
Abstract
In the past two decades, significant progress has been made in the development of polygenic scores (PGSs). One specific application of PGSs is the development and potential use of pharmacogenomic- scores (PGx-scores) to identify patients who can benefit from a specific medication or are likely to experience side effects. This systematic review comprehensively evaluates published PGx-score studies in psychiatry and provides insights into their potential clinical use and avenues for future development. A systematic literature search was conducted across PubMed, EMBASE, and Web of Science databases until 22 August 2023. This review included fifty-three primary studies, of which the majority (69.8%) were conducted using samples of European ancestry. We found that over 90% of PGx-scores in psychiatry have been developed based on psychiatric and medical diagnoses or trait variants, rather than pharmacogenomic variants. Among these PGx-scores, the polygenic score for schizophrenia (PGSSCZ) has been most extensively studied in relation to its impact on treatment outcomes (32 publications). Twenty (62.5%) of these studies suggest that individuals with higher PGSSCZ have negative outcomes from psychotropic treatment — poorer treatment response, higher rates of treatment resistance, more antipsychotic-induced side effects, or more psychiatric hospitalizations, while the remaining studies did not find significant associations. Although PGx-scores alone accounted for at best 5.6% of the variance in treatment outcomes (in schizophrenia treatment resistance), together with clinical variables they explained up to 13.7% (in bipolar lithium response), suggesting that clinical translation might be achieved by including PGx-scores in multivariable models. In conclusion, our literature review found that there are still very few studies developing PGx-scores using pharmacogenomic variants. Research with larger and diverse populations is required to develop clinically relevant PGx-scores, using biology-informed and multi-phenotypic polygenic scoring approaches, as well as by integrating clinical variables with these scores to facilitate their translation to psychiatric practice.
Similar content being viewed by others


Introduction
Psychiatric disorders are significant contributors to the global disease burden and represent a major public health concern [1], highlighting the urgent need for effective prevention and treatment strategies [2]. The 2022 World Health Organization (WHO) report estimates that nearly a billion people suffer from psychiatric disorders, with an associated economic loss of $2 trillion per year and this figure is expected to rise to $6 trillion by 2030 [3,4,5,6].
Pharmacological treatments including antidepressants, antipsychotics, mood stabilizers, and anxiolytics are commonly prescribed for people suffering from psychiatric disorders [7]. However, the effectiveness of these medications varies between individuals, with some responding well while others do not show notable improvement or experience adverse effects [7]. For example, among patients with major depressive disorder (MDD), 30–40% fail to respond to the first-line pharmacological treatment options of selective serotonin reuptake inhibitors (SSRIs), and 10-–45% exhibit moderate to severe treatment-related side effects [8, 9]. Similarly, only 30% of patients with bipolar disorder (BD) show a full clinical response to first-line lithium monotherapy [10], and up to 25% of patients with first-episode schizophrenia (SCZ) are treatment-resistant to first-line antipsychotics [11]. This variability in pharmacological treatment outcomes can be attributed to the complex interplay of genetic and environmental factors, including patients’ clinical characteristics (e.g., severity, number, and duration of illness episodes), as well as sociodemographic variables [12]. For example, in individuals with MDD, genetic factors account for 42–52% of the observed differences in antidepressant treatment response, while environmental factors contribute to the remainder [13, 14].
To date, studies employing both candidate gene investigations (pharmacogenetics) and genome-wide (pharmacogenomic) approaches, have successfully pinpointed genetic variations associated with treatment outcomes in psychiatry, including response [15], remission [16], resistance [17] and adverse drug reactions [18]. For instance, the pharmacogenetic approach has uncovered genetic polymorphisms within genes encoding drug-metabolizing enzymes including those involved in the metabolism of various psychotropic drugs (e.g., CYP2D6 and CYP2C19) [19] as well as drug transporters (e.g., 5-HTTLPR), establishing their association with patients’ responses to medications [20]. This evidence is now incorporated into commercially available pharmacogenetic testing panels, aiding drug selection and dose adjustments and ultimately aiming at improving medication efficacy and tolerability [21, 22]. Similarly, the pharmacogenomics approach has revealed a number of genetic polymorphisms located within or near pharmacologically relevant candidate genes that influence individuals’ reaction to psychiatric medications [10]. For instance, Hou et al. identified four linked genetic variants on chromosome 21 associated with lithium response in a Genome-wide Association Study (GWAS) [10]. It has been challenging, however, to translate these pharmacogenomic findings into clinical practice, mainly due to the small effect size of individual genetic variants on treatment outcomes, along with a limited understanding of gene function [23].
In an effort to improve effect estimates and make pharmacogenomic findings more clinically relevant, researchers have recently adopted polygenic score methods that combine the effect of multiple genetic variants across the genome and have developed pharmacogenomic scores (PGx-scores) [24, 25]. In this systematic review, we provide a detailed account of the research undertaken to date, and of the performance, shortfalls, and future recommendations for the development of PGx-scores for the personalisation of psychiatric care.
Methods
This systematic review adhered to the PRISMA updated guidelines 2020 [26] and was registered with the International Prospective Register of Systematic Reviews (PROSPERO) on February 9, 2023 (ID = CRD42023395404). The review protocol was prepared before commencement to ensure a transparent and standardized methodology.
Search strategy, inclusion, and exclusion criteria
The literature search was performed across three databases including PubMed, EMBASE, and Web of Science databases from January 1st, 2005 to 22nd August 2023, by using search string: ((“Polygenic score*“ OR “Polygenic risk score*“ OR “Risk profile score*“ OR “Genetic risk score*“ OR “Gene score*“ OR “Genetic score*“ OR polygenic* OR “Pharmacogenomic variants” OR “Pharmacogenomic testing” OR Pharmaco-omic* OR pharmacogeno* OR “Pharmacogenetics”) AND (“Antipsychotic agents” OR antipsycho* OR “Antidepressive agents” Antidepress* OR “Anti-anxiety agents” OR Anti-anxiet* OR Valproic acid OR Valproate OR Divalproate OR Divalproex OR Carbamazepine OR Oxcarbazepine OR Risperidone OR Gabapentin OR Lamotrigine OR Licarbazepine OR Pregabalin OR Tiagabine OR Zonisamide OR Lithium)).
Our search strategy included all original studies that developed PGx-score for drug-related phenotypes such as, drug dosage, therapeutic drug response, resistance, drug-induced side-effects, relapse or hospitalisation in psychiatry. We included studies that reported weighted PGx-score for the drug-related phenotypes mentioned above while excluding publications in languages other than English, conference abstracts, case reports, editorials, notes, and systematic reviews. NTS screened the studies for inclusion under the supervision of ATA. In the final step, all studies were imported into Endnote version 20, a reference manager software. Duplicate entries were removed, and the selection of studies was carried out based on the predetermined inclusion and exclusion criteria. Supplementary Table 1 provides details of the systematic search strategies and results in each database.
Data extraction and synthesis
NTS extracted data using a customised data extraction excel sheet format, under the supervision of ATA. This excel sheet included information on the authors’ characteristics, details of the drug outcomes, characteristics of the study cohort (such as base, target, and validation cohorts), number of variants included in the polygenic score (PGS), polygenic scoring methods, and association effect estimates. We summarized the extracted data in the supplementary table 2.
The “target cohorts” describe the cohorts where the PGS was developed and tested, while “discovery cohorts” refer to the cohorts utilized to create GWAS summary statistics. “Validation cohorts” are independent cohorts where the PGSs were validated. “Variance explained” measures the proportion of phenotype variance in which the PGS can account for in a predictive model assuming linear effects. Coefficient of effect estimates, standard error, and sample size were used to calculate odd ratios if not reported in the studies.
The results were organized thematically based on the psychiatric disorders that were studied, as well as the specific phenotypes investigated, including treatment response, treatment resistance, and drug-induced side effects. Supplementary Table 3 provides the definitions and detailed description of each study’s treatment outcome.
Quality assessment
The quality of included studies was assessed using a quality assessment form adapted from previously validated and published sources [27, 28]. The assessment criteria covered various aspects of the study design, such as the rationale and methods of PGS, power calculation, inclusion and exclusion criteria, basic characteristics of the study population, availability of validation cohort, type of analysis, correction for multiple testing, and consideration of confounders in the analysis. The quality assessment was conducted by NTS under the supervision of ATA.
Results
Our initial search identified a total of 4889 studies that were potentially relevant to the research topic. After removing 1586 duplicated publications, 3,303 articles remained for the title and abstract screening. Subsequently, 3175 studies were excluded during the initial title and abstract screening phase, leaving 127 articles for full-text review. Finally, 53 studies met the predetermined inclusion criteria and were included in the final synthesis. Figure 1 presents the flowchart of the step-by-step process of study selection with reasons for exclusion.

PRISMA flow diagram showing the steps of screening studies included in this systematic review. PGS Polygenic score, PGx Pharmacogenomics.
Quality assessment
Nearly three-quarters (39/53) of studies described the rationale for the selected polygenic scoring methods, and about 20% (10/53) of studies performed a power calculation. All studies reported the inclusion and exclusion criteria for participants’ selection. Only fourteen studies used external cohorts to validate their findings. Correction for multiple testing was performed in 83.2% (44/53) of studies. Detailed results of the quality assessment are provided in the supplementary table 4.
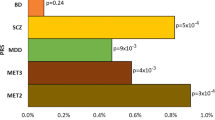
Most studies 37 (69.8%) were conducted on samples comprising individuals of European ancestry. Eleven studies (20.8%) included participants from other ancestries, such as African, African American and/or East Asian. Three studies targeted only Latin American participants and another two studies were conducted specifically on samples of East Asian ancestry. However, there was no study solely centred on samples of African ancestry. A combined analysis of both the target and discovery samples showed that 14,893,321 (90%) of participants had European descent, with an increased trend over the years 2013–2023, both in the target (Fig. 2A) and discovery cohorts (Fig. 2B).

A Target cohort; B Discovery cohort.
The sample sizes across the studies varied widely, ranging from 44 participants [29] to 12,863 participants [30] with a median sample size of 863 in the target cohorts. Three major psychiatric conditions, namely SCZ, MDD, and BD, were the focus of the included studies. In the case of SCZ, nearly 80% (21/27) of studies investigated the association between PGS and response to second-generation antipsychotics (clozapine, risperidone, lurasidone, olanzapine, aripiprazole, quetiapine, ziprasidone, and perphenazine). About half of SCZ studies (13/27) exclusively analysed clozapine treatment outcome. Nearly three-quarters of the included studies involving patients with MDD (14/19) considered the relationship between PGS and SSRIs such as citalopram or escitalopram. Six out of seven included studies of patients with BD developed PGx-scores and examined their associations with lithium treatment response.
The characteristics of the included studies and summary of the findings are presented in Tables 1–3 and described in the following sections.
The association of pharmacogenomics scores with antipsychotics treatment outcomes
Of all PGx-scores reviewed, the polygenic loading for schizophrenia (PGSSCZ) has been extensively studied (22 publications) in relation to its influence on antipsychotics treatment outcomes (see Table 1 and Fig. 3). Among these, 17 studies revealed that individuals with a higher genetic load for SCZ had a poorer treatment outcome to antipsychotics [31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47] while the remaining studies did not find significant associations [48,49,50,51,52]. For example, in patients with SCZ, a negative correlation (r = -0.05 [95%CI: -0.09– -0.01]) was found between PGSSCZ and response to second-generation antipsychotics (olanzapine, aripiprazole, risperidone, quetiapine, haloperidol, ziprasidone, perphenazine) following 6 weeks treatment [35]. Kappel et al. [34] observed a positive correlation (β = 12.21; 95%CI: 4.81–19.62) between PGSSCZ and high clozapine dosing (>600 mg/day), suggesting that individuals with a higher PGSSCZ may require increased doses of clozapine to achieve effective treatment response [34]. The patients with a higher PGSSCZ were observed to have a 3.24 times higher likelihood of having a clozapine prescription compared with healthy control (RR = 3.24 [95%CI:2.76–3.81]), indicating the clinical relevance of using PGSSCZ to personalize psychotic treatment [33]. In patients treated with risperidone, those who had a higher PGSSCZ reported more depressive symptoms [44], and worsened positive and negative psychotic symptoms [45]. A higher PGSSCZ was associated with a poor response to olanzapine or risperidone OR = 1.43 [95%CI:1.19–1.67] [42] and an increase of one standard deviation in PGSSCZ was associated with an ~11% increase in the risk of developing treatment-resistant schizophrenia (TRS) (OR = 1.11 [95%CI: 1.00–1.24]) [43].

Green line represents the positive associations of pharmacogenomic scores with treatment outcomes; Gray line indicates negative associations between PGx-scores and treatment outcomes. A wider (thick) line represents a stronger association. MDD Major depressive disorders, ADHD Attention Deficit Hyperactivity Disorders, BMI Body mass index, CRP C-reactive Protein.
Moreover, a PGSSCZ was associated with antipsychotic treatment resistance and side-effects in patients with SCZ. Specifically, patients with a higher polygenic load for SCZ were 1.22 times [95%CI: 1.05–1.41; R2 = 2.03%] more likely to be resistant to clozapine [37], had 1.50 times [95%CI: 1.13–1.96; R2 = 1.70%] higher odds to experience resistance to other antipsychotics [40] and were more likely to develop antipsychotic-induced weight gain (AIWG) [31, 32].
In decile-based comparisons, patients in the top three PGSSCZ deciles had a 2.42 [95%CI: 1.35–3.49; R2 ~ 2.00%] times higher odds of poor response to various antipsychotic medications (olanzapine, risperidone, quetiapine, and clozapine) [36] and the odds of treatment resistance for those in the 8th PGSSCZ decile was 6.50 times [95%CI: 1.47–28.80] higher than for patients in the 1st decile [36]. Patients with a higher PGSSCZ had 1.48 times [95%CI: 1.10–1.97; R2 = 2.70%] higher odds of psychiatric hospitalizations and were hospitalized longer [39]. Interestingly, in a study by Okhuijsen-Pfeifer et al. [38], patients treated with clozapine who were in the highest PGSSCZ tertile group were 1.94-fold more likely to experience low (i.e., more favourable) symptom severity [95%CI: 1.33–2.81; R2 = 1.85%], compared to those in the lowest PGSSCZ tertile group [38].
Additionally, the polygenic scores for body mass index (BMI), cognitive function, BD, and cardiometabolic traits have been assessed for their potential to predict antipsychotic treatment outcomes. These studies showed statistically significant associations. For example, a higher genetic loading for general cognitive ability was associated with better cognitive function following antipsychotic treatment [53]. Patients with SCZ carrying a greater genetic load for higher BMI were at a higher risk of being resistant to clozapine treatment [54], while those with a higher genetic load for myocarditis were more likely to develop clozapine-induced myocarditis [41] and a higher genetic loading for myocardial infarction was associated with increased antipsychotic-induced QTc interval prolongation [46]. The higher genetic loading for QT interval was also significantly associated with QT interval prolongation in schizophrenic patients taking antipsychotics [47]. The polygenic loading for BD (PGSBD) was also found to be significantly associated with clozapine metabolic ratio [29]: a measure of how clozapine is metabolized within the body, which may impact treatment response or adverse effects. In patients with first-episode psychosis, higher genetic loadings for HDL, LDL, and total cholesterol predicted antipsychotic-induced metabolic disturbance [55].
It is important to note that in the majority of studies, polygenic scores were developed using disease-specific genetic variants, and pharmacogenomic variants were considered in only a few studies. Using pharmacogenomic variants, O’Connell and colleagues developed a PGx-score for clozapine resistance which was significantly associated with TRS, accounting for ~5.0% of the variance [54]. Table 1 provides a summary of the association findings between PGx-score and antipsychotic treatment outcomes in patients with schizophrenia.
The association of pharmacogenomic scores with antidepressants treatment outcomes
In patients with MDD, studies have revealed the association of polygenic scores for psychiatric disorders, personality traits, and physical illnesses with antidepressant treatment outcomes (see Table 2 and Fig. 3). For instance, a study by Pain et al. identified that a higher PGSSCZ has been associated with poorer response to antidepressants (OR = 0.97 [95%CI: 0.96–0.98; R2 ~ 0.01%]) [56]. In a study by the Genome-Based Therapeutic Drugs for Depression (GENDEP) investigators, the polygenic loading for MDD (PGSMDD) was significantly associated with response and remission to SSRIs and tricyclic antidepressants (TCAs) treatment, although the direction of association was not reported [57]. A study that assessed the relationship between PGS for various personality traits and response to SSRIs (citalopram, escitalopram, fluvoxamine) [58] found that a higher genetic loading for openness personality trait was associated with a better SSRIs treatment response after 8 weeks of treatment OR = 1.58 [95%CI, 1.10–2.90] while the PGS for neuroticism was negatively associated with SSRIs treatment response [58]. The negative association between PGS for neuroticism and antidepressant treatment outcome was also reported in Ward et al's study [59]. Genetic loading for cardiometabolic diseases has also shown associations with response to antidepressant treatment: Marshe et al. [60] used a PGS for cardioembolic stroke to predict response to venlafaxine, an antidepressant of the serotonin-norepinephrine reuptake inhibitors (SNRI) class, after 12 week treatment. They found that a one standard deviation increase in PGS for cardioembolic stroke was associated with a decreased probability of remission (OR = 0.63 [95%CI:0.48 to 0.83]) or worsened disease symptoms (Montgomery-Asberg Depression Rating Scale (MADRS), β = -5.51 [95%CI: -9.45– -1.57]) [60]. In a different study, individuals with the highest PGSs for coronary artery disease (4th quartile) had 0.53 times ([95%CI, 0.35–0.81]) less likelihood of experiencing favourable response to SSRIs (citalopram, escitalopram, fluvoxamine) compared to those in the 1st quartile [61]. Similarly, those with higher genetic loading for obesity (4th quartile) had a 0.53 times ([95%CI, 0.32–0.88]) lower likelihood of achieving a positive response to SSRIs treatment [61]. In individuals with MDD (n = 5218) treated with SSRIs (citalopram, escitalopram) or a TCA (nortriptyline), the PGS for educational attainment was positively associated with SSRI response [56]. In a cohort of patients with psychotic depression treated with sertraline and olanzapine for 36 weeks, those who had a higher polygenic loading for Alzheimer’s disease had a decreased likelihood of relapse (OR = 0.38; [95%CI: 0.18–0.80]) during the study period [62]. Higher PGS for chronic pain was negatively associated with treatment response to SSRIs, TCAs (mirtazapine), and SNRIs (desvenlafaxine) (OR = 0.95 [95%CI: 0.92–0.98]) [30], while a higher PGS for C-reactive protein (CRP) was associated with a better response to escitalopram (OR = 2.92 [95%CI: 1.30–6.49]), but worse response to nortriptyline [63]. Despite the reported significant association between PGx-scores for psychiatric conditions, Nohr et al. [64], Garcia-Gonzalez et al. [65], Li et al. [66], and Tansey et al. [67] did not find any significant association.
In contrast to the above studies in which PGx-scores were developed based on diseases or related phenotype-specific variants to predict antidepressants treatment outcomes, a few recent studies used pharmacogenomic variants to calculate PGx-scores, directly indexing treatment outcome phenotypes. In patients with psychotic depression treated with sertraline and olanzapine, those with a higher genetic loading for antidepressant remission and response had 1.95 times [95%CI: 1.20–3.17] higher odds of reaching remission after 36 weeks [62]. In a similar context, PGS for response to SSRIs (escitalopram, sertraline, venlafaxine) predicted antidepressant treatment response in patients with MDD [68]. A study by Guo et al. [69] utilized variants ranked by their strength of association with ketamine response, a glutamate-modulating antidepressant used in patients with Treatment-Resistant Depression (TRD), to predict scopolamine treatment response in patients with either MDD or BD who had a current major depressive episode [69]. Findings indicated that patients with higher genetic loadings for ketamine response had better responses to scopolamine, an emerging antidepressant with effects on acetylcholine (Ach) neurotransmission [69]. Table 2 provides a summary of the pharmacogenomics score with antidepressants treatment outcomes in patients with major depressive disorders.
The association of pharmacogenomic scores with lithium treatment response
Studies have also found the association of polygenic scores for psychiatric disorders or related phenotypes with lithium treatment response in patients with BD (see Table 3 and Fig. 3). For example, those with a low PGSMDD (first decile) were 1.54 times [95%CI: 1.18–2.01; R2 = 0.91%] more likely to respond favourably to lithium than those who had high MDD genetic loading (10th decile) [70]. A study by Amare et al. showed that a higher PGSSCZ was associated with poorer response to lithium (OR = 0.29 [95%CI: 0.12–0.70; R2 = 0.80]) [71]. Similarly, a higher PGS for ADHD was associated with an unfavourable lithium response (OR = 0.86 [95%CI: 0.77–0.95], R2 = 0.18) [72]. Further studies using the same dataset have shown that a combined analysis of the PGSs of multiple phenotypes and PGS with patients’ clinical data can improve the predictive capacity of polygenic models. For example, a meta-analysis of the association results of the PGSSCZ and PGSMDD provided improved response prediction compared to single disorder PGS [73]. By applying machine learning methods, the PGSSCZ and PGSMDD were combined with clinical data, which resulted in an explained variance of 13.7% in lithium treatment response [74]. In a recent study, lithium clearance, an essential parameter for maintaining therapeutic levels of lithium and adjusting dosage, was positively associated with the PGSs for BMI and estimated glomerular filtration rate (eGFR), while it was negatively associated with the PGSs blood urea nitrogen (BUN) [75]. In addition to the disease-specific polygenic scores mentioned above, a PGx-score was developed for the ConLi+Gen sample using pharmacogenomic variants of lithium response (Li+RPGS), which was then evaluated in both a hold-out subsample and a smaller independent replication cohort. This analysis revealed that individuals in the highest Li+RPGS decile were 3.47 times [95%CI: 2.22–5.47, R2 = 2.60] more responsive to lithium compared to those in the lowest PGS decile, and a linear relationship was observed across the various deciles [76]. Table 3 provides a summary of pharmacogenomics scores for lithium treatment outcomes.
Discussion
In psychiatry, pharmacogenomic scores (PGx-scores) are emerging as novel tools for predicting treatment outcomes such as response, remission, resistance, side effects, or hospitalization rates. While the bench-to-bedside translation of PGx-scores has not yet been achieved, a growing body of evidence indicates their potential clinical use for treatment personalization. In this systematic review, we describe the landscape of 53 PGx-score studies in clinical psychiatry. These PGx-scores have been developed either from genetic variants associated with psychiatric or medical diagnoses (the majority of studies); or from pharmacogenomic variants associated with treatment outcome phenotypes (a few recent studies). Findings from these studies showed that individual PGx-scores account for only a small amount of variance in treatment outcomes, thus, there is insufficient evidence to support their direct clinical transition. Any future efforts toward clinical implementation need to be complemented by additional clinical data and/or biological markers.
First, we found that over 90% of PGx-scores have been developed based on genetic variants of psychiatric or medical diagnoses (e.g., SCZ, MDD, BD, ADHD, coronary artery disease (CAD)) or phenotypes related to diagnoses (e.g., cognitive function, personality traits, educational attainment, CRP level, BMI). Among these, the PGSSCZ has been most extensively studied and has consistently shown an association with pharmacotherapeutic outcomes across drug classes, including antipsychotics, antidepressants, and lithium, explaining as much as 3.2% of interindividual variability in some treatment outcomes [42]. The consistent association of the PGSSCZ and treatment outcomes may be attributed to two factors. First, SCZ has a strong genetic basis with a heritability estimate of 80-85% [77] and it is possible that PGSSCZ captures a substantial amount of the phenotypic variance of the disorder. Previous studies have shown a direct correlation between a higher phenotypic heritability and a better predictive power of PGS [78]. Second, SCZ GWASs are well-powered, including cases and controls of diverse ancestral background [79, 80], leading to more accurate PGSs [81]. The size of GWAS discovery samples has been associated with better accuracy and predictive power of PGSs [81]. For example, the Psychiatric Genomics Consortium (PGC in 2009) found that common genetic variants explained only 3% of the total variance in risk to SCZ in a sample of 3322 individuals with SCZ and 3587 controls of European ancestry [82]. In a follow-up study (in 2014) with expanded sample size and diversity (36,989 cases, 113,075 controls, multiple cohorts of East Asian ancestral background), the variance explained by PGSSCZ substantially increased to around 18% [83, 84].
It is important to highlight that in most of the reviewed studies, high PGSSCZ was associated with poor treatment response [33,34,35, 38, 42, 44, 45, 56, 71, 73, 74], more treatment resistance [36, 37, 40, 43, 54], more antipsychotic-induced side effects [31, 32, 41] or more psychiatric hospitalizations [39]. A notable exception was a positive association with lower symptom burden in SCZ patients treated with clozapine [38]. A possible explanation is that high polygenic loadings for SCZ may index individuals with a higher neurodevelopmental contribution to mental disorder aetiology. Neurodevelopmental hypotheses are well established in SCZ; for instance, excessive synaptic pruning is linked to complement system genotype [85]. Psychosis prodrome and onset [86, 87] and TRS [88] have been linked to reduced brain volume and connectivity. These ‘hard-wired’ brain characteristics may be more difficult to influence therapeutically through first-line (e.g., non-clozapine) pharmacological strategies [88].
The review also identified polygenic associations between cardiometabolic disorders [60, 61], personality traits [58], and treatment outcomes. Higher PGSs for CAD, obesity, and neurotic personality were associated with poor response to antidepressants [58, 61], while a positive association was found with the PGS for openness personality [58]. This is possibly due to shared biological mechanisms, for example, a genetic overlap between major psychiatric disorders and cardiometabolic diseases [89,90,91,92], neuroticism [93], or openness personality traits [94] and also associated multimorbidity across these disorders [95] that might impact patients’ treatment outcomes. Personality traits have an impact on medication adherence, with neuroticism linked to non-adherence and openness to compliance [96]. These findings indicated that disease-related PGSs may help us understand underlying pathology and identify drug targets.
Second, from our review, it is clear that there is a major research gap regarding PGx-scores developed from pharmacogenomic variants [54, 62, 68, 69, 76]. The lack of these studies is associated with the limited availability of well-powered GWAS summary statistics on treatment outcomes (target sample) and challenges to collecting genetic and clinical data from patients of specific diagnoses treated with similar medications (discovery sample). Currently, large-scale GWASs leverage biobank datasets, where there is limited phenotyping on medication and missing standardized data on treatment outcomes. Although the current cohort sizes for PGx-score development are much smaller than those of large-scale diagnosis-based GWASs, promising initiatives are underway to achieve deeper phenotyping for medications such as lithium [97], clozapine [37, 98], and antidepressants [56]. For instance, the ConLi+Gen cohort, which aimed to study the genetics of lithium treatment response in individuals with BD, currently has a sample size of 2367 patients of European ancestry and 220 patients of Asian ancestry with current efforts underway for a larger more diverse cohort and more detailed phenotyping [97]. By expanding current efforts, there may be opportunities to develop PGx-scores with improved accuracy for clinical use.
The third finding from this review is that the PGx-score alone falls short of explaining adequate variance in treatment outcomes for clinical translation. Notably, the highest reported explained variance solely attributed to PGx-score, by leveraging genetic variants of TRS and BMI, was 5.6% in resistance to clozapine. To address this shortfall, the combination of PGx-scores with clinical data could potentially enhance clinical use. For instance, a study modelled PGSSCZ + PGSMDD with patients’ clinical characteristics using machine learning, was able to explain 13.7% of the variance in lithium treatment response [74]. A further example is a multimodal model combining PGS with sociodemographic, clinical, biomarkers and structural imaging to predict rehospitalization risk showed a negative predictive value of 81.57% compared with a PGS-only model (54.83%) [99]. Similarly, a study that modelled polygenic scores of SCZ, MDD, and BD, along with proxy DNA methylation data and clinical symptom variables showed good regression performance for the prediction of response to multiple antipsychotic drugs (ROC = 0.87 [95% CI: 0.87–0.88]) [35]. In patients with type 2 diabetes, combining PGS with clinical data such as smoking status, BMI, blood lipid levels, blood pressure, and the use of anti-hypertensive and lipid-lowering medications substantially improved the accuracy in classifying individuals into low-, moderate-, and high-risk categories for cardiovascular events to 83%, whereas accuracy was 58% with PGSs alone (29 optimized univariable PGS) [100]. It is evident from these studies that PGx-score can be clinically useful if prediction models are refined based on a combination of PGx-scores and clinical data.
Limitations
Some of the limitations of the present systematic review should be highlighted. First, the study participants of the included studies were predominantly drawn from European populations which limits the ability to apply the study’s conclusion to non-European populations and raises concerns about the generalizability of the findings to more diverse populations. Second, the inconsistent reporting of the polygenic model parameters across studies makes it challenging to compare PGx-score in predicting pharmacological treatment outcomes. Third, a significant portion of the included studies lack sufficient statistical power to draw conclusive results to the broader populations. Finally, the lack of a standard definition of pharmacological treatment outcomes, differences in participants’ characteristics, and the use of multiple medications across the different studies make it difficult to compare findings and to perform meta-analysis.
Where associations between PGx-scores and treatment outcomes were established, effect size estimates (betas, odds ratios, hazard ratios) and measures of explained variance (R2) varied widely. For instance, the R2 of PGx-score models for predicting resistance to clozapine treatment with PGSSCZ in TRS individuals ranged from 2.03% [37] to 5.62% [54]. Similarly, the reported odds ratios for clozapine response ranged from 1.94 [95%CI: 1.33–2.81] [38] to 6.50 [95%CI: 1.47–28.80] [36]. These inconsistent findings can partly be explained by phenotypic heterogeneity, evident in diverse definitions and measurements of treatment outcomes and by differences in the sample size of these studies. As an example, the definition of TRS and TRD varies widely across studies [37, 43, 61, 101,102,103,104]. Achieving uniformity in phenotype characterization and harmonizing assessments across studies would help improve the reliability of the PGx-score in treatment outcomes.
Variations in sample size can also affect the size of individual study effect estimates and their statistical significance. Studies with small target or discovery samples have limited statistical power to detect significant associations. Choi et al. have demonstrated that in a discovery cohort of 100,000 samples, 200–500 samples in the target cohort are requisite to achieve 80% power for predicting traits across a spectrum of heritability estimates (h2:0.11–0.23) in polygenic models [105]. Recruiting a sufficiently large and well-characterized sample of uniformly treated individuals is a common challenge in PGx-score studies [81, 106].
Future directions in pharmacogenomic scores research
While PGx-scores hold promise for predicting treatment outcomes, they currently account for only a small proportion of the variance in treatment outcomes. This systematic review highlights the lack of well-defined phenotypes and small sample sizes that limit our ability to adequately quantify the genetic complexity associated with medication response. In this context, the following future directions may improve the predictive capacity of the PGx-score and move us closer to their clinical utilization in psychiatry.
Biologically informed pharmacogenomic scores
Previous PGx-score studies have been developed based on conventional polygenic modelling approaches, where the effect of genetic variants across the entire genome are aggregated, without taking into account the biological significance of these variants on the phenotype of interest [84, 107]. A biology-informed polygenic score (B-PGS) model was introduced very recently as a novel approach to improve both the predictive capability and biological meaning of polygenic scores, while also reducing sequencing costs [108, 109]. For example, in a study to predict psychosis, a pathway-specific PGS that was restricted to genomic locations within “nervous system development” and “regulation of neuron differentiation”, explained a variance of 6.9% in the risk of psychosis, outperforming the conventional PGS where genome-wide SCZ variants accounted for only 3.7% [110]. Biology-informed polygenic score potentially increases the polygenic signal-to-noise ratio by excluding variants with little association with pharmacogenomic outcomes and also enhances the clinical interpretability of polygenic models by focusing on specific molecular pathways [111]. There is emerging evidence elsewhere in medicine that B-PGS may be useful for the identification of new drug targets, for instance, in inflammatory bowel disease [112].
Multi-trait pharmacogenomic score
By leveraging the genetic correlation between multiple phenotypes, the multi-trait PGS approach aggregates genetic information across traits with the aim to improve the prediction power of PGx-scores [113,114,115]. For example, in patients with BD, the polygenic scores of SCZ or MDD explained 0.80% [71] and 0.91% [70] of the variance in lithium response, respectively. Interestingly, combining the polygenic scores of SCZ and MDD resulted in a better model, with an explained variance of 1.85% in lithium treatment response [73], indicating that multi-trait PGS outperforms single-trait PGS.
Combining multimodal data and machine learning optimization
Researchers have begun to combine PGS with other data modalities, for example, with clinical and imaging data to improve model accuracy [74, 116]. Machine learning methods are progressively being adopted for the analysis of multimodal or complex data comprising PGx-scores, socio-demographic, behavioural and clinical information [117, 118]. This approach, exemplified in a few studies included in our review [35, 74], holds promising results for clinical translation. Nevertheless, replication of these complex studies is lacking and interpretation of machine learning algorithms could be difficult for clinicians, potentially limiting their acceptance [119, 120]. To overcome this barrier, data scientists and clinicians need to collaborate at an early stage of model development to ensure that these models are not only clinically useful but also calibrated and valid for local conditions and easily understandable for end users [121,122,123].
Validation of polygenic models
Given the complexity of pharmacogenomic models, current sampling issues and the associated risks of false discovery and poor generalizability across different populations, external replication and validation of these models is critical for future implementation [25, 124,125,126]. Only 26.4% of studies included in this systematic review employed external validation [34, 35, 37, 42, 44, 45, 54, 56, 60, 61, 68, 70, 76, 127].
Multi-ancestry pharmacogenomic score
Nearly 90% of samples in the target and discovery cohorts of studies included in our systematic review were of European descent. Genetic variations and their effect on treatment outcomes can vary significantly among different populations. Given the complex pattern of linkage disequilibrium (short genetic regions) and the significant difference in the frequency of genetic variants between populations, the PGx-score constructed from one ancestral cohort may have a lower prediction in another cohort [124, 128, 129]. For instance, in cardiovascular medicine, a Brazilian-specific warfarin PGx-score used in a warfarin dosing algorithm was more accurate in Brazil than the one developed in the European population [130]. Conversely, polygenic models that incorporate information from ancestrally diverse populations, improve prediction performance, particularly in underrepresented non-European populations [131,132,133,134,135]. Diverse sampling is required to develop and validate more generalizable and transferable PGx-scores across diverse populations [84, 129]. These limitations hamper the translation of research findings into clinical practice and raise health disparity concerns. Thus, improving diversity in pharmacogenomic research is an essential step in creating polygenic models with broader applications.
Clinical implications of pharmacogenomic score
While it is clear that further development is required to improve the accuracy of the PGx-scores, and alone they have low clinical utility, findings are advancing our knowledge of pharmacogenomics toward better personalization of treatment. For instance, the genetic loading for SCZ demonstrates some capability to stratify individuals based on lithium treatment response in BD [71, 73, 74] and clozapine dosage in individuals with TRS [34]. Drawing parallels from other disciplines, such as cardiovascular medicine, PGS for coronary artery disease has been used to reclassify patients from intermediate into high-risk categories translating into stronger statin use recommendations [136, 137]. Similarly, genome-wide PGS in cardiovascular research has identified individuals with a four-fold increased risk, prompting recommendations for aggressive cholesterol-lowering therapy [138]. Such evidence indicates that the polygenic scores have the potential to stratify patients, predict treatment outcomes, and inform therapeutic decision-making based on the genetic variation of population variation among different ancestral populations. Figure 4 shows the potential use of pharmacogenomic scores in precision psychiatry.

DNA Deoxyribonucleic acid.
Conclusions
In summary, this systematic review highlights that larger and more diverse target sample sizes, focussed on well-defined and standardized pharmacogenomic outcomes, with robust replication are required to optimize the development of PGx-scores. Currently, the variance explained by these models is too small for effective clinical translation. However, new techniques, such as B-PGS and the use of multivariate modelling combining multiple traits PGS with clinical data look promising to increase accuracy. Large-scale consortia focused on pharmacogenomics are required to improve sample size and diversity.
Data availability
The original contributions presented in this study are included in the manuscript or supplementary tables. Further inquiries can be directed to the corresponding author.
References
Cuijpers P, Javed A, Bhui K. The WHO world mental health report: a call for action. Br J Psychiatry. 2023;222:227–9.
GBD Mental Disorders Collaborators. Global, regional, and national burden of 12 mental disorders in 204 countries and territories, 1990–2019: a systematic analysis for the global burden of disease study 2019. Lancet Psychiatry. 2022;9:137–50.
Smith K. Mental health: a world of depression. Nature. 2014;515:181.
Patel V, Saxena S, Lund C, Thornicroft G, Baingana F, Bolton P, et al. The lancet commission on global mental health and sustainable development. Lancet. 2018;392:1553–98.
Health TLG. Mental health matters. Lancet Glob Health. 2020;8:e1352.
WHO. World mental health report: transforming mental health for all. Executive Summary. Geneva: WHO; 2022. https://www.reliefweb.int. Accessed 16 Jan 2022.
Nathan PE, Gorman JM. A guide to treatments that work. 4th ed. Oxford University Press; 2015.
Hirschfeld RM. Efficacy of SSRIs and newer antidepressants in severe depression: comparison with TCAs. J Clin Psychiatry. 1999;60:326–35.
Cipriani A, Furukawa TA, Salanti G, Chaimani A, Atkinson LZ, Ogawa Y, et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. Focus. 2018;16:420–9.
Hou L, Heilbronner U, Degenhardt F, Adli M, Akiyama K, Akula N, et al. Genetic variants associated with response to lithium treatment in bipolar disorder: a genome-wide association study. Lancet. 2016;387:1085–93.
Siskind D, Orr S, Sinha S, Yu O, Brijball B, Warren N, et al. Rates of treatment-resistant schizophrenia from first-episode cohorts: systematic review and meta-analysis. Br. J. Psychiatry. 2022;220:115–20.
McMahon FJ. Prediction of treatment outcomes in psychiatry–where do we stand. Dialogues Clin Neurosci. 2014;16:455–64.
Tansey KE, Guipponi M, Hu X, Domenici E, Lewis G, Malafosse A, et al. Contribution of common genetic variants to antidepressant response. Biol Psychiatry. 2013;73:679–82.
Crisafulli C, Fabbri C, Porcelli S, Drago A, Spina E, De Ronchi D, et al. Pharmacogenetics of antidepressants. Front Pharmacol. 2011;2:6.
Pardiñas AF, Owen MJ, Walters JTR. Pharmacogenomics: A road ahead for precision medicine in psychiatry. Neuron. 2021;109:3914–29.
Kang HJ, Kim KT, Yoo KH, Park Y, Kim JW, Kim SW, et al. Genetic markers for later remission in response to early improvement of antidepressants. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21144884.
Lally J, Gaughran F, Timms P, Curran SR. Treatment-resistant schizophrenia: current insights on the pharmacogenomics of antipsychotics. Pharmgenomics Pers Med. 2016;9:117–29.
Jeiziner C, Wernli U, Suter K, Hersberger KE, Meyer zu Schwabedissen HE. HLA‐associated adverse drug reactions‐scoping review. Clin Transl Sci. 2021;14:1648–58.
Bousman CA, Jaksa P, Pantelis C. Systematic evaluation of commercial pharmacogenetic testing in psychiatry: a focus on CYP2D6 and CYP2C19 allele coverage and results reporting. Pharmacogenet Genomics. 2017;27:387–93.
Licinio J, Wong ML. Pharmacogenomics of antidepressant treatment effects. Dialogues Clin Neurosci. 2011;13:63–71.
Fagerness J, Fonseca E, Hess GP, Scott R, Gardner KR, Koffler M, et al. Pharmacogenetic-guided psychiatric intervention associated with increased adherence and cost savings. Am J Manag Care. 2014;20:e146–56.
Relling MV, Dervieux T. Pharmacogenetics and cancer therapy. Nat Rev Cancer. 2001;1:99–108.
Lauschke VM, Ingelman-Sundberg M. Emerging strategies to bridge the gap between pharmacogenomic research and its clinical implementation. npj Genom Med. 2020. https://doi.org/10.1038/s41525-020-0119-2.
Crouch DJ, Bodmer WF. Polygenic inheritance, GWAS, polygenic risk scores, and the search for functional variants. Proc Natl Acad Sci. 2020;117:18924–33.
Lee JW, Aminkeng F, Bhavsar A, Shaw K, Carleton B, Hayden M, Ross C. The emerging era of pharmacogenomics: current successes, future potential, and challenges. Clin Genet. 2014;86:21–8.
Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. Int J Surg. 2021;88:105906.
Van Vugt L, Van den Reek J, Coenen M, de Jong E. A systematic review of pharmacogenetic studies on the response to biologics in patients with psoriasis. Br J Dermatol. 2018;178:86–94.
Meerman JJ, Ter Hark SE, Janzing JGE, Coenen MJH. The potential of polygenic risk scores to predict antidepressant treatment response in major depression: a systematic review. J Affect Disord. 2022;304:1–11.
Mayen-Lobo YG, Martinez-Magana JJ, Perez-Aldana BE, Ortega-Vazquez A, Genis-Mendoza AD, De Montellano D, et al. Integrative genomic-epigenomic analysis of clozapine-treated patients with refractory psychosis. Pharmaceuticals. 2021;14:16.
Campos AI, Ngo TT, Medland SE, Wray NR, Hickie IB, Byrne EM, et al. Genetic risk for chronic pain is associated with lower antidepressant effectiveness: converging evidence for a depression subtype. Aust N Z J Psychiatry. 2022;56:1177–86.
Yoshida K, Marshe VS, Elsheikh SSM, Maciukiewicz M, Tiwari AK, Brandl EJ, et al. Polygenic risk scores analyses of psychiatric and metabolic traits with antipsychotic-induced weight gain in schizophrenia: an exploratory study. Pharmacogenomics J. 2023. https://doi.org/10.1038/s41397-023-00305-y.
Muntane G, Vazquez-Bourgon J, Sada E, Martorell L, Papiol S, Bosch E, et al. Polygenic risk scores enhance prediction of body mass index increase in individuals with a first episode of psychosis. Eur Psychiatry. 2023;66:e28.
Lin BD, Pinzon-Espinosa J, Blouzard E, van der Horst MZ, Okhuijsen-Pfeifer C, van Eijk KR, et al. Associations between polygenic risk score loading, psychosis liability, and clozapine use among individuals with schizophrenia. JAMA Psychiatry. 2023;80:181–5.
Kappel DB, Legge SE, Hubbard L, Willcocks IR, O’Connell KS, Smith RL, et al. Genomic stratification of clozapine prescription patterns using schizophrenia polygenic scores. Biol Psychiatry. 2023;93:149–56.
Guo L-K, Su Y, Zhang Y-Y-N, Yu H, Lu Z, Li W-Q, et al. Prediction of treatment response to antipsychotic drugs for precision medicine approach to schizophrenia: randomized trials and multiomics analysis. Military Med Res. 2023;10:24.
Talarico F, Costa GO, Ota VK, Santoro ML, Noto C, Gadelha A, et al. Systems-level analysis of genetic variants reveals functional and spatiotemporal context in treatment-resistant schizophrenia. Mol Neurobiol. 2022;59:3170–82.
Pardinas AF, Smart SE, Willcocks IR, Holmans PA, Dennison CA, Lynham AJ, et al. Interaction testing and polygenic risk scoring to estimate the association of common genetic variants with treatment resistance in schizophrenia. JAMA Psychiatry. 2022;79:260–9.
Okhuijsen-Pfeifer C, van der Horst MZ, Bousman CA, Lin B, van Eijk KR, Ripke S, et al. Genome-wide association analyses of symptom severity among clozapine-treated patients with schizophrenia spectrum disorders. Transl Psychiatr. 2022;12:145.
Facal F, Arrojo M, Paz E, Paramo M, Costas J. Association between psychiatric hospitalizations of patients with schizophrenia and polygenic risk scores based on genes with altered expression by antipsychotics. Acta Psychiatr Scand. 2022;146:139–50.
Werner MCF, Wirgenes KV, Haram M, Bettella F, Lunding SH, Rodevand L, et al. Indicated association between polygenic risk score and treatment-resistance in a naturalistic sample of patients with schizophrenia spectrum disorders. Schizophr Res. 2020;218:55–62.
Lacaze P, Ronaldson KJ, Zhang EJ, Alfirevic A, Shah H, Newman L, et al. Genetic associations with clozapine-induced myocarditis in patients with schizophrenia. Transl Psychiatr. 2020;10:37.
Zhang JP, Robinson D, Yu J, Gallego J, Fleischhacker WW, Kahn RS, et al. Schizophrenia polygenic risk score as a predictor of antipsychotic efficacy in first-episode psychosis. Am J Psychiat. 2019;176:21–8.
Gasse C, Wimberley T, Wang Y, Mors O, Borglum A, Als TD, et al. Schizophrenia polygenic risk scores, urbanicity and treatment-resistant schizophrenia. Schizophr Res. 2019;212:79–85.
Santoro ML, Ota V, de Jong S, Noto C, Spindola LM, Talarico F, et al. Polygenic risk score analyses of symptoms and treatment response in an antipsychotic-naive first episode of psychosis cohort. Transl Psychiatr. 2018;8:174.
Li J, Yoshikawa A, Brennan MD, Ramsey TL, Meltzer HY. Genetic predictors of antipsychotic response to lurasidone identified in a genome wide association study and by schizophrenia risk genes. Schizophr Res. 2018;192:194–204.
Lu Z, Zhang Y, Yan H, Su Y, Guo L, Liao Y, et al. ATAD3B and SKIL polymorphisms associated with antipsychotic-induced QTc interval change in patients with schizophrenia: a genome-wide association study. Transl Psychiatry. 2022;12:56.
Hommers L, Scherf-Clavel M, Stempel R, Roth J, Falter M, Deckert J, et al. Antipsychotics in routine treatment are minor contributors to QT prolongation compared to genetics and age. J Psychopharmacol. 2021;35:1127–33.
Hettige NC, Cole CB, Khalid S, De Luca V. Polygenic risk score prediction of antipsychotic dosage in schizophrenia. Schizophrenia Res. 2016;170:265–70.
Wimberley T, Gasse C, Meier SM, Agerbo E, MacCabe JH, Horsdal HT. Polygenic Risk Score for Schizophrenia and Treatment-Resistant Schizophrenia. Schizophr Bull. 2017;43:1064–9.
Martin AK, Mowry B. Increased rare duplication burden genomewide in patients with treatment-resistant schizophrenia. Psychol Med. 2016;46:469–76.
Kowalec K, Lu Y, Sariaslan A, Song J, Ploner A, Dalman C, et al. Increased schizophrenia family history burden and reduced premorbid IQ in treatment-resistant schizophrenia: a Swedish National Register and Genomic Study. Mol Psychiatry. 2021;26:4487–95.
Morgenroth CL, Kleymann P, Ripke S, Awasthi S, Wagner E, Oviedo-Salcedo T, et al. Polygenetic risk scores and phenotypic constellations of obsessive-compulsive disorder in clozapine-treated schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2024;274:181–93.
Blackman RK, Dickinson D, Eisenberg DP, Gregory MD, Apud JA, Berman KF. Antipsychotic medication-mediated cognitive change in schizophrenia and polygenic score for cognitive ability. Schizophrenia Res Cognition. 2022;27:7.
O’Connell KS, Koch E, Lenk HC, Akkouh IA, Hindley G, Jaholkowski P, et al. Polygenic overlap with body-mass index improves prediction of treatment-resistant schizophrenia. Psychiatry Res. 2023;325:115217.
Segura ÀG, Martínez-Pinteño A, Gassó P, Rodríguez N, Bioque M, Cuesta MJ, et al. Metabolic polygenic risk scores effect on antipsychotic-induced metabolic dysregulation: a longitudinal study in a first episode psychosis cohort. Schizophr Res. 2022;244:101–10.
Pain O, Hodgson K, Trubetskoy V, Ripke S, Marshe VS, Adams MJ, et al. Identifying the common genetic basis of antidepressant response. Biol Psychiatry Glob Open Sci. 2022;2:115–26.
Gendep Investigators, Investigators M, Investigators SD. Common genetic variation and antidepressant efficacy in major depressive disorder: a meta-analysis of three genome-wide pharmacogenetic studies. Am J Psychiatry. 2013;170:207–17.
Amare AT, Schubert KO, Tekola-Ayele F, Hsu YH, Sangkuhl K, Jenkins G, et al. Association of the polygenic scores for personality traits and response to selective serotonin reuptake inhibitors in patients with major depressive disorder. Front Psychiatry. 2018;9:65.
Ward J, Graham N, Strawbridge RJ, Ferguson A, Jenkins G, Chen W, et al. Polygenic risk scores for major depressive disorder and neuroticism as predictors of antidepressant response: meta-analysis of three treatment cohorts. PloS one. 2018;13:e0203896.
Marshe VS, Maciukiewicz M, Hauschild AC, Islam F, Qin L, Tiwari AK, et al. Genome-wide analysis suggests the importance of vascular processes and neuroinflammation in late-life antidepressant response. Transl Psychiatr. 2021;11:127.
Amare AT, Schubert KO, Tekola-Ayele F, Hsu YH, Sangkuhl K, Jenkins G, et al. The association of obesity and coronary artery disease genes with response to SSRIs treatment in major depression. J Neural Transm. 2019;126:35–45.
Men X, Marshe V, Elsheikh SS, Alexopoulos GS, Marino P, Meyers BS, et al. Genomic investigation of remission and relapse of psychotic depression treated with sertraline plus olanzapine: the STOP-PD II study. Neuropsychobiology. 2023;82:168–78.
Zwicker A, Fabbri C, Rietschel M, Hauser J, Mors O, Maier W, et al. Genetic disposition to inflammation and response to antidepressants in major depressive disorder. J Psychiatric Res. 2018;105:17–22.
Nøhr AK, Forsingdal A, Moltke I, Howes OD, Vitezic M, Albrechtsen A, Dalby M. Polygenic heterogeneity in antidepressant treatment and placebo response. Transl Psychiatry. 2022;12:456.
García-González J, Tansey KE, Hauser J, Henigsberg N, Maier W, Mors O, et al. Pharmacogenetics of antidepressant response: A polygenic approach. Prog Neuropsychopharmacol Biol Psychiatry. 2017;75:128–34.
Li QS, Wajs E, Ochs-Ross R, Singh J, Drevets WC. Genome-wide association study and polygenic risk score analysis of esketamine treatment response. Sci Rep. 2020;10:12649.
Tansey KE, Guipponi M, Domenici E, Lewis G, Malafosse A, O’Donovan M, et al. Genetic susceptibility for bipolar disorder and response to antidepressants in major depressive disorder. Am J Med Genet B Neuropsychiatr Genet. 2014;165b:77–83.
Meijs H, Prentice A, Lin BD, De Wilde B, Van Hecke J, Niemegeers P, et al. A polygenic-informed approach to a predictive EEG signature empowers antidepressant treatment prediction: a proof-of-concept study. Eur Neuropsychopharmacol. 2022;62:49–60.
Guo W, Machado-Vieira R, Mathew S, Murrough JW, Charney DS, Gruenbaum M, et al. Exploratory genome-wide association analysis of response to ketamine and a polygenic analysis of response to scopolamine in depression. Transl Psychiatry. 2018;8:9.
Amare AT, Schubert KO, Hou L, Clark SR, Papiol S, Cearns M, et al. Association of polygenic score for major depression with response to lithium in patients with bipolar disorder. Mol Psychiatr. 2021;26:2457–70.
Amare AT, Schubert KO, Hou L, Clark SR, Papiol S, et al. Association of polygenic score for schizophrenia and HLA antigen and inflammation genes with response to lithium in bipolar affective disorder: a genome-wide association study. JAMA. 2018;75:65–74.
Coombes BJ, Millischer V, Batzler A, Larrabee B, Hou L, Papiol S, et al. Association of attention-deficit/hyperactivity disorder and depression polygenic scores with lithium response: a consortium for lithium genetics study. Complex Psychiatry. 2021;7:80–9.
Schubert KO, Thalamuthu A, Amare AT, Frank J, Streit F, Adl M, et al. Combining schizophrenia and depression polygenic risk scores improves the genetic prediction of lithium response in bipolar disorder patients. Transl Psychiatr. 2021;11:606.
Cearns M, Amare AT, Schubert KO, Thalamuthu A, Frank J, Streit F, et al. Using polygenic scores and clinical data for bipolar disorder patient stratification and lithium response prediction: machine learning approach—CORRIGENDUM. Br J Psychiatry. 2022;221:494.
Millischer V, Matheson GJ, Bergen SE, Coombes BJ, Ponzer K, Wikström F, et al. Improving lithium dose prediction using population pharmacokinetics and pharmacogenomics: a cohort genome-wide association study in Sweden. Lancet Psychiatry. 2022;9:447–57.
Amare AT, Thalamuthu A, Schubert KO, Fullerton JM, Ahmed M, Hartmann S, et al. Association of polygenic score and the involvement of cholinergic and glutamatergic pathways with lithium treatment response in patients with bipolar disorder. Mol Psychiatry. 2023. https://doi.org/10.21203/rs.3.rs-2580252/v1.
Cardno AG, Gottesman II. Twin studies of schizophrenia: from bow‐and‐arrow concordances to star wars Mx and functional genomics. Am J Med Genetics. 2000;97:12–17.
Yang J, Zeng J, Goddard ME, Wray NR, Visscher PM. Concepts, estimation and interpretation of SNP-based heritability. Nat Genetics. 2017;49:1304–10.
Trubetskoy V, Pardiñas AF, Qi T, Panagiotaropoulou G, Awasthi S, Bigdeli TB, et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature. 2022;604:502–8.
Ripke S, Neale BM, Corvin A, Walters JTR, Farh K-H, Holmans PA, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7.
Dudbridge F. Power and predictive accuracy of polygenic risk scores. PLoS genetics. 2013;9:e1003348.
Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–52.
Pantelis C, Papadimitriou GN, Papiol S, Parkhomenko E, Pato MT, Paunio T, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7.
Duncan L, Shen H, Gelaye B, Meijsen J, Ressler K, Feldman M, et al. Analysis of polygenic risk score usage and performance in diverse human populations. Nat Commun. 2019;10:3328.
Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–83.
Satterthwaite TD, Wolf DH, Calkins ME, Vandekar SN, Erus G, Ruparel K, et al. Structural brain abnormalities in youth with psychosis spectrum symptoms. JAMA Psychiatry. 2016;73:515–24.
Vissink CE, Winter-van Rossum I, Cannon TD, Fusar-Poli P, Kahn RS, Bossong MG. Structural brain volumes of individuals at clinical high risk for psychosis: a meta-analysis. Biol Psychiatry Glob Open Sci. 2022;2:147–52.
Mouchlianitis E, McCutcheon R, Howes OD. Brain-imaging studies of treatment-resistant schizophrenia: a systematic review. Lancet Psychiatry. 2016;3:451–63.
Hagenaars SP, Coleman JRI, Choi SW, Gaspar H, Adams MJ, Howard DM, et al. Genetic comorbidity between major depression and cardio-metabolic traits, stratified by age at onset of major depression. Am J Med Genet B Neuropsychiatr Genet. 2020;183:309–30.
Amare AT, Schubert KO, Klingler-Hoffmann M, Cohen-Woods S, Baune BT. The genetic overlap between mood disorders and cardiometabolic diseases: a systematic review of genome wide and candidate gene studies. Transl psychiatry. 2017;7:e1007.
Willer C, Speliotes E, Loos R, Li S, Lindgren C, Heid I, et al. Genetic investigation of anthropometric traits consortium six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25.
Maccarrone G, Ditzen C, Yassouridis A, Rewerts C, Uhr M, Uhlen M, et al. Psychiatric patient stratification using biosignatures based on cerebrospinal fluid protein expression clusters. J Psychiatric Res. 2013;47:1572–80.
Okbay A, Baselmans BML, De Neve J-E, Turley P, Nivard MG, Fontana MA, et al. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat Genetics. 2016;48:624–33.
Lo MT, Hinds DA, Tung JY, Franz C, Fan CC, Wang Y, et al. Genome-wide analyses for personality traits identify six genomic loci and show correlations with psychiatric disorders. Nat Genet. 2017;49:152–6.
Hayward M, Moran P. Comorbidity of personality disorders and mental illnesses. Psychiatry. 2008;7:102–4.
Kohli R. A systematic review to evaluate the association between medication adherence and personality traits. Value Health. 2017;20:A686.
Schulze TG, Alda M, Adli M, Akula N, Ardau R, Bui ET, et al. The International Consortium on Lithium Genetics (ConLiGen): an initiative by the NIMH and IGSLI to study the genetic basis of response to lithium treatment. Neuropsychobiology. 2010;62:72–8.
Pardiñas AF, Kappel DB, Roberts M, Tipple F, Shitomi-Jones LM, King A, et al. Pharmacokinetics and pharmacogenomics of clozapine in an ancestrally diverse sample: a longitudinal analysis and genome-wide association study using UK clinical monitoring data. Lancet Psychiatry. 2023;10:209–19.
Cearns M, Opel N, Clark S, Kaehler C, Thalamuthu A, Heindel W, et al. Predicting rehospitalization within 2 years of initial patient admission for a major depressive episode: a multimodal machine learning approach. Transl Psychiatr. 2019;9:285.
Dziopa K, Chaturvedi N, Vugt M, Gratton J, Maclean R, Hingorani A, et al. Combining stacked polygenic scores with clinical risk factors improves cardiovascular risk prediction in people with type 2 diabetes. medRxiv 2022. https://doi.org/10.1101/2022.09.01.22279477.
Fanelli G, Domschke K, Minelli A, Gennarelli M, Martini P, Bortolomasi M, et al. A meta-analysis of polygenic risk scores for mood disorders, neuroticism, and schizophrenia in antidepressant response. Eur Neuropsychopharmacol. 2022;55:86–95.
Taylor RW, Coleman JRI, Lawrence AJ, Strawbridge R, Zahn R, Cleare AJ. Predicting clinical outcome to specialist multimodal inpatient treatment in patients with treatment resistant depression. J Affect Disord. 2021;291:188–97.
Fanelli G, Benedetti F, Kasper S, Zohar J, Souery D, Montgomery S, et al. Higher polygenic risk scores for schizophrenia may be suggestive of treatment non-response in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2021;108:110170.
Wigmore EM, Hafferty JD, Hall LS, Howard DM, Clarke TK, Fabbri C, et al. Genome-wide association study of antidepressant treatment resistance in a population-based cohort using health service prescription data and meta-analysis with GENDEP. Pharmacogenomics J. 2020;20:329–41.
Choi SW, Mak TS, O’Reilly PF. Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc. 2020;15:2759–72.
Blagec K, Swen JJ, Koopmann R, Cheung K-C, Crommentuijn-van Rhenen M, Holsappel I, et al. Pharmacogenomics decision support in the U-PGx project: results and advice from clinical implementation across seven European countries. PloS one. 2022;17:e0268534.
Wand H, Lambert SA, Tamburro C, Iacocca MA, O’Sullivan JW, Sillari C, et al. Improving reporting standards for polygenic scores in risk prediction studies. Nature. 2021;591:211–9.
Hari Dass SA, McCracken K, Pokhvisneva I, Chen LM, Garg E, Nguyen TTT, et al. A biologically-informed polygenic score identifies endophenotypes and clinical conditions associated with the insulin receptor function on specific brain regions. EBioMedicine. 2019;42:188–202.
Scharfe CPI, Tremmel R, Schwab M, Kohlbacher O, Marks DS. Genetic variation in human drug-related genes. Genome Med. 2017;9:117.
Tubbs JD, Leung PBM, Zhong Y, Zhan N, Hui TCK, Ho KKY, et al. Pathway-specific polygenic scores improve cross-ancestry prediction of psychosis and clinical outcomes. medRxiv 2023. https://doi.org/10.1101/2023.09.01.23294957.
Bennett D, O’Shea D, Ferguson J, Morris D, Seoighe C. Controlling for background genetic effects using polygenic scores improves the power of genome-wide association studies. Sci Reports. 2021;11:19571.
Bodea CA, Macoritto M, Liu Y, Zhang W, Karman J, King EA, et al. Pathway specific polygenic risk scores identify pathways and patient clusters associated with inflammatory bowel disease risk, severity and treatment response. medRxiv. 2021. https://doi.org/10.1101/2023.09.01.23294957.
Krapohl E, Patel H, Newhouse S, Curtis CJ, von Stumm S, Dale PS, et al. Multi-polygenic score approach to trait prediction. Mol Psychiatr. 2018;23:1368–74.
Maier RM, Zhu Z, Lee SH, Trzaskowski M, Ruderfer DM, Stahl EA, et al. Improving genetic prediction by leveraging genetic correlations among human diseases and traits. Nat commun. 2018;9:989.
Albiñana C, Zhu Z, Schork AJ, Ingason A, Aschard H, Brikell I, et al. Multi-PGS enhances polygenic prediction by combining 937 polygenic scores. Nat Commun. 2023;14:4702.
Wang M, Hu K, Fan L, Yan H, Li P, Jiang T, Liu B. Predicting treatment response in schizophrenia with magnetic resonance imaging and polygenic risk score. Front Genet. 2022;13:848205.
Zou J, Huss M, Abid A, Mohammadi P, Torkamani A, Telenti A. A primer on deep learning in genomics. Nat Genetics. 2019;51:12–8.
Eraslan G, Avsec Ž, Gagneur J, Theis FJ. Deep learning: new computational modelling techniques for genomics. Nat Rev Genetics. 2019;20:389–403.
Wagner MW, Namdar K, Biswas A, Monah S, Khalvati F, Ertl-Wagner BB. Radiomics, machine learning, and artificial intelligence—what the neuroradiologist needs to know. Neuroradiology 2021;63:1957–67.
Ngiam KY, Khor W. Big data and machine learning algorithms for health-care delivery. Lancet Oncology. 2019;20:e262–73.
Cearns M, Hahn T, Baune BT. Recommendations and future directions for supervised machine learning in psychiatry. Transl Psychiatry. 2019;9:271.
Winter NR, Cearns M, Clark SR, Leenings R, Dannlowski U, Baune BT, Hahn T. From multivariate methods to an AI ecosystem. Mol Psychiatry. 2021;26:6116–20.
Jarrett D, Stride E, Vallis K, Gooding MJ. Applications and limitations of machine learning in radiation oncology. Br J Radiol. 2019;92:20190001.
Mostafavi H, Harpak A, Agarwal I, Conley D, Pritchard JK, Przeworski M. Variable prediction accuracy of polygenic scores within an ancestry group. elife. 2020;9:e48376.
Kerminen S, Martin AR, Koskela J, Ruotsalainen SE, Havulinna AS, Surakka I, et al. Geographic variation and bias in the polygenic scores of complex diseases and traits in Finland. Am J Hum Genetics. 2019;104:1169–81.
Moons KG, Kengne AP, Woodward M, Royston P, Vergouwe Y, Altman DG, et al. Risk prediction models: I. Development, internal validation, and assessing the incremental value of a new (bio) marker. Heart. 2012;98:683–90.
Maciukiewicz M, Tiwari AK, Zai CC, Gorbovskaya I, Laughlin CP, Nurmi EL, et al. Genome-wide association study on antipsychotic-induced weight gain in Europeans and African-Americans. Schizophrenia Res. 2019;212:204–12.
Kuchenbaecker K, Telkar N, Reiker T, Walters RG, Lin K, Eriksson A, et al. The transferability of lipid loci across African, Asian and European cohorts. Nat Commun. 2019;10:4330.
Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genetics. 2019;51:584–91.
Perini J, Struchiner C, Silva‐Assunção E, Santana I, Rangel F, Ojopi E, et al. Pharmacogenetics of warfarin: development of a dosing algorithm for Brazilian patients. Clin Pharmacol Therapeutics. 2008;84:722–8.
Weissbrod O, Kanai M, Shi H, Gazal S, Peyrot WJ, Khera AV et al. Leveraging fine-mapping and non-European training data to improve cross-population polygenic risk scores. MedRxiv 2021. https://doi.org/10.1101/2021.01.19.21249483.
Márquez‐Luna C, Loh PR, Consortium SATD, Consortium STD, Price AL. Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genetic Epidemiol. 2017;41:811–23.
Weissbrod O, Kanai M, Shi H, Gazal S, Peyrot WJ, Khera AV, et al. Leveraging fine-mapping and multipopulation training data to improve cross-population polygenic risk scores. Nat Genetics. 2022;54:450–8.
Ruan Y, Lin Y-F, Feng Y-CA, Chen C-Y, Lam M, Guo Z, et al. Improving polygenic prediction in ancestrally diverse populations. Nat Genetics. 2022;54:573–80.
Patel AP, Wang M, Ruan Y, Koyama S, Clarke SL, Yang X, et al. A multi-ancestry polygenic risk score improves risk prediction for coronary artery disease. Nat Med. 2023;29:1793–803.
Kullo IJ, Jouni H, Austin EE, Brown S-A, Kruisselbrink TM, Isseh IN, et al. Incorporating a genetic risk score into coronary heart disease risk estimates: effect on low-density lipoprotein cholesterol levels (the MI-GENES clinical trial). Circulation. 2016;133:1181–8.
Natarajan P, Young R, Stitziel NO, Padmanabhan S, Baber U, Mehran R, et al. Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation. 2017;135:2091–101.
Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–24.
Acknowledgements
We appreciate Dr. Muktar Beshir Ahmed and Dr. Tesfa Dejenie Habtewold for providing valuable feedback.
Funding
This work is supported by the National Health and Medical Research Council (NHMRC) Emerging Leadership (EL1) Investigator Grant to AT Amare (APP2008000) and the 2021 NHMRC-EU Collaborative Research Grant funding to KO Schubert, SR Clark, and AT Amare (APP2018165). NT Sharew is a recipient of the University of Adelaide Research Scholarship.
Author information
Authors and Affiliations
Contributions
NTS and ATA designed the study, and/or conducted the literature search and screening. They also performed data extraction, interpreted the results, and drafted the manuscript. ATA, SRC and KOS obtained funding and critically revised the manuscript for important intellectual content. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41398_2024_2998_MOESM1_ESM.docx
Complete search strategies for the association of pharmacogenomic polygenic scores and treatment outcomes in psychiatry practice
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sharew, N.T., Clark, S.R., Schubert, K.O. et al. Pharmacogenomic scores in psychiatry: systematic review of current evidence. Transl Psychiatry 14, 322 (2024). https://doi.org/10.1038/s41398-024-02998-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-024-02998-6
This article is cited by
-
A stratified treatment algorithm in psychiatry: a program on stratified pharmacogenomics in severe mental illness (Psych-STRATA): concept, objectives and methodologies of a multidisciplinary project funded by Horizon Europe
European Archives of Psychiatry and Clinical Neuroscience (2024)
