
Abstract
The adenovirus (Ad) E4orf4 protein contributes to efficient progression of virus infection. When expressed alone E4orf4 induces p53- and caspase-independent cell-death, which is more effective in cancer cells than in normal cells in tissue culture. Cancer selectivity of E4orf4-induced cell-death may result from interference with various regulatory pathways that cancer cells are more dependent on, including DNA damage signaling and proliferation control. E4orf4 signaling is conserved in several organisms, including yeast, Drosophila, and mammalian cells, indicating that E4orf4-induced cell-death can be investigated in these model organisms. The Drosophila genetic model system has contributed significantly to the study of cancer and to identification of novel cancer therapeutics. Here, we used the fly model to investigate the ability of E4orf4 to eliminate cancer tissues in a whole organism with minimal damage to normal tissues. We show that E4orf4 dramatically inhibited tumorigenesis and rescued survival of flies carrying a variety of tumors, including highly aggressive and metastatic tumors in the fly brain and eye discs. Moreover, E4orf4 rescued the morphology of adult eyes containing scrib− cancer clones even when expressed at a much later stage than scrib elimination. The E4orf4 partner protein phosphatase 2A (PP2A) was required for inhibition of tumorigenesis by E4orf4 in the system described here, whereas another E4orf4 partner, Src kinase, provided only minimal contribution to this process. Our results suggest that E4orf4 is an effective anticancer agent and reveal a promising potential for E4orf4-based cancer treatments.
Similar content being viewed by others

Introduction
The adenovirus E4orf4 protein is a multifunctional viral regulator, which contributes to regulation of the progression of viral infection1,2 and to inhibition of the cellular DNA damage response (DDR), thus increasing the efficiency of adenovirus replication3,4. When expressed alone E4orf4 induces a p53- and caspase-independent mode of cell-death, which can lead to classical caspase-dependent apoptosis in some cell lines5,6,7,8. E4orf4 cell-death signaling is conserved in several organisms, including yeast, Drosophila, and mammalian cells9,10,11,12. Our studies in normal Drosophila tissues demonstrated that E4orf4 induced both caspase-dependent and –independent cell-death in the fly, but also inhibited classical apoptosis, thereby causing minimal tissue damage and a marginal effect on fly survival12. Studies in mammalian cells revealed that E4orf4-induced cell-death was more efficient in oncogene-transformed cells than in normal cells13, indicating that investigation of E4orf4 signaling may have practical implications for cancer therapy. The cancer selectivity of E4orf4-induced cell-death may result from a combination of several E4orf4 activities that interfere with various pathways of cell regulation1,2.
Several E4orf4 cellular partners that contribute to E4orf4-induced cell-death have been described, including protein phosphatase 2A (PP2A) and Src kinases2. E4orf4 binds the heterotrimeric PP2A holoenzyme through direct association with its regulatory B55 subunit14,15.
The model organism Drosophila melanogaster has contributed considerably to the study of cancer and to identification of novel cancer therapeutics16,17,18,19,20,21. Various mutations are known to cause tumorigenesis in Drosophila including those affecting tumor suppressor genes that are required for normal cell polarity and asymmetric cell divisions, such as scribble (scrib)22. Human homologs of these genes are associated with the formation of diverse types of cancers23,24. When scrib mutant cells are surrounded by similar cells they develop tumors, but when surrounded by normal cells they do not25, unless supplemented by constitutive activation of the Ras pathway, which confers a proliferation advantage and leads to formation of aggressive, metastatic tumors25,26. Activated Ras (RasV12) also cooperates with activation of the phosphoinositide 3-kinase (PI3K) pathway to generate more aggressive tumors27.
Here, we show that E4orf4 expression dramatically inhibited the development of various aggressive tumors without greatly affecting healthy tissues, and could restore normal tissue morphology even when expressed at a late stage. The role of E4orf4 partners in this process is also described.
Results
E4orf4 counteracts Ras V12-induced impairment of tissue differentiation and fly survival in a dose-dependent manner
The consequences of RasV12 expression in the presence or absence of E4orf4 were initially examined in eye imaginal discs. RasV12 was expressed together with either E4orf4 or a control GFP gene using the UAS-GAL4 system driven by the eyeless (ey) promoter, which drives expression in the eye disc, as well as in regions of the brain28,29. GAL4-driven expression rises with increasing temperatures, facilitating investigation of dose-dependent effects. As seen in Fig. 1a, the expression of RasV12 together with GFP at 24 oC resulted in loss of normal eye disc morphology and a significant increase in disc size indicating induction of uncontrolled proliferation and differentiation defects. The differentiation defects were also visualized by the unorganized staining of the neuronal differentiation marker ELAV. The expression of E4orf4 counteracted the RasV12-induced over-proliferation of cells and the loss of normal differentiation as evident by the smaller size and normal shape of the E4orf4 RasV12 expressing discs.

a Eye-antennal imaginal discs expressing RasV12 under the regulation of ey-GAL4 together with GFP (ey-GAL4 > UAS-RasV12; FRT82B, UAS-GFP) or E4orf4 (ey-GAL4 > UAS-RasV12; FRT82B, UAS-E4orf4-17.22) are shown. Discs were dissected from age-matched 3rd-instar larvae grown at 18 oC or 24 oC. The discs were stained with antibodies to E4orf4 (blue) and ELAV (red) and were analyzed by confocal microscopy. All images were taken at the same magnification (x40) and represent projections of multiple sections. A representative eye disc is shown in each picture. The 50 μm scale bar applies to all the pictures. It should be noted that age-matched larvae were utilized, but E4orf4-expressing larvae were consistently slower to differentiate. b Percentage of adult eclosion (“Live”) and of non-eclosed pupae (“Dead”) was determined in flies expressing GFP (labeled as WT) or RasV12 under the regulation of ey-GAL4 together with another copy of GFP (ey-GAL4 > UAS-GFP;FRT82B, UAS-GFP, and ey-GAL4 > UAS-RasV12;FRT82B, UAS-GFP) or E4orf4. Two different fly strains harboring an E4orf4 transgene were tested. These strains express the viral protein to different levels: “strong”: UAS-E4orf4-17.22 (ey-GAL4 > UAS-GFP;FRT82B,UAS-E4orf4-17.22 and ey-GAL4 > UAS-RasV12;FRT82B,UAS-E4orf4-17.22) and “weak”: UAS-E4orf4-attP2, also called “weak1” in c below (ey-GAL4 > UAS-GFP;UAS-E4orf4-attP2 and ey-GAL4 > UAS-RasV12;UAS-E4orf4-attP2). The flies were grown at the indicated temperatures. N > 300, collected from three independent experiments. c Protein extracts were prepared from ten 3rd instar larvae expressing “strong” E4orf4 and two types of “weak” E4orf4 under the regulation of ey-GAL4 (ey-GAL4 > UAS-E4orf4-17.22, ey-GAL4 > UAS-E4orf4-attP2 (weak#1), and ey-GAL4 > UAS-E4orf4-attP40 (weak #2)). A representative western blot is shown, stained with antibodies to E4orf4 and to Tubulin, which served as a loading control
In the experiments described in Fig. 1a, we noticed a reduced proportion of adult eclosion upon RasV12 expression at high temperatures, possibly resulting from ey-driven RasV12 effects in the brain. We therefore examined the ability of E4orf4 to rescue the RasV12-induced lethality at various temperatures (Fig. 1b). Two different UAS-E4orf4 fly strains were utilized, one expressing higher E4orf4 levels than the other when driven by ey-GAL4 (Fig. 1c). In a WT background (ey > GFP), even high levels of E4orf4 (“strong”) had an insignificant effect on adult fly viability at 18 oC and 24 oC (p > 0.1) and reduced viability by 13% at most at 29 oC (p = 0.013). These results are consistent with our previous findings showing that expression of E4orf4 in normal fly tissues caused only mild effects12. In contrast, strong E4orf4 expression rescued the 100% lethality induced by ey > RasV12 at 24 oC and 29 oC to 89% and 24% viability, respectively (p < 0.012). Weaker E4orf4 expression led to rescue of 50% viability at 24 oC (p < 0.0001), but could not rescue the RasV12-induced lethality at 29 oC. These findings confirm that the dose-dependence of E4orf4 activity, which was previously reported in WT flies12 is also sustained in a RasV12 background, and reveal that E4orf4 counteracts RasV12-induced lethality.
E4orf4 inhibits the formation of highly aggressive tumors in the fly
The preliminary experiments described in Fig. 1 investigated RasV12 and E4orf4 effects occurring when these genes were expressed broadly in the developing eye. However, since tumor growth is significantly influenced by the microenvironment, additional investigation of E4orf4 effects on tumorigenesis was conducted in clonally induced tumors. For this purpose, the mosaic analysis with a repressible cell marker (MARCM) method was utilized as described previously26 to induce mitotic GFP-labeled clones in the eye discs and brain. The overexpression of RasV12 induces non-invasive tumors in flies26,30. Since we wished to examine the ability of E4orf4 to eliminate various types of tumors, including highly aggressive, metastatic tumors, we analyzed mitotic clones carrying genetic alterations including RasV12 activation and/or inactivation of the tumor suppressor scribbled (scrib), a regulator of cell polarity23. The clones were produced by ey-driven FLP recombinase, which mediated recombination between chromosome arms, leading to homozygous loss of scrib and to loss of the UAS-GAL4 system inhibitor Gal80, facilitating GAL4-driven expression of UAS-regulated genes within the clones26. Flies generated using this system carried one of four types of mitotic clones, including WT clones, scrib− clones, clones expressing RasV12, and scrib− clones that expressed RasV12. Each of these clones also expressed either the strong E4orf4 transgene or a control RFP gene.
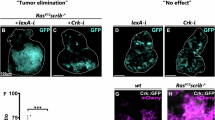
As shown in Fig. 2a, eye discs and brains of 3rd instar larvae containing WT or scrib− clones were similar in size and morphology, and E4orf4 expression did not greatly affect organ size, although it reduced clone sizes in the scrib− background. Brains and eye discs of some larvae with RasV12-overexpressing clones were slightly enlarged, but the alterations were not significant in comparison with larvae carrying RasV12, E4orf4-coexpressing clones. There were also no dramatic changes in the overall organ morphology. In contrast, RasV12-overexpression in combination with scrib deletion resulted in massive growth of brain and eye discs and loss of recognizable morphology, creating a huge tumor mass. Furthermore, whereas the GFP-marked mitotic clones with other genotypes were restricted to parts of the eye discs and to limited areas in the brain hemispheres, the RasV12, scrib− mitotic clones colonized the whole eye disc and brain area and invaded the ventral nerve cord (VNC), as described previously26,30. E4orf4 expression had a remarkable effect on these highly aggressive tumors, resulting in complete restoration of normal size and morphology of both eye discs and brains, as well as nearly complete elimination of GFP-marked clones, reducing their size to that of WT clones.

a Brains and eye-antennal discs harboring ey-FLP-induced clones were dissected from 3rd-instar larvae grown at 24 oC and analyzed by confocal microscopy. WT and mutant clones of the indicated genotypes are marked by GFP fluorescence (middle column); the presence of E4orf4 (blue) or control RFP (red) was detected by immunostaining or RFP fluorescence (left column). The morphology of brains and eye-antennal discs is shown in the right column together with GFP fluorescence to demonstrate the degree of colonization of these organs by the GFP-marked clones. Fluorescent images in the left and middle columns represent projections of multiple sections and the images in the right column show a single plane. The discs and brains shown are representative of several samples. All images were taken at the same magnification (x10) and the 200 μm scale bar in the bottom right applies to all the pictures. The females in all crosses were of the genotype eyFLP;Act5C,GAL4,UAS-GFP;FRT82B,tubGal80 and the males were, from top to bottom, UAS-RFP-attP40; FRT82B, UAS-RFP-attP2 (WT, RFP), UAS-RFP-attP40; FRT82B, UAS-E4orf4-17.22 (WT, E4orf4), UAS-RFP attP40; FRT82B, scribble1/TM6B (scrib−, RFP), UAS-RFP-attP40; FRT82B, UAS-E4orf4-17.22, scribble1/TM6B (scrib− E4orf4), UAS-RasV12, UAS-RFP-attP40; FRT82B (RasV12, RFP), UAS-RasV12; FRT82B, UAS-E4orf4-17.22 (RasV12, E4orf4), UAS-RasV12, UAS-RFP-attP40; FRT82B, scribble1/TM6B (RasV12, scrib−, RFP), UAS-RasV12; FRT82B, UAS-E4orf4-17.22, scribble1/TM6B (RasV12, scrib−, E4orf4). b The percentage of adult eclosion, signifying fly survival, was determined for flies carrying clones of the indicated genotypes, expressing E4orf4 (E4orf4:+) or a control RFP gene (E4orf4: −) as described in a. N > 300, collected from a minimum of three independent experiments. Error bars represent standard deviation. c Representative eyes harboring clones of the indicated genotypes are shown. The skull symbols represent pupal-lethality. The 50 μm scale bar in the bottom right applies to all the pictures
Next, we determined how generation of the mitotic clones, with or without E4orf4, affected fly viability at 24 oC. As seen in Fig. 2b, 100% of flies with WT mitotic clones were viable, and E4orf4 expression reduced viability in the WT background by only 3%. The generation of scrib− homozygous clones reduced fly viability by 12% in the absence of E4orf4, but the flies were 96.5% viable when E4orf4 was expressed in these clones. Clonal RasV12 overexpression led to a complete loss of fly viability in both WT and scrib− backgrounds. However, E4orf4 expression could rescue 97% of flies with RasV12-expressing clones and 80% of flies with RasV12, scrib− mitotic clones.
The eye phenotypes of adult flies that emerged from this experiment demonstrated an improvement in eye morphology in flies with scrib− mitotic clones expressing E4orf4, and similar eye morphologies were evident in flies expressing E4orf4 in RasV12 and RasV12, scrib− mitotic clones (Fig. 2c).
Altogether, the results shown in Fig. 2 indicate that E4orf4 is highly potent in preventing the development of even very aggressive tumors, and consequently reduces the ensuing lethality and eye deformities caused by the tumors.
As E4orf4 was reported to induce cell-death in flies and mammalian cells12,13, we examined whether cancer clone elimination could potentially result from E4orf4-induced cell-death manifested by activation of the Dcp1 caspase. Dispersed caspase activation, which was not concentrated near the strongest GFP-marked clones, was evident in control WT clones, as well as in RasV12, scrib− clones (Fig. 3a, c). Dcp1 activation was observed around strong E4orf4-expressing clones in the WT background (Fig. 3b), but the staining was somewhat dispersed in comparison to a robust accumulation of Dcp1 staining around RasV12, scrib− clones with strong E4orf4 expression (Fig. 3d). It should be noted that most of the E4orf4-expressing RasV12, scrib− clones have already been eliminated by this stage (Fig. 2a) and therefore it is likely that only remnants of the E4orf4-induced cell-death are still evident. Nonetheless, the results suggest that cell-death could explain, at least in part, the elimination of the large RasV12, scrib− tumor clones.

Third-instar larvae were collected at 24 oC from crosses between eyFLP;Act5C,GAL4,UAS-GFP;FRT82B,tubGal80 females and males of the following genotypes: FRT82B, UAS-GFP (a), FRT82B, UAS-E4orf4-17.22 (b), UAS-RasV12; FRT82B, scrib−, UAS-GFP (c), UAS-RasV12; FRT82B, scrib−, UAS-E4orf4-17.22 (d). Eye-antennal discs were dissected from these larvae and analyzed by confocal microscopy. GFP-expressing clones were visualized by GFP fluorescence and clones expressing E4orf4 were stained with E4orf4-specific antibodies (blue). Active (cleaved) Dcp1 staining is shown in red. All images represent projections of multiple sections. A representative eye disc is shown in each picture with a 50 μm scale bar. An enlarged inset of E4orf4-expressing RasV12, scrib− clones surrounded by concentrated active Dcp1 staining is displayed in d
In addition to the highly tumorigenic collaboration between activated RasV12 and deletion of cell polarity-regulating genes, a collaboration between RasV12 and activation of the insulin-PI3K pathway induces significantly more tumorigenesis than RasV12 alone, possibly due to increased metabolism that supports cancer cell proliferation27,31. Activation of the insulin-PI3K pathway in Drosophila can be achieved by overexpression of Chico, the fly insulin receptor substrate, or by reduced expression of PTEN, the PI3K pathway inhibitor. To determine how E4orf4 affects PI3K-regulated tumorigenesis, E4orf4 or a control RFP transgene were expressed under ey-GAL4 regulation together with various combinations of RasV12, chico, and PTEN interfering RNA (IR), and fly viability was assayed. To detect differences between the effects of RasV12 alone and RasV12 supplemented by activation of PI3K, we counted separately empty pupal cases from which adult flies have eclosed, pupae containing pharate adults that failed to eclose, and pupae in which no metamorphosis took place. Figure 4a demonstrates that chico overexpression with or without concomitant expression of E4orf4 did not reduce significantly fly viability. Co-expression of RasV12 with two copies of the RFP transgene caused 100% lethality at 24 oC, whereas RasV12, E4orf4 co-expression facilitated adult eclosion from 80% of the pupae. Co-expression of chico with RasV12 aggravated the effect of RasV12, increasing the percent of non-metamorphosing pupae from 83% to 94% (p = 0.005). However, even in this background, E4orf4 expression dramatically increased adult eclosion up to 74%.

a The percentage of adult eclosion was determined for flies expressing RFP, or chico, or RasV12 and RFP, or RasV12 and chico, together with E4orf4 or RFP. The females in all crosses were of the genotype ey-GAL4 and the males were, from left to right, RFP RFP: UAS-RFP-attP40; UAS-RFP-attP2; E4orf4 RFP: UAS-E4orf4-attP40; UAS-RFP-attP2; RFP chico: UAS-RFP- attP40; UAS-chico; E4orf4 chico: UAS-E4orf4-attP40; UAS-chico; RFP RasV12 RFP: UAS-RasV12, UAS-RFP-attP40; UAS-RFP-attP2; E4orf4 RasV12 RFP: UAS-RasV12, UAS-E4orf4-attP40; UAS-RFP-attP2; RFP RasV12 chico: UAS-RasV12, UAS-RFP-attP40; UAS-chico; E4orf4 RasV12 chico: UAS-RasV12, UAS-E4orf4-attP40; UAS-chico. All crosses were maintained at 24 oC. Non-eclosed pupae were divided into two categories: those that have undergone metamorphosis and those that have not. N > 300, collected from three independent experiments. Error bars represent standard deviation. b The percentage of adult eclosion was determined for flies expressing RFP, or PTEN-directed RNAi construct (PTEN-IR), or RasV12 and RFP, or RasV12 and PTEN-IR, together with E4orf4 or RFP. The females in all crosses were of the genotype ey-GAL4 and the males were of the following genotypes: RFP RFP: UAS-RFP- attP40; UAS-RFP-attP2; E4orf4 RFP: UAS-RFP- attP40; UAS-E4orf4-17.22; RFP PTEN-IR: UAS-PTEN-IR; UAS- RFP-attP2; E4orf4 PTEN-IR: UAS-PTEN-IR; UAS-E4orf4-17.22; RFP RasV12 RFP: UAS-RasV12, UAS-RFP-attP40; UAS-RFP-attP2; E4orf4 RasV12 RFP: UAS-RasV12, UAS-RFP-attP40; UAS-E4orf4-17.22; RFP RasV12 PTEN-IR: UAS-RasV12, UAS-PTEN-IR; UAS- RFP-attP2; E4orf4 RasV12 PTEN-IR: UAS-RasV12, UAS-PTEN-IR; UAS-E4orf4-17.22. N > 300, collected from three independent experiments. Error bars represent standard deviation. c Representative eyes of flies obtained as described in a are shown. The pictures were captured at a magnification of x5. The skull symbol represents pupal-lethality and the absence of pupae that have undergone metamorphosis
Similar results were obtained when activation of the PI3K pathway was induced by knockdown of PTEN (PTEN-IR, Fig. 4b). Whereas PTEN-IR alone reduced fly viability to 76%, co-expression of E4orf4 rescued viability to 94% (p = 0.01). The concomitant expression of PTEN-IR aggravated the effect of RasV12, increasing the percentage of non-metamorphosing pupae from 83% to 100% (p = 0.002). Here too, co-expression of E4orf4 resulted in a dramatic increase in adult eclosion to 85% in this background, very similar to a rescue to 88% eclosion in flies expressing ey > RasV12 without PTEN-IR.
To investigate the effect of E4orf4 on tumor tissues, adult eyes were examined. As shown in Fig. 4c, the phenotype of RasV12, E4orf4-expressing eyes was milder than that of eyes expressing RasV12 alone, which were visualized in 0.6% adults that have eclosed. The ability of E4orf4 to rescue the eye phenotype of RasV12, chico expressing flies could not be evaluated due to pupal-lethality of the RasV12, chico genotype. As previously reported, E4orf4 expression caused a minor rough eye phenotype in a WT or chico backgrounds.
The results summarized in Fig. 4 confirm that E4orf4 is a potent inhibitor of tumorigenesis.
E4orf4 partners that contribute to cancer elimination by E4orf4
We previously reported that two E4orf4 partners, PP2A and Src, contributed in an additive manner to the mild E4orf4-induced cell-death in normal Drosophila tissues12. We now tested whether these partners contributed similarly to the E4orf4-induced elimination of cancer tissues. The flies used in this experiment carried a UAS-RasV12 transgene on the 2nd chromosome and an additional transgene integrated in the attP2 site on the 3rd chromosome containing a UAS-driven cDNA of RFP, WT E4orf4, or E4orf4 mutants that cannot bind Src (“Src−”) or PP2A (“PP2A−”), or both (“Src−, PP2A−”)12. Integration of all transgenes at the same chromosomal site minimizes position effects on their expression. Flies of these genotypes were crossed to ey > GAL4 flies at 24 oC. As shown in Fig. 5a, ey-GAL4-driven RasV12 expression in the presence of a control RFP gene reduced fly viability and only 2% of the flies reached adulthood. Despite the relatively low expression levels of WT E4orf4 integrated at the attP2 site (Fig. 1c), E4orf4 was still able to increase viability of ey > RasV12-expressing flies to 60%. The Src− E4orf4 mutant that is unable to bind Src kinase was only slightly less efficient than WT E4orf4 in rescuing the viability of ey > RasV12-expressing flies, leading to 50% adult eclosion. In contrast, the PP2A− E4orf4 mutant, which could not bind PP2A, lost completely the ability to counteract RasV12 activity, and fly viability remained 0.7%. Although the WT E4orf4 protein accumulated at higher levels than the E4orf4 mutants, the dissimilarity in function of the two mutants cannot be explained by differences in protein levels, as the levels of the PP2A− mutant were consistently higher than the levels of the Src− mutant (Fig. 5b). The double mutant, PP2A− Src−, which lost the ability to bind both Src and PP2A was also unable to increase fly viability, resulting in 0% adult eclosion.

a Percentage of adult eclosion and non-eclosed pupae was determined in flies expressing ey-GAL4 > RasV12 together with control RFP (marked as E4orf4-None: UAS-RFP-attP40; UAS-RFP-attP2) or WT E4orf4 (UAS-RFP-attP40; UAS-E4orf4-attP2), or the E4orf4 mutants that do not bind Src (Src−: UAS-RFP-attP40; UAS-E4orf4 R73/74/75A-attP2), or PP2A (PP2A−: UAS-RFP- attP40; UAS-E4orf4 R81F84A -attP2, or the double mutant that does not bind both Src and PP2A (Src−, PP2A−: UAS-RFP-attP40; UAS-E4orf4 R73/74/75A +R81F84A-attP2). b Protein extracts were prepared from ten 3rd instar larvae expressing RFP, WT E4orf4, or the E4orf4 mutants described in a, using the ey driver at 24 oC. A representative western blot is shown, stained with antibodies to E4orf4 and to Tubulin, which served as a loading control. c Eye-antennal imaginal discs obtained from crosses as described in a were dissected from age-matched 3rd-instar larvae grown at 24 oC. The discs were stained with antibodies to ELAV and were analyzed by confocal microscopy. All images were taken at the same magnification (x40) and represent projections of multiple sections. A representative eye disc is shown in each picture with a 50 μm scale bar. d Representative eyes of flies harboring clones of the indicated genotypes and reared at 18 oC or 24 oC are shown. Pictures were captured at a magnification of x5. A skull symbol represents pupal-lethality. The females in all crosses were of the genotype eyFLP;Act5C,GAL4,UAS-GFP;FRT82B,tubGal80 and the males were, in the upper row WT: FRT82B,UAS-GFP, tws: FRT82B,tws60/TM6B, RasV12: UAS-RasV12;FRT82B,UAS-GFP, RasV12, tws: UAS-RasV12;FRT82B,tws60/TM6B and in the lower panel, WT: FRT82B,UAS-E4orf4-17.22, tws: FRT82B,tws60,UAS-E4orf4-17.22/TM6B, RasV12:UAS-RasV12;FRT82B,UAS-E4orf4-17.22, RasV12, tws: UAS-RasV12;FRT82B,tws60,UAS-E4orf4-17.22/TM6B. It should be noted that even though flies with the tws mutation did not express a control GFP transgene to match the E4orf4 transgene whereas the other flies contained this control, potential competition for the GAL4 driver by UAS-E4orf4 did not lead to rescue of eye morphology in the mutant background
In addition to examining the ability of E4orf4 mutants to rescue the viability of ey > RasV12-expressing flies, we evaluated their ability to rescue eye disc differentiation. Eye discs of 3rd instar larvae that expressed RasV12 together with RFP or E4orf4 under ey > GAL4 regulation were stained with anti-ELAV antibodies. As seen before (Fig. 1a), expression of RasV12 in the absence of E4orf4 resulted in loss of normal differentiation marked by irregular ELAV staining (Fig. 5c). WT E4orf4 expression (the “weak1” transgene) enhanced normal differentiation, as did expression of the Src−E4orf4 mutant. In contrast, the PP2A− E4orf4 mutant was much less efficient in recovering normal ommatidia patterns, further confirming its reduced ability to counteract RasV12 effects.
To validate the contribution of PP2A to the ability of E4orf4 to counteract RasV12 activities, the influence of E4orf4 on the RasV12 induced eye phenotype was compared in WT eyes and in eyes lacking tws, the fly PP2A-B55 subunit that mediates the E4orf4-PP2A interaction13,14. Figure 5d demonstrates that E4orf4 caused a minor rough eye phenotype in both WT and tws backgrounds, possibly through Src binding. RasV12 expression at 24 oC was lethal and no adult progeny eclosed. Concomitant expression of E4orf4 rescued fly viability and produced eyes with some defects, which retained regular differentiation in large areas. In contrast, the tws mutation reduced RasV12-induced lethality and adult eyes with a typical “raspberry-like” appearance were observed. However, E4orf4 expression did not improve eye morphology in this background. To overcome the RasV12-induced lethality, flies were additionally reared at 18 oC. At this temperature, ey > RasV12 flies could eclose and they presented rough, overproliferating eye tissues and some small growths, whereas E4orf4 expression reverted eye morphology to near normal. In the tws background, the ey > RasV12 eyes remained abnormal upon E4orf4 expression.
The findings shown in Fig. 5 demonstrate that the interaction with PP2A is crucial for the ability of E4orf4 to rescue flies from RasV12–induced tumorigenesis and lethality, but surprisingly, the interaction of E4orf4 with Src has only a minor contribution to counteracting RasV12-induced lethality. This differs from the situation in normal fly tissues, in which PP2A and Src contribute additively to E4orf4 function12.
E4orf4 rescues effects of early scrib elimination even when expressed at a late stage
In the previous experiments, the concomitant expression of RasV12 and E4orf4 was induced by the ey driver (Figs. 1, 4, and 5), or was induced following ey > FLP activation together with scrib deletion (Fig. 2). Thus, the results of these experiments show that E4orf4 inhibits tumorigenesis when expressed simultaneously with cancer induction. To examine the consequences of late E4orf4 expression, scrib− GFP-marked homozygous clones were induced under the regulation of ey > FLP, which is expressed in the eye disc as early as the end of the 1st instar larval stage33. In contrast, the expression of E4orf4 or a GFP control gene was driven by GMR-GAL4, which is activated in the eye disc only at the 3rd instar larval stage34. As seen in Fig. 6a, the eyes of flies with mitotic clones lacking scrib and expressing the control GFP gene were abnormal, with small growths and necroses. However, GMR > E4orf4 expression resulted in a significant suppression of this adult eye phenotype. We have previously shown that, in WT background, GMR > E4orf4 on its own induced only a mild rough eye phenotype12.

a Representative eyes carrying homozygous scrib mutant clones induced by ey > FLP and expressing either GFP or WT E4orf4 under the regulation of GMR-GAL4 are shown. Flies of the genotype yw,eyFLP;19A,UAS-med8GFP2A;FRT82B,tubGAL80/TM3 were crossed to flies of the following genotypes: GMR-GAL4; FRT82B, UAS-GFP, scribble1/TM6B (scrib−, GFP), GMR-GAL4; FRT82B, UAS-E4orf4-17.22, scribble1/TM6B (scrib−, E4orf4). The 50 μm scale bar in the bottom right applies to both pictures. b Eye-antennal discs were dissected from 3rd-instar larvae grown at 24 oC and analyzed by confocal microscopy. Flies with scrib− clones were as described in a. Flies with WT clones were generated by a cross between yw,eyFLP;19A,UAS-med8GFP2A;FRT82B,tubGAL80/TM3 and GMR-GAL4; FRT82B,UAS-GFP (WT, GFP) or GMR-GAL4; FRT82B, UAS-E4orf4-17.22 (WT, E4orf4). All images were taken at the same magnification (x40) and represent projections of multiple sections. WT and scrib− clones, as indicated in the figure, are marked by GFP fluorescence (green) or stained with antibodies to E4orf4 (blue) (left column). The discs were also stained with ELAV-specific antibodies (red)(middle column) to visualize their state of differentiation. A merge of all stains is seen in the right column. A representative eye disc is shown in each picture. The 50 μm scale bar in the bottom right applies to all the pictures. c The sizes of GFP-marked clones from eye-antennal discs with WT or scrib− mutant backgrounds were detected by GFP fluorescence (present also in E4orf4-expressing clones, but not shown in b) and quantified using ImageJ software. Average pixel numbers are shown. All the discs used here were age-matched. N > 8 from > 3 independent experiments, error bars represent the standard deviation. Statistical significance was determined by a student t-test. *p < 0.05, NS: not significant
To evaluate the size and state of differentiation of scrib-deficient clones, the eye discs of 3rd instar larvae containing GFP-marked mitotic clones were co-stained for ELAV and E4orf4. As seen in Fig. 6b, GFP-marked scrib-deficient clones were relatively large and ELAV staining in these clones was irregular compared with WT clones, indicating abnormal differentiation. In contrast, scrib-deficient clones that expressed E4orf4 were smaller and the pattern of ELAV staining resembled more closely the staining in WT clones, although E4orf4 had some effect on photoreceptor differentiation as the spacing between the ommatidia appeared increased. It should be noted that because the expression of E4orf4 and GFP was driven by GMR-GAL4, it was seen only in the ey > FLP- generated clones, in which the GAL4 system inhibitor Gal80 was removed by recombination. Figure 6c demonstrates that whereas E4orf4 reduced the size of WT clones to 72%, it reduced the size of scrib-deficient clones to 43% compared to the GFP-expressing control. Statistical analysis (Summary Independent-Samples Test) further demonstrated that the decrease in cancer clone sizes induced by E4orf4 was significantly larger than the E4orf4-induced decrease in WT clone sizes (p = 0.012). These findings indicate that E4orf4 can specifically eliminate scrib− clones and rescue adult eye morphology even when expressed at a much later stage than scrib elimination, and that its toxic effects on WT cells are much milder than its toxic effects in a cancer tissue.
Discussion
E4orf4 effects in various types of tumors
Previous work in tissue culture revealed that E4orf4-induced cell-death was more efficient in mammalian cancer cells than in normal cells13. By utilizing the power of fly genetics, the present work demonstrates that E4orf4 can also strongly suppress the tumorigenic process in a whole organism with minimal damage to normal tissues.
Various types of tumors were used in this study: Hyperplastic RasV12-induced tumors, which over-proliferate and cause imaginal disc overgrowth without metastasizing; Neoplastic scrib− tumors, which lose the monolayered organization and correct apical-basal polarity of wild-type epithelia35 and are deficient in normal differentiation as well as prone to metastasis; and highly aggressive and metastatic RasV12, scrib− tumors. In addition, we examined the effects of E4orf4 on tumors induced by combined activation of the Ras and insulin-PI3K pathways. The tumor types investigated here are highly relevant to human tumors. Activation of Ras signaling occurs in ~30% of human cancers36 and deregulation of scrib, as well as of many other cell polarity-regulating genes contributes to many types of human tumors37. Disruption of insulin-PI3K signaling is also quite common in human cancers38.
This work shows that E4orf4 was efficient in inhibiting tumorigenesis in representatives of all types of tumor tissues described above. Especially exciting was the ability of E4orf4 to prevent formation of the massive metastatic tumors induced by the combination of RasV12 overexpression and scrib deletion (Fig. 2). E4orf4 succeeded in preventing both overgrowth and the ability of cancer cells to totally colonize the tissue and invade the VNC. The mild effects of E4orf4 on normal tissues were reaffirmed.
E4orf4 induction of cell-death is responsible, at least in part, for its anti-tumorigenic effect. Although technical difficulties prevented the visualization of E4orf4-induced cell-death in early 2nd instar larvae shortly after initiation of E4orf4 expression in the highly aggressive RasV12, scrib− tumors, Dcp1 activation was evident around clones with strong E4orf4 expression in 3rd instar larvae (Fig. 3). This observation suggests that induction of cell-death played a role in the elimination of tumorous clones. E4orf4 is also known to kill normal cells, but to a lesser degree12, and thus the elimination of aggressive RasV12, scrib− cancer clones as well as of scrib− clones was more efficient than removal of WT clones (Figs. 2a and 6c). Effects on cell proliferation could also potentially contribute to cancer elimination by E4orf4 but we found no evidence supporting this possibility.
The ability of E4orf4 to revert the effects of scrib deletion when expressed at a later stage (Fig. 6) is also interesting. scrib− clones were shown to be eliminated by their WT neighbors and by JNK-dependent apoptosis, causing eye deformities30,39,40. It is therefore possible that E4orf4, which inhibits classical apoptosis in normal cells12, could also inhibit apoptosis in the scrib− clones, thus rescuing normal eye morphology. It is also possible, however, that E4orf4 reverted the neoplastic identity of these clones through other mechanisms.
The contribution of E4orf4 partners
Two E4orf4 partners, PP2A and Src kinase contribute additively to E4orf4-induced cell-death both in mammalian transformed cell lines32 and in normal Drosophila tissues12. The major surprising finding here was that PP2A binding was crucial for the ability of E4orf4 to inhibit Ras carcinogenesis in vivo, whereas Src binding played a marginal role (Fig. 5). Several types of PP2A holoenzymes act as tumor suppressors, and mutations in the scaffolding A subunit, as well as in some regulatory B subunits, were found in various types of human cancer41. Moreover, PP2A activation by small molecules was shown to inhibit growth of some tumors42,43. In contrast, in some cellular contexts the PP2A B55 subunit was reported to promote tumorigenesis44 and certain PP2A inhibitors are tested as chemotherapeutics43. Thus, PP2A activity must be carefully balanced in the cellular context. The mechanisms by which PP2A supports E4orf4-induced cell-death are still debated. Although one report suggested that inhibition of PP2A activity contributed to this process45, several findings indicate that the E4orf4-PP2A complex possesses PP2A phosphatase activity14 and that PP2A contributes positively to E4orf4-induced cell-death13,15. Furthermore, PP2A is recruited by E4orf4 to several substrates involved in its functions, including induction of cell-death2,46. Thus, recruitment of PP2A to novel substrates or stabilization of existing interactions may shift the balance in several E4orf4-targeted cellular pathways. Cancer cells may be more sensitive to these changes than normal cells, leading to cell-death. As one example, E4orf4 inhibits some branches of the DDR in a PP2A-dependent manner3. Many cancer cells have deficiencies in DNA damage signaling and may rely more heavily on their remaining intact DDR pathways. Targeting the remaining pathways by a DDR inhibitor such as E4orf4 may therefore be selectively toxic to these cancer cells.
The potential of E4orf4 for cancer therapy
This work demonstrated the remarkable activity of E4orf4 as an anticancer agent in vivo, providing a strong incentive for development of E4orf4-based cancer treatments. The potential advantages of E4orf4-based cancer therapy include the following: (1) E4orf4 kills several types of tumor cell lines but not normal cells6,8,13,47,48. (2) E4orf4 induces p53-independent cell-death8,49, which could be effective in p53-deficient tumors. (3) E4orf4 induces caspase-independent cell-death5, which could be useful in cancers with deficiencies in classical apoptosis. (4) As E4orf4 induces a unique mode of programmed cell-death by targeting a combination of several cellular pathways2,3, it may provide alternative solutions in cases of therapy-resistant disease. Therefore, design of E4orf4-based cancer therapies would be highly beneficial. Cell polarity genes as well as the PI3K pathway are associated with the formation of diverse types of cancer23,24,38, and providing appropriate therapy would be beneficial. Moreover, despite more than three decades of research, no anti-Ras therapies have become available50. Therefore, finding effective treatment of cancers with Ras activation36 would in itself be a satisfactory result of using E4orf4-based cancer therapy.
Materials and methods
Fly culture
Fly stocks (Table 1) were cultured on standard cornmeal/yeast media. Crosses were maintained at 18 °C, 24 °C, or 29 °C depending on the required experimental conditions. Each cross included ten virgin females and five males. Parents were allowed to mate for 3 days and were then transferred to a new vial for additional 3 days and were then discarded.
Adult eclosion test
The number of empty pupal cases from which adult flies have eclosed and of pupae that died before or after metamorphosis were counted in each vial for up to 3 weeks after pupa formation. Eclosed adults were removed daily. The ratio between empty pupal cases and the total number of pupae was calculated.
Phenotypic characterization of adult eyes
Adult flies were anesthetized with CO2. Equal numbers of female eyes were examined for each genotype (n > 20 unless otherwise stated). The eyes were photographed using AxioCam MRc and Zeiss Discovery V8 binocular. To represent the depth of field in full, a series of images with different foci were taken, and the images were stacked using the Helicon Focus software.
Western blots
Whole-cell extracts were prepared from 3rd-instar larvae in lysis buffer [50 mM Tris·HCl (pH 7.4), 250 mM NaCl, 5 mM EDTA, 0.1% Triton X-100, 0.5% Nonidet P-40, and a 1/10 volume of Complete protease inhibitor mixture (Roche, Basel, Switzerland)]. Proteins were analyzed by western blots using antibodies to E4orf48 and alpha-Tubulin (Sigma, Rehovot, Israel).
Staining imaginal discs
Larval tissues were dissected and stained following standard procedures. Primary antibodies used: mouse monoclonal anti-E4orf4 (#16, 1:100)8; rat monoclonal anti-ELAV (hybridoma bank, 9F8A9), rabbit activated (cleaved) Dcp1-specific antibody (Cell Signaling Technology, Danvers, MA, USA). Secondary fluorescent antibodies for fluorescent staining were Cy2, Cy3, or Cy5-conjugated anti-rabbit/mouse/rat (1:100) (Jackson Immunoresearch, West Grove, PA, USA). Samples were mounted with a DakoCytomation mounting medium (Agilent-Dako, Santa Clara, CA, USA).
Clone size quantification
The size of GFP-positive clones was measured in pixels using the ImageJ software (NIH). Briefly, images of all samples were captured using the same magnification and acquisition conditions in a confocal microscope. A Z-stack was formed from all eye-antennal discs and a maximum intensity projection was created to include the entire depth of the discs. Eye disc size was quantified by increasing the brightness/contrast level marking the disc margins and measuring its area. The area of GFP-marked clones with intensities above a defined threshold were measured and summarized.
Data analysis
The statistical significance of differences in eclosion rates or clone sizes between the various fly groups were calculated by an unpaired t-test. Summary Independent-Samples Test was used to assess whether the differences between E4orf4-induced changes in WT and scrib− clone sizes were significant. This statistical analysis was carried out using the SPSS software package (Release 23.0.0.0, SPSS Inc., 2014).
References
Kleinberger, T. Induction of cancer-specific cell-death by the adenovirus e4orf4 protein. Adv. Exp. Med. Biol. 818, 61–97 (2014).
Kleinberger, T. Mechanisms of cancer cell killing by the adenovirus E4orf4 protein. Viruses 7, 2334–2357 (2015).
Brestovitsky, A., Nebenzahl-Sharon, K., Kechker, P., Sharf, R. & Kleinberger, T. The adenovirus E4orf4 protein provides a novel mechanism for inhibition of the DNA damage response. PLoS Pathog. 12, e1005420 (2016).
Nebenzahl-Sharon, K. et al. Biphasic Functional Interaction between the Adenovirus E4orf4 Protein and DNA-PK. J. Virol. 93, https://doi.org/10.1128/JVI.01365-18 (2019).
Lavoie, J. N., Nguyen, M., Marcellus, R. C., Branton, P. E. & Shore, G. C. E4orf4, a novel adenovirus death factor that induces p53-independent apoptosis by a pathway that is not inhibited by zVAD-fmk. J. Cell Biol. 140, 637–645 (1998).
Livne, A., Shtrichman, R. & Kleinberger, T. Caspase activation by adenovirus E4orf4 protein is cell line-specific and is mediated by the death receptor pathway. J. Virol. 75, 789–798 (2001).
Marcellus, R. C. et al. The early region 4orf4 protein of human adenovirus type 5 induces p53-independent cell-death by apoptosis. J. Virol. 72, 7144–7153 (1998).
Shtrichman, R. & Kleinberger, T. Adenovirus type 5 E4 open reading frame 4 protein induces apoptosis in transformed cells. J. Virol. 72, 2975–2982 (1998).
Kornitzer, D., Sharf, R. & Kleinberger, T. Adenovirus E4orf4 protein induces PP2A-dependent growth arrest in S. cerevisiae and interacts with the anaphase promoting complex/cyclosome. J. Cell Biol. 154, 331–344 (2001).
Maoz, T., Koren, R., Ben-Ari, I. & Kleinberger, T. YND1 interacts with CDC55 and is a novel mediator of E4orf4-induced toxicity. J. Biol. Chem. 280, 41270–41277 (2005).
Roopchand, D. E. et al. Toxicity of human adenovirus E4orf4 protein in Saccharomyces cerevisiae results from interactions with the Cdc55 regulatory B subunit of PP2A. Oncogene 20, 5279–5290 (2001).
Pechkovsky, A., Lahav, M., Bitman, E., Salzberg, A. & Kleinberger, T. E4orf4 induces PP2A- and Src-dependent cell-death in Drosophila melanogaster and at the same time inhibits classic apoptosis pathways. Proc. Natl Acad. Sci. USA 110, E1724–1733 (2013).
Shtrichman, R., Sharf, R., Barr, H., Dobner, T. & Kleinberger, T. Induction of apoptosis by adenovirus E4orf4 protein is specific to transformed cells and requires an interaction with protein phosphatase 2A. Proc. Natl Acad. Sci. USA 96, 10080–10085 (1999).
Kleinberger, T. & Shenk, T. Adenovirus E4orf4 protein binds to protein phosphatase 2A, and the complex down regulates E1A-enhanced junB transcription. J. Virol. 67, 7556–7560 (1993).
Shtrichman, R., Sharf, R. & Kleinberger, T. Adenovirus E4orf4 protein interacts with both Bα and B’ subunits of protein phosphatase 2A, but E4orf4-induced apoptosis is mediated only by the interaction with Bα. Oncogene 19, 3757–3765 (2000).
Yadav, A. K., Srikrishna, S. & Gupta, S. C. Cancer drug development using drosophila as an in vivo tool: From bedside to bench and back. Trends Pharmacol. Sci. 37, 789–806 (2016).
Sonoshita, M. & Cagan, R. L. Modeling human cancers in Drosophila. Curr. Top. Dev. Biol. 121, 287–309 (2017).
Markstein, M. et al. Systematic screen of chemotherapeutics in Drosophila stem cell tumors. Proc. Natl Acad. Sci. USA 111, 4530–4535 (2014).
Willoughby, L. F. et al. An in vivo large-scale chemical screening platform using Drosophila for anti-cancer drug discovery. Dis. Model Mech. 6, 521–529 (2013).
Gonzalez, C. Drosophila melanogaster: a model and a tool to investigate malignancy and identify new therapeutics. Nat. Rev. 13, 172–183 (2013).
Brumby, A. M. & Richardson, H. E. Using Drosophila melanogaster to map human cancer pathways. Nat. Rev. 5, 626–639 (2005).
Hariharan, I. K. & Bilder, D. Regulation of imaginal disc growth by tumor-suppressor genes in Drosophila. Annu. Rev. Genet. 40, 335–361 (2006).
Humbert, P. O. et al. Control of tumourigenesis by the Scribble/Dlg/Lgl polarity module. Oncogene 27, 6888–6907 (2008).
Pearson, H. B. et al. The polarity protein Scrib mediates epidermal development and exerts a tumor suppressive function during skin carcinogenesis. Mol. Cancer 14, 169 (2015).
Menendez, J., Perez-Garijo, A., Calleja, M. & Morata, G. A tumor-suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proc. Natl Acad. Sci. USA 107, 14651–14656 (2010).
Pagliarini, R. A. & Xu, T. A genetic screen in Drosophila for metastatic behavior. Science New York, NY 302, 1227–1231 (2003).
Read, R. D., Cavenee, W. K., Furnari, F. B. & Thomas, J. B. A drosophila model for EGFR-Ras and PI3K-dependent human glioma. PLoS Genet. 5, e1000374 (2009).
Callaerts, P. et al. Drosophila Pax-6/eyeless is essential for normal adult brain structure and function. J. Neurobiol. 46, 73–88 (2001).
Halder, G., Callaerts, P. & Gehring, W. J. Induction of ectopic eyes by targeted expression of the eyeless gene in Drosophila. Science New York, NY 267, 1788–1792 (1995).
Brumby, A. M. & Richardson, H. E. scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 22, 5769–5779 (2003).
Hirabayashi, S., Baranski, T. J. & Cagan, R. L. Transformed Drosophila cells evade diet-mediated insulin resistance through wingless signaling. Cell 154, 664–675 (2013).
Champagne, C., Landry, M. C., Gingras, M. C. & Lavoie, J. N. Activation of adenovirus type 2 early region 4 ORF4 cytoplasmic death function by direct binding to Src kinase domain. J. Biol. Chem. 279, 25905–25915 (2004).
Kumar, J. P. & Moses, K. EGF receptor and Notch signaling act upstream of Eyeless/Pax6 to control eye specification. Cell 104, 687–697 (2001).
Hay, B. A., Wolff, T. & Rubin, G. M. Expression of baculovirus P35 prevents cell-death in Drosophila. Development (Cambridge, England) 120, 2121–2129 (1994).
Gateff, E. & Schneiderman, H. A. Neoplasms in mutant and cultured wild-tupe tissues of Drosophila. Natl. Cancer Inst. Monogr. 31, 365–397 (1969).
Fernandez-Medarde, A. & Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2, 344–358 (2011).
Halaoui, R. & McCaffrey, L. Rewiring cell polarity signaling in cancer. Oncogene 34, 939–950 (2015).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501 (2014).
Ohsawa, S. et al. Elimination of oncogenic neighbors by JNK-mediated engulfment in Drosophila. Dev. Cell 20, 315–328 (2011).
Uhlirova, M., Jasper, H. & Bohmann, D. Non-cell-autonomous induction of tissue overgrowth by JNK/Ras cooperation in a Drosophila tumor model. Proc. Natl Acad. Sci. USA 102, 13123–13128 (2005).
Ruvolo, P. P. The broken “Off” switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA Clin. 6, 87–99 (2016).
Sangodkar, J. et al. Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J Clin. Invest. 127, 2081–2090 (2017).
Mazhar, S., Taylor, S. E., Sangodkar, J. & Narla, G. Targeting PP2A in cancer: Combination therapies. Biochim. Biophys. Acta Mol. Cell Res. 1866, 51–63 (2019).
Hein, A. L. et al. PR55alpha subunit of protein phosphatase 2A supports the tumorigenic and metastatic potential of pancreatic cancer cells by sustaining hyperactive oncogenic signaling. Cancer Res. 76, 2243–2253 (2016).
Li, S. et al. The adenovirus E4orf4 protein induces G2/M arrest and cell-death by blocking PP2A activity regulated by the B55 subunit. J. Virol. 83, 8340–8352 (2009).
Brestovitsky, A., Sharf, R., Mittelman, K. & Kleinberger, T. The adenovirus E4orf4 protein targets PP2A to the ACF chromatin-remodeling factor and induces cell-death through regulation of SNF2h-containing complexes. Nucl. Acids Res. 39, 6414–6427 (2011).
Branton, P. E. & Roopchand, D. E. The role of adenovirus E4orf4 protein in viral replication and cell killing. Oncogene 20, 7855–7865 (2001).
Lavoie, J. N., Champagne, C., Gingras, M.-C. & Robert, A. Adenovirus E4 open reading frame 4-induced apoptosis involves dysregulation of Src family kinases. J. Cell Biol. 150, 1037–1055 (2000).
Marcellus, R. C. et al. Induction of p53-independent apoptosis by the adenovirus E4orf4 protein requires binding to the Balpha subunit of protein phosphatase 2A. J. Virol. 74, 7869–7877 (2000).
Hobbs, G. A., Der, C. J. & Rossman, K. L. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 129, 1287–1292 (2016).
Acknowledgements
We are grateful to T. Xu, D. Builder, G. Halder, H. Stocker, and T. Uemura for gifts of fly stains. This research was supported in part by grants from the Israel Science Foundation and from the Rappaport Faculty of Medicine and Research Institute, Technion—Israel Institute of Technology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by R. Aqeilan
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rosen, H., Sharf, R., Pechkovsky, A. et al. Selective elimination of cancer cells by the adenovirus E4orf4 protein in a Drosophila cancer model: a new paradigm for cancer therapy. Cell Death Dis 10, 455 (2019). https://doi.org/10.1038/s41419-019-1680-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-019-1680-4
This article is cited by
-
Identification of the Potential Role of the E4orf4 Protein in Adenovirus A, B, C, and D Groups in Cancer Therapy: Computational Approaches
Molecular Biotechnology (2024)
-
Prospects for the use of viral proteins for the construction of chimeric toxins
Archives of Virology (2024)
