
Abstract
Cardiometabolic traits pose a major global public health burden. Large-scale genome-wide association studies (GWAS) have identified multiple loci accounting for up to 30% of the genetic variance in complex traits such as cardiometabolic traits. However, the contribution of parent-of-origin effects (POEs) to complex traits has been largely ignored in GWAS. Family-based studies enable the assessment of POEs in genetic association analyses. We investigated POEs on a range of complex traits in 3 family-based studies. The discovery phase was carried out in large pedigrees from the Kibbutzim Family Study (n = 901 individuals) and in 872 parent–offspring trios from the Jerusalem Perinatal Study. Focusing on imprinted genomic regions, we examined parent-specific associations with 12 complex traits (i.e., body-size, blood pressure, lipids), mostly cardiometabolic risk traits. Forty five of the 11,967 SNPs initially found to have POE were evaluated for replication (p value < 1 × 10−4) in Framingham Heart Study families (max n = 8000 individuals). Three common variants yielded evidence of POE in the meta-analysis. Two variants, located on chr6 in the HLA region, showed a paternal effect on height (rs1042136: βpaternal = −0.023, p value = 1.5 × 10−8 and rs1431403: βpaternal = −0.011, p value = 5.4 × 10−6). The corresponding maternally-derived effects were statistically nonsignificant. The variant rs9332053, located on chr13 in RCBTB2 gene, demonstrated a maternal effect on hip circumference (βmaternal = −4.24, p value = 9.6 × 10−6; βpaternal = 1.29, p value = 0.23). These findings provide evidence for the utility of incorporating POEs into association studies of cardiometabolic traits, especially anthropometric traits. The study highlights the benefits of using family-based data for deciphering the genetic architecture of complex traits.
Similar content being viewed by others


Introduction
Cardiometabolic disease pose a major global public health burden [1]. Cardiometabolic risk traits consist of measures such as lipid and lipoproteins, glucose and insulin resistance, adiposity, blood pressure, and inflammation markers. These quantitative cardiometabolic traits are strong predictors of overall risk for coronary heart disease, diabetes, and related disorders [2]. Prior studies based on twins and families have shown substantial heritability for cardiometabolic quantitative traits (30–80%) [3]. Over the past decade thousands of genome-wide association studies (GWAS) have identified numerous loci that account for the interindividual variation in these cardiometabolic traits. The most recent GWAS meta-analyses include >100,000 individuals and have begun to explain a larger proportion of complex traits heritability (up to 30%) [4]. Tens of thousands of trait-SNP associations were discovered, consistent with heritability models that suggest a polygenic or omnigenic model of inheritance [5]. However, the vast majority of these studies are based on unrelated individuals, which do not take into consideration parent-of-origin genetic effects (POEs).
POE analysis using family-based data, can further add to explaining heritability by uncovering genetic effects controlled by more complex genetic mechanisms [6]. POE refers to a class of genetic effects that are transmitted from parents to offspring whereby the expression of the phenotype in the offspring depends upon whether the transmission originated from the mother or father [7]. The main mechanism underlying POEs is genomic imprinting in which imprinted genes show parent-specific expression regulated by epigenetic mechanisms such as DNA methylation [8]. Around 80% of imprinted genes are found in clusters, regulated by imprint control regions whose epigenetic state controls expression of all genes in the cluster [9]. More than 150 imprinted genes have been described in humans, listed in geneimprint and the Otago imprinting databases, but there are likely many more imprinted genes that have yet to be validated [10]. Many of the imprinted genes are critical for normal fetal growth and neurodevelopment, metabolism, and adult behavior [11]. The haploid expression state of imprinted genes makes them more vulnerable to being either inactivated or over-expressed, thus causing a range of developmental abnormalities such as fetal growth restriction, overgrowth (macrosomia), or cancer [12].
The earliest and strongest evidence for imprinting effects on cardiometabolic traits comes from syndromic causes of obesity and diabetes, e.g., Prader-Willi, Silver-Russell and Temple syndromes that involve disruption to known imprinted regions [13, 14]. Recent studies have shown POEs on cardiometabolic traits in known imprinted genes [15, 16] and in candidate genes [17,18,19,20]. For example Kong et al. demonstrated POE on type 2 diabetes (T2D) and cancer in Icelandic families showing strengthening of effects for previously reported associations and revealing novel associations with T2D [20]. Two other studies in Icelandic families, detected POEs on age at menarche and thyroid-stimulating hormone levels, traits that are associated with cardiometabolic disease risk [21, 22]. As a proof of concept, our group has demonstrated on a limited dataset of mothers–offsrping pairs and a limited set of variants, a maternal-specific effect on adiposity of a common variant in APOB, a gene previously known to be associated with dyslipidemia [23]. The effect was replicated in additional cohorts from other ancestries [23]. These results reveal that the observed effects from GWAS do not necessarily reflect only the direct effects of the variants. Thus, incorporating POEs within an association analysis can further reveal more details parsing the genetic effects and deepen the understanding of the genetic architecture of these traits [6].
A recent study explored genome-wide POE of 21 quantitative phenotypes in a large Hutterite pedigree [24]. The authors identified POEs with 11 phenotypes, most of which are risk factors for cardiovascular disease. Other than this report and despite accumulating evidence of POEs on cardiometabolic traits, we are unaware of other studies assessing the contribution of POEs to a range of cardiometabolic traits, using a family-based approach and genome-wide data.
To comprehensively examine POEs on a wide range of cardiometabolic traits we leveraged genome-wide array data in two unique Israeli family studies; the Kibbutzim Family Study (KFS) [25] comprising large families and the Jerusalem prenatal study (JPS) [26] comprising young adults and their parents. Focusing on imprinted genomic intervals spanning more than 50 M base pairs, we examined the associations of maternally-and paternally-derived minor alleles with 12 cardiometabolic traits. We hypothesized that incorporating the parental source of genetic variants into the analyses of putatively imprinted regions would reveal associations with cardiometabolic traits that might have been missed or underestimated in standard genetic association analyses of nonfamilial studies. Replication of significant findings was evaluated in extended families from the Framingham Heart Study (FHS).
Methods
Datasets
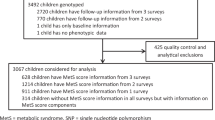
The KFS consists of 1033 participants from 150 extended families of Jewish ancestry, ranging from 2 to 55 individuals recruited during two visits in the 1990s from six Kibbutzim in Northern Israel, the majority (85%) from Ashkenazi origin [25]. Genome-wide genetic data were available for 901 of the participants.
The JPS consists of young adults (mean age 32 years at time of measurement) born in the 1970s in Jerusalem and their parents. Approximately 50% are of Ashkenazi Jewish ancestry, and the other half are of Middle-Eastern and North African Jewish ancestries [26]. It is a sample stratified by maternal pre-pregnancy body mass index (BMI) and birth weight, with oversampling of over-weight and obese mothers and both low- and -high-birth weight offspring [26]. Genome-wide genetic data were available for 872 offsprings and at least one of their parents.
Replication was conducted in an additional family study, the FHS. The FHS enrolled the Original cohort in 1948, the Offspring cohort (offspring of the Original cohort and the offsprings’ spouses) in 1971 and the Third Generation cohort (grandchildren of the Original cohort and children of the Offspring cohort) in 2002. Individuals were restricted to European ancestry in this analysis. Genome-wide genetic data were available for ~8000 of the participants.
KFS and JPS were both approved by the Institutional Review Board of the Hadassah-Hebrew University Medical Center and the National Genetic IRB committee. JPS was also approved by the University of Washington Human Subject Review Committee. The FHS was approved by the Boston University Medical Campus IRB. In all the studies, a signed informed consent was obtained from all participants.
Data collection and cardio-metabolic outcomes
In all studies, all participants completed a socio-demographic and health questionnaire and underwent a physical examination [26,27,28,29,30]. Peripheral blood samples (25 ml) were collected for biochemical measurements and DNA extraction.
The following cardiometabolic traits were measured in all studies: Height (m), Weight (kg), body mass index (BMI, calculated by dividing weight (kg) by squared height (m2)), waist circumference (cm), hip circumference (cm), waist to hip ratio (WHR), low-density lipoprotein cholesterol (LDL-C (mg/dL)), high-density lipoprotein cholesterol (HDL-C (mmol/L)), triglycerides (TG (mg/dL, (inverse normal transformed)), systolic and diastolic blood pressure ((SBP and DBP, mmHg) mean of three consecutive measures in JPS and KFS, and mean of two measurements 10 min apart in FHS), and heart rate (beats per 30 s). All traits were treated as continuous variables.
Genotyping and quality control
Genotyping in KFS was performed using Illumina HumanCoreExome BeadChip array comprising of 525,794 variants. Genotyping in JPS was performed using Affymetrix Biobank array comprising of 587,021 variants. We used the following criteria in both datasets for exclusion of genetic variants: Missingness rate > 0.1, Mendelian error > 100, and zero minor allele frequency (MAF) (monomorphic variant). The following criteria were used to exclude individual samples in both datasets: missingness rate > 0.05, unresolved gender mismatch, and Mendelian error rate > 0.05. We confirmed the reported familial relationships in KFS and JPS using IBD analysis. Variants and individuals were filtered using Plink [31]. In KFS, a total of 323,708 variants and 901 individuals passed QC and were used in POE downstream analyses. In JPS corresponding numbers were 398,491 variants and 872 individuals (658 complete trios, 192 parent–offspring duos and 22 offspring only). Imputation was done separately for each cohort using a combined reference panel of the 1000 genomes and The Ashkenazi Genome Consortium (n = 128) [25, 32]. We considered imputed variants with MAF ≥ 1% and a strict imputation quality score ≥0.9 (hard calls).
Genotyping in the FHS was previously described in Tsao et al. [33]. Genotypes from Affymetrix 500K SNP arrays supplemented with the MIPS 50K array were available. A subset of 378,163 SNPs passing stringent filters including a MAF ≥ 0.01, SNP call rate ≥ 0.97, HWE p value ≥ 1e−6, differential missingness (a test to determine if missingness depends on genotypes based on flanking inferred haplotypes) p value ≥ 1e-9 and <100 Mendelian errors were used for imputation (hard calls) based on the haplotypes of the Haplotype Reference Consortium using the MACH software [34].
KFS and JPS data reported in this paper can be found in the European Genome-phenome Archive (EGA), https://ega-archive.org/, under accession numbers EGAS00001002782 and EGAS00001004075, respectively. FHS data can be found in dbGaP, https://www.ncbi.nlm.nih.gov/gap/ under Study Accession: phs000007.v30.p11.
Genetic variants selection
In the reported analyses we focused on a set of 50 putatively human imprinted regions, spanning ~54 M bases of the genome (Supplementary Table 1). We hypothesized that if variants in these regions are associated with cardiometabolic traits, then these variants are more likely to be associated with the traits in a parent-specific manner that may be undetected when parental source of alleles is not taken into account. The regions were initially defined based mainly on the geneimprint database, the Catalog of Parent of Origin Effects and several additional papers on POE [35,36,37,38]. We further extended each transcribed region to include 500 kb upstream and downstream, to allow the inclusion of regulatory regions and genes with undetermined imprinting status [20] (Supplementary Table 1). We identified 6305 and 7287 genotyped variants (MAF > 0.05) in these regions in KFS and JPS, respectively, of which 1527 are overlapping. We used imputed data to complete the information when a variant was present in one study and absent from the other study. Overall, we analyzed 11,967 variants in these regions and examined the associations of maternally-and paternally-derived minor alleles with 12 cardiometabolic traits.
Parent of origin association analysis
KFS and FHS
We used the quantitative transmission disequilibrium test (QTDT) software [39] to test the genotype effect and the associations of maternally- and paternally-derived minor alleles using information from the extended families, adjusting for proband sex, age, study-specific covariate (i.e., visit in KFS and cohort in FHS), and first ten genotypic principal components (PCs). Principal component analysis was performed using PC-AiR [40], which is robust to known or cryptic relatedness as described elsewhere [25]. Specifically, the command “qtdt -at –ot” was used to test for POE (maternal effect = paternal effect), and “qtdt -at –om” and “qtdt -at –op” were used to test maternal and paternal effects, respectively (modified to adjust for the other parent’s contribution). QTDT software assigns parental origins to the alleles and uses a linear mixed effect model to account for familial correlations. Lipid-lowering medication was accounted for by introducing into the model a dichotomous covariate for medication use and blood pressure lowering medication was adjusted for by adding 10 and 5 mmHg to systolic (SBP) and diastolic (DBP) blood pressures, respectively [41]. Because the QTDT approach does not provide estimates of standard errors (SEs), SEs were estimated by converting p values into a z-statistic and setting: SE = beta/z.
JPS
Analyses in JPS were carried out using an approach similar to the one we previously applied using mother–offspring pairs [23]. To assess the separate contribution of the maternally-and paternally-inherited variants to offspring cardiometabolic phenotypes, we used genotype data based on parent–offspring triads to determine the parental origin of the offspring minor alleles. For every given variant we constructed two indicators: maternally- (M-D) and paternally-derived (P-D) minor allele indicators. These indicators count the number of minor alleles inherited from mother and/or father; assigning values of either 0 or 1 (Supplementary Table 2). When offspring and parents were heterozygous at a given SNP the source of the minor allele was ambiguous, and both indicators were designated as 0.5. We used linear regression models to examine each SNP-trait association. We first assessed the associations of offspring genotype with trait, using an additive genetic model. Then the parental-specific association with the trait was assessed by including the M-D and P-D indicators together in a single model, as described [23]. The following mean models were used:
where Y denotes trait, G0 = offspring genotype, GMD = indicator of maternally-derived minor allele, GPD = indicator of paternally-derived allele, and Z = other covariates. Existence of POE in model 2 was assessed via a test of the following null hypotheses: (1) βMD = 0; (2) βPD = 0; (3) βMD = βPD. P values for difference between maternally- and paternally-derived effects were calculated using an F-test. All models were adjusted for offspring sex and first 10 genotypic PCs calculated by PC-AiR [40], similarly to the KFS PCs calculations [25]. Offspring who reported using lipid-lowering medication (n = 13) or blood pressure-lowering medication (n = 11) were excluded from the analyses. All analyses were carried out using Stata 12.0 (StataCorp, College Station TX). To account for the stratified sampling (aforementioned), we used Stata’s ‘pweight’ option to weight estimates by individuals’ inverse probability of being sampled [23].
Meta-analysis and replication
We meta-analyzed the results from the two studies, KFS and JPS, and combined them together to form the discovery stage. We conducted a z-based meta-analysis for the p value from the F-test in JPS and the test for POE (-ot option) in KFS to determine the combined significance of the difference between maternally- and paternally-derived effects. We also conducted a fixed-effect meta-analysis of regression estimates and SEs for both studies (JPS and KFS), using an inverse-variance weighting approach as implemented in META software which includes verification of strand alignment across studies. Associations that meet the following two criteria for any one of the examined outcomes were moved forward for replication in FHS extended families 1) meta-analysis p value for the difference between maternally- and paternally-derived effects ≤0.05; and 2) combined p value for the maternally- or paternally-derived effect was ≤0.0001 (a liberal threshold was selected to decrease false negatives in the discovery stage, as is often done in GWAS [42]).
Meta-analysis of FHS results with results from the discovery stage (meta-analyzed KFS and JPS estimates) was conducted. We conducted LD (linkage disequilibrium) pruning in the JPS dataset, using the --indep-pairwise command in Plink (window size 50 kb, a shift of 10 variants at each step, and LD between variants r2 > 0.8) to determine the number of variants and the effective number of tests (8403). Our criteria for significance was 1) meta-analysis p value for the difference between maternally- and paternally-derived effects ≤0.05; and 2) meta-analysis p value for the maternally- or the paternally-derived effect using a stringent Bonferroni correction 0.05/8403, leading to a p value threshold of ≤5.95 × 10−6 [43].
Bioinformatics
Using publically available databases and tools, we performed bioinformatics analyses for top-hits in the POE analyses. Variant annotations was done by combining information from GWAVA, RegulomeDB, UCSC, HaploReg 4.1, and GTEx Portal. Associations of variants and genes with traits and diseases were explored using the GWAS catalog, Phegen, and DisGenet. Supporting evidence for imprinting and monoallelic expression (MAE) of genes was assessed using two databases: geneimprint and Catalog of Parent-of-Origins Effects, a recent paper on MAE [35], and differentially methylated regions (DMRs) using the Epigenome Browser.
Results
Distributions of age, sex, and cardio-metabolic traits among KFS, JPS, and FHS are summarized in Supplementary Table 3, and are comparable between the studies. Forty-five of the 11,967 SNPs initially tested for POE in the discovery study were moved forward for replication in FHS. POE effects of all 45 replicated variants on all 12 cardiometabolic traits are summarized in Supplementary Table 4, and their corresponding genotype effects are summarized in Supplementary Table 5 (all variants were in HWE).
The joint analysis of findings from the discovery stage (KFS and JPS) and FHS, revealed two variants showing significant POEs on height and another variant demonstrating a borderline significant POE on hip circumference (Table 1).
The two variants associated with height, rs1042136 and rs1431403, are located in chr6p21.32 in the HLA region. Linkage disequilibrium (R2) between these two SNPs is 0.617 in JPS and 0.47 in Europeans. In the discovery stage conducted in KFS and JPS, rs1042136 demonstrated significant associations with offspring height when the minor allele was inherited from the father (p value = 1.1 × 10−7) (Table 1). The corresponding maternally-derived effects were nonsignificant (p value > 0.78), and the genotype effect was nominally significant (p value = 1.0 × 10−2) (Table 1). As expected in the setting of POEs, the effect of the paternally-derived minor allele was stronger than the genotype effect (βpaternal = −0.038 vs. βgenotype effect = −0.018) (Table 1). Testing for POEs in the replication stage, also showed a significant paternally-derived effect on height in FHS, and in all three studies combined (βpaternal = −0.023, p value = 1.5 × 10−8) (Table 1). Effect sizes for the paternal-specific associations in KFS, JPS, and FHS were comparable and negative. These results indicate that mean height of offspring who inherit the paternal minor allele are ~2 cm lower compared with those who did not inherit the paternal minor allele. P values for genotype effect of this variant were only nominally significant for the combined analyses (p value = 1.8 × 10−2) (Table 1), demonstrating the benefit of including the POE component to the association analysis. The other closely-located significant SNP, rs1431403, presented a similar, yet slightly weaker, paternal effect (discovery stage βpaternal = −0.03, p value = 4.7 × 10−6, combined all studies βpaternal = −0.011, p value = 5.1 × 10−6).
The variant demonstrating suggestive evidence of POE on hip circumference, rs9332053, is located in chr13q14.2 in the RCBTB2 gene. In the discovery stage, rs9332053 demonstrated significant POEs on hip circumference (Table 1). Specifically, statistically significant associations were demonstrated when the minor allele was inherited from the mother (βmaternal = −4.793, p value = 3.2 × 10−5). The corresponding paternally-derived effects were statistically nonsignificant (p value > 0.92). The maternally-derived minor allele had a much stronger effect than the genotype effect (βmaternal = −4.793 vs. βgenotype effect = 0.14) (Table 1). Testing for POEs in the replication stage, also showed a significant maternally-derived effect in FHS on hip circumference, and in all three studies combined (βmaternal = −4.241, p value = 9.6 × 10−6) (Table 1). The effect sizes for the maternal-specific associations in KFS, JPS, and FHS were all comparable and negative. These results indicate that an offspring who inherits the maternal minor allele, on average will have a slimmer hip circumference (by ~4 cm) compared to an offspring who did not inherit a maternal minor allele. P values for genotype effect of this variant were not statistically significant (Table 1), meaning the POE analysis revealed a novel association.
We further examined whether there is evidence for POEs for these three top-hits on other cardiometabolic traits (Supplementary Tables 6–8). For the two variants showing a paternally-derived effect on height, we did not find similar effects on any other cardiometabolic trait (Supplementary Tables 6–7). However, the variant showing a maternally-derived effect on hip circumference also demonstrated nominaly significant effects on BMI (βmaternal = –1.054, p value = 2.3 × 10–2), height (βmaternal = –0.023, p value = 1.0 × 10–3), waist circumference (βmaternal = –4.883, p value = 5.1 × 10–5) and weight (βmaternal = –4.771, p value = 5.0 × 10–4) (Supplementary Tables 8). However, p value for difference between maternal and paternal effects were nominally significant only for waist circumference (p value = 1.8 × 10–2). The corresponding paternally-derived effects and genotype effects were non-significant.
A complete summary of the bioinformatics analyses for POE top-hits is presented in Supplementary Table 9. Bioinformatics annotation based on the GTEx portal revealed that the two variants associated with height are located in genes (HLA-DPA1, HLA-DPB1) expressed mostly in the lung, spleen, tibial nerve, whole blood, and small intestine (Supplementary Table 6). DNA methylation data in the Epigenome Browser [44] was available for three of these tissues; small intestine, lung, and spleen. They all showed a ~50% methylation pattern for the highest top-hit associated with height, rs1042136 (Fig. 1).

Bioinformatics annotation for rs1042136 located within the second exon of the HLA-DPB1 gene, and the promoter region of the HLA-DPA1 gene, using the Epigenome Browser (http://epigenomegateway.wustl.edu/). The genes are shown in the blue track and the focal SNP with a yellow bar. Levels of Differentially Methylated Regions (DMRs) were plotted in three relevant tissues: small Intestine, lung and spleen. Methylation ranging from 0 to 1.
Discussion
POE has the potential to add a new level of information to the genetic architecture of common traits, contributing to certain extent to the broad research efforts aimed at uncovering a larger portion of the missing heritability of these traits. We have identified three novel parent-of-origin association effects by incorporating POE to the association analysis of 12 cardiometabolic traits; two adjacent variants in the HLA region on chr6p21.32 showing a paternal effect on height, and one variant on chr13q14.2 showing a maternal effect on hip circumference. Corresponding genotype effects, not taking into account the parental source of the variant, showed borderline or nonsignificant results. These results were identified in two family based studies, the KFS and the JPS, composed mostly of Ashkenazi Jewish origin, and replicated in a third European family based study, the FHS.
The two top-hits showing paternal effect on height, rs1042136 and rs1431403 are located in an intron and an exon, respectively, 1,864 bp apart, the first in the HLA-DPB1 gene and the second in both HLA-DPA1 and HLA-DPB1 genes. These genes belong to the major histocompatibility complex (MHC) also referred to as the HLA complex. This is a gene complex residing on a 3 Mbp stretch within chromosome 6p21 which encodes cell-surface proteins that are responsible for the regulation of the immune system in humans. The HLA locus encompasses the strongest genetic risk variants for many autoimmune and inflammatory diseases [45], though it is not known to be associated with height or other body size measurements. However, a potential indirect association through autoimmune diseases was recently presented in a study showing a complex genetic relationship between growth and immune phenotypes, revealing the genetic background of their correlation in the context of pleiotropy [46]. For example, they observed statistically significant inverse effects of immune cell count phenotypes on human height, and a slight but significant negative influence of human height on allergic disease. This region is not a known imprinted region: however, several studies have investigated parental specific associations in the HLA locus. Maternal POEs have been demonstrated for HLA haplotypes in multiple sclerosis (MS) [47], however studies of POEs on susceptibility to other diseases (e.g., type 1 diabetes, systemic lupus erythematosus) at this locus are less conclusive, some studies suggesting POE while others reporting the absence of these effects [47].
Bioinformatics annotation showed a ~50% methylation pattern for our highest top-hit associated with height, rs1042136 (Fig. 1). It is known that imprinted genes and imprinting control regions show predominant intermediate methylation in adult somatic tissues [48, 49]. Although a ~50% methylation pattern is not necessarily a result of parental specific expression, it may still provide some support for the suggested POE association between this variant and height.
Two other studies have identified POE on height; a variant in the imprinted KCNQ1 gene reduces height when maternally inherited in a Sardinian population [50], and three variants within two imprinted regions (IGF2-H19 and DLK1-MEG3) reduce height when paternally inherited in an Icelandic population [51]. Replication attempts of some of these top-hit variants in the KFS and JPS datasets were not successful (data not shown). This could be due to the very different genetic background of the studies’ populations, as well as the distinct environmental factors characterizing each population or due to chance as our sample is of a modest size.
The third top-hit, rs9332053, showing maternal effect on hip circumference and other obesity traits, is located in an intron of the RCBTB2 gene. The RCBTB2 gene is located nearby the retinoblastoma 1 (RB1) gene, a tumor suppressor gene which was found to be preferentially expressed from the maternal allele [52]. This close-by known maternal expression supports our finding and it may indicate the imprinting in this region is more comprehensive than originally thought. This RCBTB2 gene encodes a protein that is related to regulation of chromosome condensation. This gene encompasses human quantitative trait loci (QTLs) for asthma and osteoporosis [53], and rat QTLs for cholesterol, renal function, bone structure and strength and blood pressure.
The most widely accepted hypothesis for the evolution of genomic imprinting is the “parental conflict hypothesis” [54]. It states that the parents have differing interests in terms of evolutionary fitness of their genes. Paternally expressed genes tend to be growth promoting assisting their chances of surviving, and maternally expressed genes tend to be growth limiting, so the mother can save resources for her other offsprings. Overall, the current findings showing parental effects on offspring body size (height and hip circumference), support the hypothesis claim for an association between parental effects and growth traits.
Because most genetic association studies, conducted to date, overlook POE due to lack of family data, limited association data in the literature is available to support the current findings. In addition, parent-of-origin molecular mechanisms may be apparent in specific tissues or stages of development. Therefore, bioinformatics data, such as expression and methylation, provide only limited resources for follow-up analyses aimed to provide support for parental-specific statistical associations. Adding to this challenge is suggestive evidence that non-imprinted genes can generate POEs by interacting with imprinted loci in so-called imprinted gene networks (IGN) [55, 56]. All these raise difficulties in the identification of genomic loci or genes that show POEs. Our findings provide some support for the potential importance of POEs on cardiometabolic complex traits, especially anthropometric traits, however further investigation is needed to determine these results.
The major strengths of our study are the combination of two unique family-based studies, mainly from European Ashkenazi Jewish origin, and the opportunity to replicate our findings in another large family study of European descent. Notably, the family design minimizes confounding due to population stratification. In addition, focusing on putatively known imprinted regions takes advantage of available scientific knowledge as well as reduces the problem of multiple comparisons. There are also several limitations to our study. First, the variants identified have not been previously reported in large GWAS consortia as related with cardiometabolic traits. Although this is likely the result of GWAS typically ignoring the parental source of alleles due to lack of family data, there is a possibility that our POE results, although replicated, reflect false positive findings. Allele specific methylation data of different human tissues at different developmental stages could potentially ratify our results. Second, we may have missed true findings (false negative) that are exclusive to Jewish or Ashkenazi populations yet could not be replicated in the FHS European population. Lastly, the moderate sample sizes and ethnic narrowness of discovery samples may also lead to missed POEs that could be found in diverse or larger datasets.
In summary, we have demonstrated that taking into account the parental origin of offspring alleles compared to examining the offspring genotype as a whole may enhance our understanding of genetic associations with consortia traits. Our results highlight the potential contribution of POEs to uncovering complex relations between genetic variants and common traits, and motivate further research in this area, including assessment of the impact of POEs on common traits and investigation of the mechanisms underlying these associations. Exploring parent of origin genetic effects of known cardiometabolic traits, can also have a great influence on predicting personal risk scores for individuals, consider personal prevention strategies and influence development of therapies based on novel biological insights.
Web resources
https://www.cog-genomics.org/plink2.
http://csg.sph.umich.edu/abecasis/mach/index.html.
http://www.geneimprint.com/site/genes-by-species.
http://csg.sph.umich.edu/abecasis/qtdt/.
https://rdrr.io/bioc/GENESIS/man/pcair.html.
https://mathgen.stats.ox.ac.uk/genetics_software/meta/meta.html.
https://www.sanger.ac.uk/sanger/StatGen_Gwava.
http://www.regulomedb.org/.https://genome.ucsc.edu/.
https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php.https://gtexportal.org/home/.
https://www.ncbi.nlm.nih.gov/gap/phegeni.
References
Roth GA, Johnson C, Abajobir A, Abd-Allah F, Abera SF, Abyu G, et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70:1–25.
Lacey B, Herrington WG, Preiss D, Lewington S, Armitage J. The role of emerging risk factors in cardiovascular outcomes. Curr Atheroscler Rep. 2017;19:1–10.
Whitfield JB. Genetic insights into cardiometabolic risk factors. Clin Biochem Rev. 2014;35:15–36.
Torkamani A, Wineinger NE, Topol EJ. The personal and clinical utility of polygenic risk scores. Nat Rev Genet. 2018;19:581–90.
Boyle EA, Li YI, Pritchard JK. An expanded view of complex traits: from polygenic to omnigenic. Cell. 2017;169:1177–86.
Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–50.
Rampersaud E, Mitchell BD, Naj AC, Pollin TI. Investigating parent of origin effects in studies of type 2 diabetes and obesity. Curr Diabetes Rev. 2008;4:329–39.
Koerner MV, Barlow DP. Genomic imprinting—an epigenetic gene-regulatory model. Curr Opin Genet Dev. 2010;20:164–70.
Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2:21–32.
Cuellar Partida G, Laurin C, Ring SM, Gaunt TR, Relton CL, Smith GD, et al. Imprinted loci may be more widespread in humans than previously appreciated and enable limited assignment of parental allelic transmissions in unrelated individuals Genome-wide survey of parent-of-origin effects on DNA methylation identifies candidate imp. https://www.biorxiv.org/content/10.1101/161471v1.abstract. 2017.
Smith FM, Garfield aS, Ward a. Regulation of growth and metabolism by imprinted genes. Cytogenet Genome Res. 2006;113:279–91.
Ishida M, Moore GE. The role of imprinted genes in humans. Mol Asp Med. 2013;34:826–40.
Geoffron S, Habib WA, Chantot-Bastaraud S, Dubern B, Steunou V, Azzi S, et al. Chromosome 14q32.2 imprinted region disruption as an alternative molecular diagnosis of Silver-Russell Syndrome. J Clin Endocrinol Metab. 2018;103:2436–46.
Yang A, Kim J, Cho Y, Jin D-K. Prevalence and risk factors for type 2 diabetes mellitus with Prader–Willi syndrome: a single center experience. Orphanet J rare Dis. 2017;12:1–9.
Weinstein LS, Xie T, Qasem A, Wang J, Chen M. The role of GNAS and other imprinted genes in the development of obesity. Int J Obes. 2010;34:6–17.
Horikoshi M, Beaumont RN, Day FR, Warrington NM, Kooijman MN, Fernandez-Tajes J, et al. Genome-wide associations for birth weight and correlations with adult disease. Nature. 2016;538:248–52.
Wermter A-K, Scherag A, Meyre D, Reichwald K, Durand E, Nguyen TT, et al. Preferential reciprocal transfer of paternal/maternal DLK1 alleles to obese children: first evidence of polar overdominance in humans. Eur J Hum Genet. 2008;16:1126–34.
Liu X, Hinney A, Scholz M, Scherag A, Tönjes A, Stumvoll M, et al. Indications for potential parent-of-origin effects within the FTO gene. PLoS ONE. 2015;10:1–12.
Li A, Robiou-du-Pont S, Anand SS, Morrison KM, McDonald SD, Atkinson SA, et al. Parental and child genetic contributions to obesity traits in early life based on 83 loci validated in adults: the FAMILY study. Pediatr Obes. 2018;13:133–40.
Kong A, Steinthorsdottir V, Masson G, Thorleifsson G, Sulem P, Besenbacher S, et al. Parental origin of sequence variants associated with complex diseases. Nature. 2009;462:868–74.
Gudbjartsson DF, Helgason H, Gudjonsson SA, Zink F, Oddson A, Gylfason A, et al. Large-scale whole-genome sequencing of the Icelandic population. Nat Genet. 2015;47:435–44.
Perry JRB, Day F, Elks CE, Sulem P, Thompson DJ, Ferreira T, et al. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nature. 2014;514:92–7.
Hochner H, Allard C, Granot-hershkovitz E, Chen J, Sitlani M, Sazdovska S, et al. Parent-of-origin effects of the APOB gene on adiposity in young adults. Plos Genet. 2015;10:1–23.
Mozaffari SV, DeCara JM, Shah SJ, Sidore C, Fiorillo E, Cucca F, et al. Parent-of-origin effects on quantitative phenotypes in a large Hutterite pedigree. Commun Biol. 2019;2:28.
Granot-Hershkovitz E, Karasik D, Friedlander Y, Rodriguez-Murillo L, Dorajoo R, Liu J, et al. A study of Kibbutzim in Israel reveals risk factors for cardiometabolic traits and subtle population structure. Eur J Hum Genet. 2018;26:1848–58.
Lawrence GM, Siscovick DS, Calderon-Margalit R, Enquobahrie DA, Granot-Hershkovitz E, Harlap S, et al. Cohort profile: the Jerusalem perinatal family follow-up study. Int J Epidemiol. 2015;45:343–52.
Lemaitre RN, Siscovick DS, Berry EM, Kark JD, Friedlander Y. Familial aggregation of red blood cell membrane fatty acid composition: the Kibbutzim Family Study. Metab. 2008;57:662–8.
Dawber TR, Meadors GF, Moore FE Jr. Epidemiological approaches to heart disease: the Framingham Study. Am J Public Health Nations Health. 1951;41:279–81.
Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families. The Framingham offspring study. Am J Epidemiol. 1979;110:281–90.
Splansky GL, Corey D, Yang Q, Atwood LD, Cupples LA, Benjamin EJ, et al. The third generation cohort of the national heart, lung, and blood institute’s framingham heart study: design, recruitment, and initial examination. Am J Epidemiol. 2007;165:1328–35.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7.
Carmi S, Hui KY, Kochav E, Liu X, Xue J, Grady F, et al. Sequencing an Ashkenazi reference panel supports population-targeted personal genomics and illuminates Jewish and European origins. Nat Commun. 2014;5:4835.
Tsao CW, Vasan RS. Cohort profile: the Framingham Heart Study (FHS): overview of milestones in cardiovascular epidemiology. Int J Epidemiol. 2015;44:1800–13.
Grove ML, Yu B, Cochran BJ, Haritunians T, Bis JC, Taylor KD, et al. Best Practices and Joint Calling of the HumanExome BeadChip: the CHARGE Consortium. PLoS ONE;8:e68095.
Savova V, Chun S, Sohail M, McCole RB, Witwicki R, Gai L, et al. Genes with monoallelic expression contribute disproportionately to genetic diversity in humans. Nat Genet. 2016;48:231–7.
Yu Y, Luo R, Lu Z, Wei Feng W, Badgwell D, Issa J, et al. Biochemistry and biology of ARHI (DIRAS3), an imprinted tumor suppressor gene whose expression is lost in ovarian and breast cancers. Methods Enzymol. 2006;407:455–68.
Gertz J, Varley KE, Reddy TE, Bowling KM, Pauli F, Parker SL, et al. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 2011;7:e1002228.
Court F, Tayama C, Romanelli V, Martin-Trujillo A, Iglesias-Platas I, Okamura K, et al. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 2014;24:554–69.
Abecasis GR, Cookson WO, Cardon LR. Pedigree tests of transmission disequilibrium. Eur J Hum Genet. 2000;8:545–51.
Conomos MP, Reiner AP, Weir BS, Thornton TA. Model-free estimation of recent genetic relatedness. Am J Hum Genet. 2016;98:127–48.
Cui JS, Hopper JL, Harrap SB. Antihypertensive treatments obscure familial contributions to blood pressure variation. Hypertension. 2003;41:207–10.
Kuo KHM. Multiple testing in the context of gene discovery in sickle cell disease using genome-wide association studies. Genomics Insights. 2017;10:1178631017721178.
Fadista J, Manning AK, Florez JC, Groop L. The (in)famous GWAS P-value threshold revisited and updated for low-frequency variants. Eur J Hum Genet. 2016;24:1202–5.
Zhou X, Maricque B, Xie M, Li D, Sundaram V, Martin EA, et al. The human epigenome browser at Washington University. Nat Methods. 2011;12:989–90.
Matzaraki V, Kumar V, Wijmenga C, Zhernakova A. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. 2017;18:76.
Zhang Z, Ma P, Li Q, Xiao Q, Sun H, Olasege BS, et al. Exploring the genetic correlation between growth and immunity based on summary statistics of genome-wide association studies. Front Genet. 2018;9:393.
Chao MJ, Herrera BM, Ramagopalan SV, Deluca G, Handunetthi L, Orton SM, et al. Parent-of-origin effects at the major histocompatibility complex in multiple sclerosis. Hum Mol Genet. 2010;19:3679–89.
Prickett AR, Oakey RJ. A survey of tissue-specific genomic imprinting in mammals. Mol Genet Genomics. 2012;287:621–30.
Pervjakova N, Kasela S, Morris AP, Kals M, Metspalu A, Lindgren CM, et al. Imprinted genes and imprinting control regions show predominant intermediate methylation in adult somatic tissues. Epigenomics. 2016;8:789–99.
Zoledziewska M, Sidore C, Chiang CWK, Sanna S, Mulas A, Steri M, et al. Height-reducing variants and selection for short stature in Sardinia. Nat Genet. 2015;47:1352–6.
Benonisdottir S, Oddsson A, Helgason A, Kristjansson RP, Sveinbjornsson G, Oskarsdottir A, et al. Epigenetic and genetic components of height regulation. Nat Commun. 2016;7:1–10.
Buiting K, Kanber D, Horsthemke B, Lohmann D. Imprinting of RB1 (the new kid on the block). Brief Funct Genomics. 2010;9:347–53.
Lutz SM, Cho MH, Young K, Hersh CP, Castaldi PJ, McDonald M-L, et al. A genome-wide association study identifies risk loci for spirometric measures among smokers of European and African ancestry. BMC Genet. 2015;26:1–11.
Moore T, Haig D. Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet. 1991;7:45–9.
Mott R, Yuan W, Kaisaki P, Gan X, Cleak J, Edwards A, et al. The architecture of parent-of-origin effects in mice. Cell. 2014;156:332–42.
Ruhrmann S, Stridh P, Kular L, Jagodic M. Genomic imprinting: a missing piece of the multiple sclerosis puzzle? Int J Biochem Cell Biol. 2015;67:49–57.
Acknowledgements
We are grateful to the study participants, recruiters, interviewers and nurses.
Funding
This study was supported by Israeli Science Foundation grants 201/98-1 and 407/17; partially by NUS-HUJ 370062002, NIH R01HL088884, Samson Family, NHLBI contract HHSN268201500001I, NIDDK R01 DK078616 and K24 DK080140. Genotyping, quality control and calling of the Illumina HumanExome BeadChip used to genotype some of the genetic variants in the FHS was supported by funding from the National Heart, Lung and Blood Institute Division of Intramural Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services. The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Granot-Hershkovitz, E., Wu, P., Karasik, D. et al. Searching for parent-of-origin effects on cardiometabolic traits in imprinted genomic regions. Eur J Hum Genet 28, 646–655 (2020). https://doi.org/10.1038/s41431-019-0568-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0568-1
This article is cited by
-
Obesity risk in young adults from the Jerusalem Perinatal Study (JPS): the contribution of polygenic risk and early life exposure
International Journal of Obesity (2024)
-
Parent-of-Origin inference for biobanks
Nature Communications (2022)
