rosenbaum, A. N. & Pereira, N. L. Updates on the genetic paradigm in heart failure. Curr. Treat. Options Cardiovasc. Med. 21, 37 (2019).
Briasoulis, A., Asleh, R. & Pereira, N. in Encyclopedia of Cardiovascular Research and Medicine 1st edn (eds Sawyer, D. & Vasan, R.) 368–379 (Elsevier, 2018).
Elliott, P. Cardiomyopathy: diagnosis and management of dilated cardiomyopathy. Heart 84, 106–106 (2000).
Japp, A. G., Gulati, A., Cook, S. A., Cowie, M. R. & Prasad, S. K. The diagnosis and evaluation of dilated cardiomyopathy. J. Am. Coll. Cardiol. 67, 2996–3010 (2016).
Sweet, M. E., Taylor, M. R. & Mestroni, L. Diagnosis, prevalence, and screening of familial dilated cardiomyopathy. Expert. Opin. Orphan Drugs 3, 869–876 (2015).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Am. Coll. Med. Genet. Genomics 17, 405–424 (2015).
Cresci, S. et al. Clinical and genetic modifiers of long-term survival in heart failure. J. Am. Coll. Cardiol. 54, 432–444 (2009).
Liew, C.-C. & Dzau, V. J. Molecular genetics and genomics of heart failure. Nat. Rev. Genet. 5, 811–825 (2004).
Harakalova, M. et al. A systematic analysis of genetic dilated cardiomyopathy reveals numerous ubiquitously expressed and muscle-specific genes. Eur. J. Heart Fail. 17, 484–493 (2015).
Tayal, U., Prasad, S. & Cook, S. A. Genetics and genomics of dilated cardiomyopathy and systolic heart failure. Genome Med. 9, 1–14 (2017).
Hershberger, R. E. et al. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 20, 899–909 (2018).
Lakdawala, N. K. et al. Genetic testing for dilated cardiomyopathy in clinical practice. J. Card. Fail. 18, 296–303 (2012).
Ganesh, S. K. et al. Genetics and genomics for the prevention and treatment of cardiovascular disease: update. a scientific statement from the American Heart Association. Circulation 128, 2813–2851 (2013).
Pugh, T. J. et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 16, 601–608 (2014).
Hershberger, R. E. et al. Genetic evaluation of cardiomyopathy—a Heart Failure Society of America practice guideline. J. Card. Fail. 24, 281–302 (2009).
Adams, D. R. & Eng, C. M. Next-generation sequencing to diagnose suspected genetic disorders. N. Engl. J. Med. 379, 1353–1362 (2018).
Kushwaha, S. S., Fallon, J. T. & Fuster, V. Restrictive cardiomyopathy. N. Engl. J. Med. 336, 267–276 (1997).
Finsterer, J. Cardiogenetics, neurogenetics, and pathogenetics of left ventricular hypertrabeculation/noncompaction. Pediatr. Cardiol. 30, 659–681 (2009).
Towbin, J. A., Lorts, A. & Jefferies, J. L. Left ventricular non-compaction cardiomyopathy. Lancet 386, 813–825 (2015).
Gallego-Delgado, M. et al. Idiopathic restrictive cardiomyopathy is primarily a genetic disease. J. Am. Coll. Cardiol. 67, 3021–3023 (2016).
Haas, J. et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 36, 1123–1135 (2015).
Bennett, R. L. The Practical Guide to the Genetic Family History 2nd edn (Wiley, 2010).
Arbustini, E. et al. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: endorsed by the world heart federation. Glob. Heart 8, 355–382 (2013).
Mcnally, E. M., Golbus, J. R. & Puckelwartz, M. J. Genetic mutations and mechanisms in dilated cardiomyopathy. J. Clin. Invest. 123, 19–26 (2013).
Baig, M. K. et al. Familial dilated cardiomyopathy: cardiac abnormalities are common in asymptomatic relative and may represent early disease. J. Am. Coll. Cardiol. 31, 195–201 (1998).
Yancy, C. W. et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association task force on practice guidelines. J. Am. Coll. Cardiol. 62, e147–e239 (2013).
Charron, P. et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 31, 2715–2728 (2010).
Bozkurt, B. et al. Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 134, e579–e646 (2016).
Ackerman, M. J. et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 8, 1308–1339 (2011).
Towbin, J. A. et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm https://doi.org/10.1016/j.hrthm.2019.05.007 (2019).
Sylvius, N. et al. In vivo and in vitro examination of the functional significances of novel lamin gene mutations in heart failure patients. J. Med. Genet. 42, 639–647 (2005).
Fatkin, D. et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 341, 1715–1724 (1999).
van Berlo, J. H. et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J. Mol. Med. 83, 79–83 (2004).
Parks, S. B. et al. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am. Heart J. 156, 161–169 (2008).
van Tintelen, J. P. et al. High yield of LMNA mutations in patients with dilated cardiomyopathy and/or conduction disease referred to cardiogenetics outpatient clinics. Am. Heart J. 154, 1130–1139 (2007).
Morita, H., Seidman, J. G. & Seidman, C. E. Genetic causes of human heart failure. J. Clin. Invest. 115, 518–526 (2005).
Vikhorev, P. G. et al. Abnormal contractility in human heart myofibrils from patients with dilated cardiomyopathy due to mutations in TTN and contractile protein genes. Sci. Rep. 7, 14829 (2017).
Gramlich, M. et al. Stress-induced dilated cardiomyopathy in a knock-in mouse model mimicking human titin-based disease. J. Mol. Cell. Cardiol. 47, 352–358 (2009).
Gerull, B. et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 30, 201–204 (2002).
Herman, D. S. et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 366, 619–628 (2012).
Tayal, U. et al. Phenotype and clinical outcomes of titin cardiomyopathy. J. Am. Coll. Cardiol. 70, 2264–2274 (2017).
Jansweijer, J. A. et al. Truncating titin mutations are associated with a mild and treatable form of dilated cardiomyopathy. Eur. J. Heart Fail. 19, 512–521 (2017).
Veselka, J., Anavekar, N. S. & Charron, P. Hypertrophic obstructive cardiomyopathy. Lancet 389, 1253–1267 (2017).
Mogensen, J. et al. Frequency and clinical expression of cardiac troponin I mutations in 748 consecutive families with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 44, 2315–2325 (2004).
Moolman, J. C. et al. Sudden death due to troponin T mutations. J. Am. Coll. Cardiol. 29, 549–555 (1997).
Watkins, H. et al. Mutations in the genes for cardiac troponin t and α-tropomyosin in hypertrophic cardiomyopathy. N. Engl. J. Med. 332, 1058–1064 (1995).
Fujino, N. et al. A novel mutation Lys273Glu in the cardiac troponin T gene shows high degree of penetrance and transition from hypertrophic to dilated cardiomyopathy. Am. J. Cardiol. 89, 29–33 (2002).
Kokado, H. et al. Clinical features of hypertrophic cardiomyopathy caused by a Lys183 deletion mutation in the cardiac troponin I gene. Circulation 102, 663–669 (2000).
Regitz-Zagrosek, V., Erdmann, J., Wellnhofer, E., Raible, J. & Fleck, E. Novel mutation in the α-tropomyosin gene and transition from hypertrophic to hypocontractile dilated cardiomyopathy. Circulation 102, e112–e116 (2000).
Kamisago, M. et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 343, 1688–1696 (2000).
Daehmlow, S. et al. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 298, 116–120 (2002).
Bos, J. M. & Ackerman, M. J. Z-disc genes in hypertrophic cardiomyopathy: stretching the cardiomyopathies? J. Am. Coll. Cardiol. 55, 1136–1138 (2010).
Knöll, R. et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 111, 943–955 (2002).
Olson, T. M. et al. Metavinculin mutations alter actin interaction in dilated cardiomyopathy. Circulation 105, 431–437 (2002).
Villard, E. et al. Mutation screening in dilated cardiomyopathy: prominent role of the beta myosin heavy chain gene. Eur. Heart J. 26, 794–803 (2005).
Duboscq-Bidot, L. et al. Mutations in the Z-band protein myopalladin gene and idiopathic dilated cardiomyopathy. Cardiovasc. Res. 77, 118–125 (2008).
Dalakas, M. et al. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N. Engl. J. Med. 342, 770–780 (2000).
Li, D. et al. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation 100, 461–464 (1999).
Arbustini, E. et al. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur. J. Heart Fail. 8, 477–483 (2006).
Begay, R. L. et al. FLNC gene splice mutations cause dilated cardiomyopathy. JACC Basic Transl. Sci. 1, 344–359 (2016).
Ortiz-Genga, M. F. et al. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J. Am. Coll. Cardiol. 68, 2440–2451 (2016).
Begay, R. L. et al. Filamin C truncation mutations are associated with arrhythmogenic dilated cardiomyopathy and changes in the cell–cell adhesion structures. JACC Clin. Electrophysiol. 4, 504–514 (2018).
Selcen, D. et al. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann. Neurol. 65, 83–89 (2009).
Elliott, P. et al. Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ. Cardiovasc. Genet. 3, 314–322 (2010).
Garcia-Pavia, P. et al. Desmosomal protein gene mutations in patients with idiopathic dilated cardiomyopathy undergoing cardiac transplantation: a clinicopathological study. Heart 97, 1744–1752 (2011).
Ichida, F. et al. Novel gene mutations in patients with left ventricular noncompaction or barth syndrome. Circulation 103, 1256–1263 (2001).
Captur, G. & Nihoyannopoulos, P. Left ventricular non-compaction: genetic heterogeneity, diagnosis and clinical course. Int. J. Cardiol. 140, 145–153 (2010).
D’Adamo, P. et al. The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am. J. Hum. Genet. 61, 862–867 (1997).
Bione, S. et al. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 12, 385–389 (1996).
Brauch, K. M. et al. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 54, 930–941 (2009).
van den Hoogenhof, M. M. G. et al. RBM20 mutations induce an arrhythmogenic dilated cardiomyopathy related to disturbed calcium handling. Circulation 138, 1330–1342 (2018).
Olson, T. M. et al. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA 293, 447–454 (2005).
McNair, W. P. et al. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation 110, 2163–2167 (2004).
Brega, A., Narula, J. & Arbustini, E. Functional, structural, and genetic mitochondrial abnormalities in myocardial diseases. J. Nucl. Cardiol. 8, 89–97 (2001).
Zaragoza, M. V., Brandon, M. C., Diegoli, M., Arbustini, E. & Wallace, D. C. Mitochondrial cardiomyopathies: how to identify candidate pathogenic mutations by mitochondrial DNA sequencing, MITOMASTER and phylogeny. Eur. J. Hum. Genet. 19, 200–207 (2011).
Anan, R. et al. Cardiac involvement in mitochondrial diseases: a study on 17 patients with documented mitochondrial DNA defects. Circulation 91, 955–961 (1995).
Nair, V., Belanger, E. C. & Veinot, J. P. Lysosomal storage disorders affecting the heart: a review. Cardiovasc. Pathol. 39, 12–24 (2019).
Kamdar, F. & Garry, D. J. Dystrophin-deficient cardiomyopathy. J. Am. Coll. Cardiol. 67, 2533–2546 (2016).
Towbin, J. A. et al. X-linked dilated cardiomyopathy - molecular-genetic evidence of linkage to the Duchenne muscular-dystrophy (dystrophin) gene at the Xp21 locus. Circulation 87, 1854–1865 (1993).
Mounkes, L. C., Burke, B. & Stewart, C. L. The A-type lamins: nuclear structural proteins as a focus for muscular dystrophy and cardiovascular diseases. Trends Cardiovasc. Med. 11, 280–285 (2001).
Bonne, G. et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 21, 285–288 (1999).
Nigro, V. & Savarese, M. Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myol. 33, 1–12 (2014).
Mestroni, L. et al. Guidelines for the study of familial dilated cardiomyopathies. Eur. Heart J. 20, 93–102 (1999).
Xue, Y., Ankala, A., Wilcox, W. R. & Hegde, M. R. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. Genet. Med. 17, 444–451 (2015).
Yang, Y. et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 369, 1502–1511 (2013).
Bagnall, R. D. et al. Whole genome sequencing improves outcomes of genetic testing in patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 72, 419–429 (2018).
Biesecker, L. G. & Green, R. C. Diagnostic clinical genome and exome sequencing. N. Engl. J. Med. 370, 2418–2425 (2014).
Mohammed, S. et al. Genetic insurance discrimination in sudden arrhythmia death syndromes empirical evidence from a cross-sectional survey in North America. Circ. Cardiovasc. Genet. 10, e001442 (2017).
Joly, Y., Braker, M. & Le Huynh, M. Genetic discrimination in private insurance: global perspectives. New Genet. Soc. 29, 351–368 (2010).
Bland, A. et al. Clinically impactful differences in variant interpretation between clinicians and testing laboratories: a single-center experience. Genet. Med. 20, 369–373 (2018).
Amendola, L. M. et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the clinical sequencing exploratory research consortium. Am. J. Hum. Genet. 99, 247 (2016).
Harrison, S. M. et al. Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genet. Med. 19, 1096–1104 (2017).
Kelly, M. A. et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet. Med. 20, 351–359 (2018).
Hoffman-Andrews, L. The known unknown: the challenges of genetic variants of uncertain significance in clinical practice. J. Law Biosci. 4, 648–657 (2017).
Lv, W. et al. Functional annotation of TNNT2 variants of uncertain significance with genome-edited cardiomyocytes. Circulation 138, 2852–2854 (2018).
Stark, K. et al. Genetic association study identifies HSPB7 as a risk gene for idiopathic dilated cardiomyopathy. PLOS Genet. 6, e1001167 (2010).
Villard, E. et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur. Heart J. 32, 1065–1076 (2011).
Cappola, T. P. et al. Common variants in HSPB7 and FRMD4B associated with advanced heart failure. Circ. Cardiovasc. Genet. 3, 147–154 (2010).
Meder, B. et al. A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. Eur. Heart J. 35, 1069–1077 (2014).
Cappola, T. P. et al. Loss-of-function DNA sequence variant in the CLCNKA chloride channel implicates the cardio-renal axis in interindividual heart failure risk variation. Proc. Natl Acad. Sci. USA 108, 2456–2461 (2011).
Franaszczyk, M. et al. The BAG3 gene variants in Polish patients with dilated cardiomyopathy: four novel mutations and a genotype-phenotype correlation. J. Transl Med. 12, 192 (2014).
Parsa, A. et al. Hypertrophy-associated polymorphisms ascertained in a founder cohort applied to heart failure risk and mortality. Clin. Transl Sci. 4, 17–23 (2011).
Fox, E. R. et al. Genome-wide association study of cardiac structure and systolic function in African Americans: the Candidate Gene Association Resource (CARe) study. Circ. Cardiovasc. Genet. 6, 37–46 (2013).
Smith, N. L. et al. Association of genome-wide variation with the risk of incident heart failure in adults of European and African ancestry: a prospective meta-analysis from the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium. Circ. Cardiovasc. Genet. 3, 256–266 (2010).
Meune, C. et al. Primary prevention of sudden death in patients with lamin A/C gene mutations. N. Engl. J. Med. 354, 209–210 (2006).
Bécane, H. M. et al. High incidence of sudden death with conduction system and myocardial disease due to lamins A and C gene mutation. Pacing Clin. Electrophysiol. 23, 1661–1666 (2000).
Smith, J. G. et al. Discovery of genetic variation on chromosome 5q22 associated with mortality in heart failure. PLOS Genet. 12, e1006034 (2016).
Morrison, A. C. et al. Genomic variation associated with mortality among adults of European and African ancestry with heart failure: the Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium. Circ. Cardiovasc. Genet. 3, 248–255 (2010).
Myers, V. D. et al. Association of variants in BAG3 with cardiomyopathy outcomes in African American individuals. JAMA Cardiol. 3, 929–938 (2018).
Liggett, S. B. et al. A polymorphism within a conserved β1-adrenergic receptor motif alters cardiac function and β-blocker response in human heart failure. Proc. Natl Acad. Sci. USA 103, 11288–11293 (2006).
Bruck, H. et al. The Arg389Gly β1-adrenoceptor polymorphism and catecholamine effects on plasma-renin activity. J. Am. Coll. Cardiol. 46, 2111–2115 (2005).
Terra, S. G. et al. β1-Adrenergic receptor polymorphisms and left ventricular remodeling changes in response to β-blocker therapy. Pharmacogenet. Genomics 15, 227–234 (2005).
White, H. et al. An evaluation of the β1 adrenergic receptor Arg389Gly polymorphism in individuals at risk of coronary events: a WOSCOPS substudy. Eur. Heart J. 23, 1087–1092 (2002).
White, H. L. et al. An evaluation of the β1 adrenergic receptor Arg389Gly polymorphism in individuals with heart failure: a MERIT-HF sub-study. Eur. J. Heart Fail. 5, 463–468 (2003).
Andersson, B., Blange, I., Sylven, C. & Sylvén, C. Angiotensin-II type 1 receptor gene polymorphism and long-term survival in patients with idiopathic congestive heart failure. Eur. J. Heart Fail. 1, 363–369 (1999).
Huang, W., Xie, C., Zhou, H., Yang, T. & Sun, M. Association of the angiotensin-converting enzyme gene polymorphism with chronic heart failure in Chinese Han patients. Eur. J. Heart Fail. 6, 23–27 (2004).
McNamara, D. M. et al. Pharmacogenetic interactions between angiotensin-converting enzyme inhibitor therapy and the angiotensin-converting enzyme deletion polymorphism in patients with congestive heart failure. J. Am. Coll. Cardiol. 44, 1644–1649 (2004).
Nelveg-Kristensen, K. E. et al. Pharmacogenetic risk stratification in angiotensin-converting enzyme inhibitor-treated patients with congestive heart failure: a retrospective cohort study. PLOS ONE 10, 1–16 (2015).
Pare, G. et al. Genetic variants associated with angiotensin-converting enzyme inhibitor-associated angioedema. Pharmacogenet. Genomics 23, 470–478 (2013).
Pereira, N. L. & Weinshilboum, R. M. Cardiovascular pharmacogenomics and individualized drug therapy. Nat. Rev. Cardiol. 6, 632–638 (2009).
Merlo, M. et al. Evolving concepts in dilated cardiomyopathy. Eur. J. Heart Fail. 20, 228–239 (2017).
Van Spaendonck-Zwarts, K. Y. et al. Peripartum cardiomyopathy as a part of familial dilated cardiomyopathy. Circulation 121, 2169–2175 (2010).
Blauwet, L. A. & Cooper, L. T. Diagnosis and management of peripartum cardiomyopathy. Heart 97, 1970–1981 (2011).
Scott, E., Hasbullah, J. S., Ross, C. J. & Carleton, B. C. Reducing anthracycline-induced cardiotoxicity through pharmacogenetics. Pharmacogenomics 19, 1147–1150 (2018).
Hazebroek, M. R. et al. Prognostic relevance of gene-environment interactions in patients with dilated cardiomyopathy applying the MOGE(S) classification. J. Am. Coll. Cardiol. 66, 1313–1323 (2015).
Pepin, M. E. et al. Genome-wide DNA methylation encodes cardiac transcriptional reprogramming in human ischemic heart failure. Lab. Invest. 99, 371–386 (2018).
Zierhut, H. A., MacFarlane, I. M., Ahmed, Z. & Davies, J. Genetic counselors’ experiences and interest in telegenetics and remote counseling. J. Genet. Couns. 27, 329–338 (2018).
Du, L. & Becher, S. I. Genetic and genomic consultation: are we ready for direct-to-consumer telegenetics? Front. Genet. 9, 550 (2018).
Author information
Authors and Affiliations
Contributions
All the authors researched data for the article, wrote the manuscript and reviewed and edited it before submission. A.N.R. and N.L.P. contributed to discussions about the article content.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Cardiology thanks L. Mestroni, A. Morales, G. Sinagra and M. van den Berg for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rosenbaum, A.N., Agre, K.E. & Pereira, N.L. Genetics of dilated cardiomyopathy: practical implications for heart failure management. Nat Rev Cardiol 17, 286–297 (2020). https://doi.org/10.1038/s41569-019-0284-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41569-019-0284-0
