
Abstract
Epigenetic alternations concern heritable yet reversible changes in histone or DNA modifications that regulate gene activity beyond the underlying sequence. Epigenetic dysregulation is often linked to human disease, notably cancer. With the development of various drugs targeting epigenetic regulators, epigenetic-targeted therapy has been applied in the treatment of hematological malignancies and has exhibited viable therapeutic potential for solid tumors in preclinical and clinical trials. In this review, we summarize the aberrant functions of enzymes in DNA methylation, histone acetylation and histone methylation during tumor progression and highlight the development of inhibitors of or drugs targeted at epigenetic enzymes.
Similar content being viewed by others
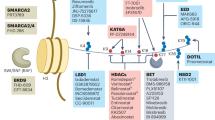
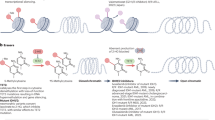
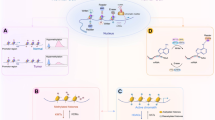
Introduction
After the discovery of DNA and the double helix structure, classic genetics has long assumed that the sequences of DNA determine the phenotypes of cells. DNA is packaged as chromatin in cells, with nucleosomes being the fundamental repeating unit. Four core histones (H2A, H2B, H3, and H4) form an octamer and are then surrounded by a 147-base-pair (bp) segment of DNA. Nucleosomes are separated by 10–60 bp DNA. Researchers have gradually found organisms that share the same genetic information but have different phenotypes, such as somatic cells from the same individual that share a genome but function completely differently. The term epigenetics was first proposed and established in 1942 when Conrad Waddington tried to interpret the connection between genotype and phenotype.1 Later, Arthur Riggs and his group interpreted epigenetics as inherited differences in mitosis and meiosis, which could explain the changes in phenotypes. They were both trying to find the link between genotype and phenotype. Epigenetics is usually referred to as a genomic mechanism that reversibly influences gene expression without altering DNA sequences. Holliday assumed that epigenetics was also mitotically and/or meiotically heritable without DNA sequence change. Aberrant DNA methylation could be repaired via meiosis, but some patterns are still transmitted to offspring.2 This phenomenon covers a wide range of cellular activities, such as cell growth, differentiation, and disease development, and is heritable.3 Generally, epigenetic events involve DNA methylation, histone modification, the readout of these modifications, chromatin remodeling and the effects of noncoding RNA. The elements involved in different modification patterns can be divided into three roles, “writer,” “reader,” and “eraser”. The “writers” and “erasers” refer to enzymes that transfer or remove chemical groups to or from DNA or histones, respectively. “Readers” are proteins that can recognize the modified DNA or histones (Fig. 1). To coordinate multiple biological processes, the epigenome cooperates with other regulatory factors, such as transcription factors and noncoding RNAs, to regulate the expression or repression of the genome. Epigenetics can also be influenced by cellular signaling pathways and extracellular stimuli. These effects are temporary and yet long-standing. Given the importance of epigenetics in influencing cell functions, a better understanding of both normal and abnormal epigenetic processes can help to understand the development and potential treatment of different types of diseases, including cancer.

Gene silencing in mammalian cells is usually caused by methylation of DNA CpG islands together with hypoacetylated and hypermethylated histones. The “writers” (DNMTs, HATs, and HMTs) and “erasers” (DNA-demethylating enzymes, HDACs, and KDMs) are enzymes responsible for transferring or removing chemical groups to or from DNA or histones; MBDs and other binding proteins are “readers” that recognize methyl-CpGs and modified histones. DNMTs, DNA methyltransferases; MBDs, methyl-CpG binding domain proteins; HATs, histone acetylases; HDACs, histone deacetylases; HMTs, histone methyltransferases; KDMs, histone-demethylating enzymes.
The etiology of cancer is quite complicated and involves both environmental and hereditary influences. In cancer cells, the alteration of genomic information is usually detectable. Like genome instability and mutation, epigenome dysregulation is also pervasive in cancer (Fig. 2). Some of the alterations determine cell function and are involved in oncogenic transformation.4 However, by reversing these mutations by drugs or gene therapy, the phenotype of cancer can revert to normal. Holliday proposed a theory that epigenetic changes are responsible for tumorigenesis. The alteration of cellular methylation status by a specific methyltransferase might explain the differences in the probability of malignant transformation.5 In clinical settings, we noticed that although cancer patients share the same staging and grade, they present totally different outcomes. In tumor tissues, different tumor cells show various patterns of histone modification, genome-wide or in individual genes, indicating that epigenetic heterogeneity exists at a cellular level.6 Likewise, using molecular biomarkers is thought to be a potential method to divide patients into different groups. It is important to note that tumorigenesis is the consequence of the combined action of multiple epigenetic events. For example, the repression of tumor suppressor genes is usually caused by methylation of DNA CpG islands together with hypoacetylated and hypermethylated histones.7 During gene silencing, several hallmarks of epigenetic events have been identified, including histone H3 and H4 hypoacetylation, histone H3K9 methylation, and cytosine methylation.8,9

Alterations in epigenetic modifications in cancer regulate various cellular responses, including cell proliferation, apoptosis, invasion, and senescence. Through DNA methylation, histone modification, chromatin remodeling, and noncoding RNA regulation, epigenetics play an important role in tumorigenesis. These main aspects of epigenetics present reversible effects on gene silencing and activation via epigenetic enzymes and related proteins. DNMTs, DNA methyltransferases; TETs, ten-eleven translocation enzymes; HATs, histone acetylases; HDACs, histone deacetylases; HMTs, histone methyltransferases; HDMs, histone-demethylating enzymes. MLL, biphenotypic (mixed lineage) leukemia.
Therefore, epigenetics enables us to investigate the potential mechanism underlying cancer phenotypes and provides potential therapy options. In this review, we focused and briefly expanded on three aspects of epigenetics in cancer: DNA methylation, histone acetylation and histone methylation. Finally, we summarized the current developments in epigenetic therapy for cancers.
DNA methylation
The DNA methylation pattern in mammals follows certain rules. Germ cells usually go through a stepwise demethylation to ensure global repression and suitable gene regulation during embryonic development. After implantation, almost all CpGs experience de novo methylation except for those that are protected.10 Normal dynamic changes in DNA methylation and demethylation based on altered expression of enzymes have been known to be associated with aging.11,12 However, inappropriate methylation of DNA can result in multiple diseases, including inflammatory diseases, precancerous lesions, and cancer.13,14,15 Of note, de novo methylation of DNA in cancer serves to prevent reactivation of repressed genes rather than inducing gene repression.16 Because researchers have found that over 90% of genes undergoing de novo methylation in cancer are already in a repressed status in normal cells.17 Nevertheless, aberrant DNA methylation is thought to serve as a hallmark in cancer development by inactivating gene transcription or repressing gene transcription and affecting chromatin stability.18
The precise mechanism by which DNA methylation affects chromatin structure unclear, but it is known that methyl-DNA is closely associated with a closed chromatin structure, which is relatively inactive.19 Hypermethylation of promoters and hypomethylation of global DNA are quite common in cancer. It is widely accepted that gene promoters, especially key tumor suppressor genes, are unmethylated in normal tissues and highly methylated in cancer tissues.20 P16, a tumor suppressor encoded by CDKN2A, has been found to gain de novo methylation in ~20% of different primary neoplasms.21 Mutations in important and well-studied tumor-suppressive genes, such as P53 and BRCA1, are frequently identified in multiple cancers.22,23,24 Studies have found that the level of methylation is positively associated with tumor size. In support of this, a whole-genome methylation array analysis in breast cancer patients found significantly increased CpG methylation in FES, P2RX7, HSD17B12, and GSTM2 coincident with increasing tumor stage and size.25 After analysis of long-range epigenetic silencing at chromosome 2q14.2, methylation of EN1 and SCTR, the first well-studied example of coordinated epigenetic modification, was significantly increased in colorectal and prostate cancers.26,27 EN1 methylation has also been observed to be elevated by up to 60% in human salivary gland adenoid cystic carcinoma.28 Of note, only ~1% of normal samples exhibited EN1 CpG island hypermethylation.26 Therefore, the significant difference between cancer cells and normal cells makes EN1 a potential cancer marker in diagnosis. In human pancreatic cancer, the APC gene, encoding a regulator of cell junctions, is hypermethylated by DNMT overexpression.29 During an analysis of colorectal disease methylation patterns, researchers found several genes that showed significant changes between precancerous diseases and cancers, including RUNX3, NEUROG1, CACNA1G, SFRP2, IGF2 DMR0, hMLH1, and CDKN2A.30 In the human colon cancer cell line HCT116, hMLH1 and CDKN2A always bear genetic mutation and hypermethylation of one allele, and this leads to inactivation of key tumor suppressors.31 It is known that p16, p15, and pax6 are usually aberrantly methylated in bladder cancer and show enhanced methylation in cell culture.32. Unlike gene promoter methylation, gene body methylation usually results in increased transcriptional activity.33 This process often occurs in CpG-poor areas and causes a base transition from C to T.34 The hypermethylation of specific CpG islands in cancer tissues is informative of mutations when the gene in normal tissues is unmethylated. One representative marker is glutathione S-transferase-π (GSTP1), which is still the most common alteration in human prostate cancer.35 Recently, DNA methylation in cancer has generally been associated with drug resistance and predicting response to treatment.36 For example, MGMT (O-6-methylguanine DNA methyltransferase) hypermethylation is still the best independent predictor of response to BCNU (carmustine) and temozolomide in gliomas because hypermethylation of MGMT makes tumor cells more sensitive to treatments and is associated with regression of tumor and prolonged overall survival.37,38 Similarly, MGMT is also a useful predictor of response to cyclophosphamide in diffuse large B-cell lymphoma39 (Table 1).
DNA methyltransferases (DNMTs)
DNA methylation is a covalent modification of DNA and is one of the best-studied epigenetic markers. It plays an important role in normal cell physiology in a programmed manner. The best-known type of DNA methylation is methylation of cytosine (C) at the 5th position of its carbon ring (5-mC), especially at a C followed by a guanine (G), so-called CpG sites. Non-CpG methylation, such as methylation of CpA (adenine) and CpT (thymine), is not common and usually has restricted expression in mammals.40 CpG islands traverse ~60% of human promoters, and methylation at these sites results in obvious transcriptional regression.41 Meanwhile, among the ~28 million CpGs in the human genome in somatic cells, 60–80% are methylated in a symmetric manner and are frequently found in promoter regions.42,43 The process of DNA methylation is regulated by the DNA methyltransferase (DNMT) family via the transfer of a methyl group from S-adenosyl-L-methionine (SAM) to cytosine.44 There are five members of the DNMT family: DNMT1, DNMT2, DNMT3a, DNMT3b, and DNMT3L. DNMT1 is responsible for the maintenance of methyl-DNA, recognizes hemimethylated DNA strands and regenerates the fully methylated DNA state of DNA during cell division.45 In a recent study, DNMT1 with Stella, a factor essential for female fertility, was responsible for the establishment of the oocyte methylome during early embryo development.46 DNMT3a and DNMT3b are regarded as de novo methylation enzymes that target unmethylated CpG dinucleotides and establish new DNA methylation patterns, but they have nonoverlapping functions during different developmental stages.47,48 DNMT2 and DNMT3L are not regarded as catalytically active DNA methyltransferases. DNMT2 functions as an RNA methyltransferase, while DNMT3L contains a truncated inactive catalytic domain and acts as an accessory partner to stimulate the de novo methylation activity of DNMT3A. The DNA methyltransferase-like protein DNMT3L can modulate DNMT3a activity as a stimulatory factor.49
During aberrant DNA methylation, DNMTs play an important role. Compared with DNMT1 and DNMT3a, DNMT3b was significantly overexpressed in tumor tissues.50 Overexpression of DNMT1, DNMT3a, and DNMT3b has been observed in multiple cancers, including AML, CML, glioma, and breast, gastric, colorectal, hepatocellular, pancreatic, prostate, and lung cancers. In cervical cancer patients, DNMT1 was expressed in more than 70% of cancer cells, whereas only 16% of normal cells expressed DNMT1. The higher level of DNMT1 expression was also associated with worse prognosis.51 The expression of DNMT1, DNMT3a, and DNMT3b has been observed to be elevated in acute myeloid leukemia (AML) and various solid cancers. These three methyltransferases do not show significant changes in the chronic phase of chronic myeloid leukemia (CML), but they are significantly increased during progression to the acute phase in CML.52,53 Notably, downregulation of DNMTs can also lead to tumorigenesis (Table 1).
Methyl-CpG recognition proteins
How DNA methylation leads to gene repression has been considered in many studies. Several hypotheses have been proposed. Three methyl-CpG binding domain protein (MeCP) families can read the established methylated DNA sequences and in turn recruit histone deacetylases, a group of enzymes responsible for repressive epigenetic modifications, to inhibit gene expression and maintain genome integrity.10,54 The first group is methyl-CpG binding domain (MBD) proteins, including MeCP2, MBD1, MBD2, and MBD4. MeCP1 is a complex containing MBD2, the histone deacetylase (HDAC) proteins HDAC1 and HDAC2, and the RbAp46 and RbAp48 proteins (also known as RBBP7 and RBBP4).55 MBD3 is unlike the other four family members and is not capable of binding to methylated DNA but instead binds to hydroxymethylated DNA.56 The zinc-finger and BTB domain-containing protein family is the second group and comprises three structurally different proteins, KAISO (ZBTB33), ZBTB4 and ZBTB38, which bind to methylated DNA via zinc-finger motifs. The third family includes two ubiquitin-like proteins with PHD and RING finger domains, UHRF1 and UHRF2, which recognize 5-mC via RING finger-associated (SRA) domains. On the other hand, methylation of DNA can also be a barrier for certain transcription factors to bind to promoter sites such as AP-2, c-Myc, CREB/ATF, E2F, and NF-kB.13
As for methyl-group binding proteins, many studies have investigated their roles in various cancers, but the mechanism underlying these alterations remains unclear. MBD proteins cooperate with other proteins to regulate gene transcription.57,58 However, the role of MBD1 and MBD2 has not been identified in human lung or colon cancer, with only limited mutations being detected.59 Furthermore, loss of MBD1 did not show any carcinogenic effect in MBD−/− mice.60 Compared with MBD1, MBD2 shows more effect on tumorigenesis. Deficiency of MBD2 strongly suppresses intestinal tumorigenesis in APCMin-background mice.61 A possible reason is that many important signaling pathways are downregulated in colorectal cancer, and loss of MBD2 leads to reexpression of these genes.62 Meanwhile, inhibition of MBD2 shows promising effects on suppression of the tumorigenesis of human lung cancer and colon cancer.63 Although MBD3 does not directly bind to methylated DNA, it regulates the methylation process via interactions with other proteins, such as MBD2 and HDAC. For example, application of an HDAC inhibitor in lung cancer cells upregulated p21 (also known as CDKN1A) and downregulated ErbB2, leading to inhibition of cancer cell growth. Silencing of MBD3 blocked the effects of an HDAC inhibitor.64 MBD3 and MBD2 form a complex, nucleosome remodeling and deacetylase (NuRD), which interacts with histone-demethylating enzymes to regulate gene expression in cancer.65 Mutation of MBD4 has been found in colorectal cancer, endometrial carcinoma and pancreatic cancer.66 Furthermore, this mutation unexpectedly affects the stability of the whole genome, not only CpG sites.67 Knockout of MBD4 indeed increased tumorigenesis in APCMin-background mice, which makes MBD4 a tumor suppressor.68 MBD4 is important in DNA damage repair, given the interaction between MBD4 and MMR.69 In contrast, the expression of MeCP2 and the UHRF family tends to promote tumor growth.70,71,72,73,74 In the KAISO family, KAISO directly binds to p120ctn, a protein with an alternative location in some cancer cells, and they together regulate cell adhesion and motility.75,76 However, deficiency of ZBTB4 contributes to tumorigenesis77 (Table 1).
DNA-demethylating enzymes
DNA methylation is a stable and highly conserved epigenetic modification of DNA in many organisms.78 However, loss of 5-mC and DNA demethylation have been identified in different biologic processes. For example, DNA demethylation is important for primordial germ cells (PGCs) to gain pluripotent ability.79,80 DNA demethylation is actively regulated by the TET protein family (ten-eleven translocation enzymes, TET1-3) via the removal of a methyl group from 5-mC. These three proteins differ from each other in terms of expression depending on the developmental stage and cell type.18 TETs oxidize 5-mC in an iterative manner and catalyze the conversion of 5-mC to 5-hydroxymethylcytosine (5-hmC), which is a key intermediate in the demethylation process.81 5-hmC, as a relatively stable intermediate substrate, is less prone to further oxidation by TET proteins than 5-mC.82 However, overexpression of only TET1 and TET2 can cause a global decrease of 5-mC.18 Stepwise oxidation of 5-hmC by TET proteins can yield two products: 5-formylcytosine (5-fC) and 5-carboxylcytosine (5-caC).83 These two molecules can be excised by thymine-DNA glycosylase (TDG) and eventually be repaired to unmodified C.84 DNA demethylation or restoration of the unmodified cytosine can also occur passively through replication-dependent dilution of 5-mC.
Disruption of normal DNA demethylation is thought to be associated with oncogenesis. TET proteins were initially associated with leukemia. Researchers have found that in a small number of AML patients, TET1 is fused to MLL via the chromosome translocation t(10;11)(q22;q23).85 Further studies found that TET2 was more widely expressed in different tissues than TET1 and TET3. Analyses revealed that mutation or deficiency of TET2 occurred in ~15% of patients with myeloid cancers, including myelodysplastic syndrome (MDS), myeloproliferative disorders, and AML.86 In patients with CML, mutation of TET2 has been detected in ~50% of patients.87 Although TET2 mutations have been found in several myeloid malignancies, their prognostic effect remains controversial. Based on the phenomenon that mutation of TET2 was elevated in patients whose disease transformed from chronic myeloid malignancy to AML, researchers considered that TET2 loss was important for cells to regain the ability to self-renew.88 The role of TET proteins has also been investigated in several solid tumors. Compared with surrounding normal tissues, 5-hmC is significantly reduced in human breast, liver, lung, pancreatic, and prostate cancers with reduced expression of TET family proteins.89 Deficiency of TET1 in prostate and breast cancer is associated with tumor cell invasion and breast xenograft tumor formation via the inhibition of the methylation of metalloproteinase (TIMP) family proteins 2 and 3.90 Loss of 5-hmC is an epigenetic hallmark of melanoma, and thus, introducing TET2 into melanoma cells results in suppression of tumor growth and increased survival in an animal model91 (Table 1).
Histone modification
Histone modification can occur to the flexible tails as well as the core domain of histones, including those sites that are buried by DNA. In particular, the flexible histone tails are enriched with basic Lys/Arg and hydroxyl group-containing Ser/Thr/Tyr residues, thereby being hotspots for hallmark histone modifications. The tails extend from the surface of the nucleosome and are readily modulated by covalent posttranslational modification (PTM). PTMs modify histones by adding or removing chemical groups and regulate many biological processes via the activation or inactivation of genes. These processes mainly include acetylation and methylation of lysines (K) and arginines (R), phosphorylation of serines (S) and threonines (T), ubiquitylation, and sumoylation of lysines. In addition to those mentioned and discussed above, histone modifications also include citrullination, ADP-ribosylation, deamination, formylation, O-GlcNAcylation, propionylation, butyrylation, crotonylation, and proline isomerization at over 60 amino acid residues.157,158 In addition to conventional PTMs, novel PTM sites are also found outside of the N-terminal tails.
Histone modifications at certain sites, such as promoters and enhancers, are thought to be largely invariant, whereas a small number of these sites remain dynamic. H3K4me1 and H3K27ac, two dynamic modifications, were identified to activate enhancers and regulate gene expression.159 H3K9ac and H3K9me3 are two common modifications at promoters.160,161 Appropriate histone modifications are important in gene expression and human biology; otherwise, alterations in PTMs may be associated with tumorigenesis. Analysis of cancer cells reveals that they exhibit aberrant histone modifications at individual genes or globally at the single-nuclei level.6,162 Understanding histone modification patterns in cancer cells can help us to predict and treat cancers. Thus far, most studies have focused on aberrant modifications within an individual site, such as H4K20me3 or H4K16ac, rather than enzymatic activity-associated abnormalities. Generally, alterations in histone modifications occur at an early stage and accumulate during tumorigenesis.163
Histone acetylation (lysine)
Histone acetylation occurs at multiple lysine residues at the N-terminus via the catalysis of histone acetyltransferases (HATs), also named lysine acetyltransferases (KATs). Histone acetylation regulates the compaction state of chromatin via multiple mechanisms, such as neutralizing the basic charge at unmodified lysine residues, and is associated with active transcription, especially at gene promoters and enhancers and the gene body; it also facilitates the recruitment of coregulators and RNA polymerase complexes to the locus.157,164 To date, HATs and histone deacetylases (HDACs) are the two of the best characterized groups of enzymes involved in histone PTMs. HATs transfer the acetyl groups from acetyl-CoA cofactors to lysine residues at histones, whereas the role of HDACs is the opposite, which makes histone acetylation a highly reversible process.
Histone acetyltransferases
HATs are predominantly located in the nucleus, but multiple lines of evidence have shown lysine acetylation in the cytoplasm, and their acetylation is associated with key cellular events.165 In addition, lysine acetylation found outside histones reminds us of the role of HATs in nonhistone PTMs.166 The first HAT was identified in yeast, and was named HAT1,167 and was then isolated from tetrahymena as HAT A by Allis and coworkers.168 In humans, HATs can be roughly divided into three groups: general control nondepressible 5 (GCN5)-related N-acetyl transferase (GNAT) (based on the protein Gcn5 found in yeast; including GCN5 and PCAF), MYST (based on the protein MOZ; including MOZ, MOF, TIP60, and HBO1), and p300/cAMP-responsive element-binding protein (CBP).169 Other HATs, including nuclear receptors and transcription factors, such as SRC1, MGEA5, ATF-2, and CLOCK, also harbor the ability to acetylate histones. Notably, a number of acetyltransferases also perform protein acetylation outside histones, such as TFIIB, MCM3AP, ESCO, and ARD1.170 Knockout of CBP/p300 is lethal for early embryonic mouse models.171,172 The acetyl group transfer strategies for each HAT subfamily are different. For the GCN5 and PCAF family, the protein crystal structure shows a conserved glutamate in the active site. Blockade of this amino leads to a significantly decreased acetylation function.173,174 Similarly, there is also a conserved glutamate plus a cysteine residue located at active sites of MYST family proteins.175 Unlike the other two families, the p300/CBP HAT subfamily has two other potential conserved residues, a tyrosine and a tryptophan.176 Generally, their catalytic mechanisms of acetyl group transfer can be divided into two groups. The GNAT family depends on a sequential ordered mechanism, whereas the members of the MYST family use a so-called ping-pong (i.e., double displacement) catalytic mechanism, which means that the acetyl groups are first transferred to a cysteine residue and then transferred to a lysine residue.177 In addition to differences in the acetyl transfer mechanism, HAT subfamilies, even different proteins in the same family, also have remarkable diversity in targeting sites.
Appropriate acetylation within cells is important since upregulation or downregulation of HATs is associated with tumorigenesis or poor prognosis.162,178 Compared with solid tumors, the association between histone modifications and cancer has been widely investigated in hematological malignancies. Germline mutation of CBP results in Rubinstein-Taybi syndrome along with an increased predisposition to childhood malignancies. Meanwhile, loss of another family member, p300, has also been associated with hematological malignancies.179,180 Therefore, both CBP and p300 seem to function as tumor suppressors. During cancer development, the expression of HAT genes can be disrupted by chromosomal translocations, although these are rare events. Generation of the fused protein CBP-MOZ is the result of the t(8,16)(p11,p13) translocation in AML.181 Translocation of t(10;16)(q22;p13) leads to the CBP-MORF chimera.182 Similarly, p300-MOZ, MLL-CBP, and MLL-p300 (MLL, mixed lineage leukemia) have also been identified in hematological malignancies.183,184,185 Generally, chromosomal rearrangements involving CBP are more common than those involving p300. Researchers have also investigated solid tumors, which are less mutated. The expression of translocated P300 in laryngeal squamous cell carcinoma (LSCC) tissue is much higher than that in adjacent normal tissue and is associated with advanced stage and poor prognosis.178 Missense point mutations in p300 are found in colorectal adenocarcinoma, gastric adenocarcinoma and breast cancer with quite low incidences.186,187 Rare inactivating mutations in CBP and PCAF have only been identified in cancer cell lines but not primary tumors.188 Based on these findings, we hypothesize that the differences between cell lines and primary tumors cannot be ignored. Amplified in breast cancer 1 (AIB1), also frequently called NCOA3 (nuclear receptor coactivator 3) or SRC3 (steroid receptor coactivator 3), is overexpressed in ~60% of human breast cancers, and increased levels of AIB1 are associated with tamoxifen resistance and decreased overall survival.189 Steroid receptor coactivator 1 (SRC1) is also associated with the chromosomal translocation t(2;2)(q35;p23), which results in PAX3–NCOA1 gene fusion in rhabdomyosarcoma without a consistent genetic abnormality during embryonic development190 (Table 2).
Acetyl-lysine recognition proteins
The bromodomain (BRD) motif is an ~110-amino-acid conserved protein module and is regarded as the first and sole histone-binding module that contains a hydrophobic pocket to identify acetyl-lysine.191 The specificity of different BRDs depends on the sequences within the loops that form the hydrophobic pocket. Therefore, each BRD has a preference for different histones.192,193 In addition to their recognition of acetyl-lysine, BRDs are also capable of interacting with other chromatin molecules, such as plant homeodomain (PHD) finger motifs or another BRD. To date, 42 proteins containing bromodomains and 61 unique bromodomains have been discovered.194,195 Based on the sequence length and sequence identity of BRDs, the human BRD family can be divided into nine groups and one additional set of outliers, which has been well illustrated in published papers.169,194 Different BRD-containing proteins contain one to six BRDs. Intriguing, the most notable and well-studied bromodomain proteins are also HATs, such as PCAF, GCN5, and p300/CBP. Yaf9, ENL, AF9, Taf14, Sas5 (YEATS), and double PHD finger (DPF) have also been discovered to be acyl-lysine reader domains.191,196 Human MOZ and DPF2 are two proteins containing the DPF domain. Mutations in the YEATS and DPF domains are associated with cancer. For example, mutation of AF9 has been found in hematological malignancies, and ENL dysregulation leads to kidney cancer.197,198
Another important family is the BRD and extraterminal domain (BET) protein family, including BRD2, BRD3, BRD4, and BRDt, and this family shares two conserved N-terminal bromodomains and a more divergent C-terminal recruitment domain.199,200 These bromodomain proteins are critical as mediators of gene transcriptional activity.201 Of note, bromodomains have also been found in some histone lysine methyltransferases, such as ASH1L and MLL. BRDs are promiscuous domains and have been discussed in other well-constructed papers.169,194 In this review, we focus on the role of BRDs in tumorigenesis.
As histone acetylation “readers”, bromodomain proteins play important roles in tumorigenesis. BRD4 recruits the positive transcription elongation factor complex (P-TEFb), a validated target in chronic lymphocytic leukemia associated with c-Myc activity.202,203,204 Chromosomal translocation of BRD4, via the t(15;19) translocation, results in the generation of the fusion protein BRD4-NUT (nuclear protein in testis), which is found in NUT midline carcinoma (NMC). Importantly, inhibition of BRD4-NUT induces differentiation of NMC cells.205 Moreover, BRD4 is required for the maintenance of AML with sustained expression of Myc206 (Table 2).
Histone deacetylases
Histone deacetylases (HDACs) have recently attracted increasing attention. In humans, the genome encodes 18 HDACs. In contrast to the function of HATs, HDACs usually act as gene silencing mediators and repress transcription. Similarly, HDACs are expressed not only in the nucleus but also in the cytoplasm, and their substrates are also not limited to histones. Based on sequence similarity, HDACs can be divided into four classes: class I HDACs, yeast Rpd3-like proteins, are transcriptional corepressors and have a single deacetylase domain at the N-terminus and diversified C-terminal regions (HDAC1, HDAC2, HDAC3, and HDAC8); class II HDACs, yeast Hda1-like proteins, have a deacetylase domain at a C-terminal position (HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, and HDAC10); class III HDACs are yeast silent information regulator 2 (Sir2)-like proteins (SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, and SIRT7); and class IV involves one protein (HDAC11). The class IV protein shares sequence similarity with both class I and class II proteins.207,208 Classes I, II, and IV are included in the histone deacetylase family, whereas class III HDACs belong to the Sir2 regulator family.209 The catalytic mechanisms for these two families are different; classes I, II, and IV are Zn2+-dependent HDACs, whereas sir2-like proteins (sirtuins) are nicotinamide adenine dinucleotide (NAD+)-dependent HDACs and are also capable of mono-ADP-ribosyltransferase activity, another pattern of histone modification.210 Intriguingly, SIRT4 is thought to have more mono-ADP-ribosyltransferase activity than HDAC activity. SIR2 and SIRT6 seem to have equal levels of both mono-ADP-ribosyltransferase and HDAC activities.211,212 Moreover, after revealing the crystal structure of SIRT5, researchers found that SIRT5 is also a lysine desuccinylase and demalonylase.213 Therefore, the diversity of the sirtuin family makes them a group of multifunctional enzymes.
So far, the major known recognition sites of each HDAC are different, and these largely remain to be uncovered. For example, HDAC3 is thought to deacetylate H4K8 and H4K12,214 but in an HDAC3-knockout HeLa cell line, the acetylation levels of H4K8 and H4K12, even the overall acetylation levels of H3 and H4, were comparable with those in wild-type cells.215 Nevertheless, HDAC1 or HDAC3 siRNA can indeed increase the acetylation levels of H3K9 and H3K18.215 Therefore, partially because of the functional complementation and diversity within HDAC families, especially in class I, II, and IV, it is difficult to identify the specific substrates of certain HDACs. However, the substrates of the sirtuin family are quite clear. It is notable that because SIRT4 and SIRT5 are only located in mitochondria, they have no effect on histones. However, nonhistone lysine acetylation is also prevalent, since more than 3600 acetylation sites on 1750 proteins have been identified.166 The tumor suppressor p53 and the cytoskeletal protein α-tubulin are two representative substrates of HDACs.216,217,218 Notably, HDACs are also capable of regulating gene transcription by deacetylating other proteins that are responsible for epigenetic events, such as DNMTs, HATs, and HDACs.166,219 Another phenomenon is that some HDACs have to form a complex along with other components to function as transcriptional corepressors, which provides ideas and methods to design novel HDAC inhibitors. The Sin3, NuRD, and CoREST complexes are three complexes containing HDAC1 and HDAC2. Studies have found that purified HDAC1 or HDAC2 without associated components shows fairly weak deacetylation activity in vitro.220 HDAC3 interacts with the corepressors SMRT/NCoR to form the functional complexes, which significantly increases HDAC3 activity. NCoR also interacts with HDAC1, HDAC2 and the class II deacetylases HDAC4, HDAC5, and HDAC7, but usually not in the form of a complex.221,222 Deleted in breast cancer 1 (DBC1) and active regulator of SIRT1 (AROS) are two proteins that are able to bind to SIRT1, whereas their interactions present opposite functions. The DBC1/SIRT1 complex inhibits the deacetylation activity of SIRT1, whereas the combination of AROS and SIRT1 stimulates the activity of SIRT1.223,224
HDACs not only are able to deacetylate histones and nonhistone proteins but also interact with other epigenetic-associated enzymes, which gives them a vital role in tumorigenesis.162,178 Alterations in HDACs in cancers usually result in aberrant deacetylation and inactivation of tumor suppressor genes. For example, hypoacetylation of the promoter of p21, a tumor suppressor encoded by CDKN1A, can be reversed by HDAC inhibitors, resulting in an antitumor effect.225 A screen of the mutations in several HATs and HDACs, such as CBP, PCAF, HDAC1, HDAC2, HDAC5, HDAC7, and SIRT1, in more than 180 cancer samples including primary tumors and cancer cells indicated that the expression profiles of HDAC1, HDAC5, HDAC7, and SIRT1 are distinctive for colorectal cancers and normal colorectal mucosa, and the expression profiles of HDAC4 and CBP are capable of distinguishing breast cancer tissue from normal tissues226 (Table 2).
Histone methylation (lysine and arginine)
Similar to the process of histone acetylation, histone methylation also consists of three important components: “writers”, histone methyltransferases (HMTs), “readers”, histone methylation-recognizing proteins, and “erasers”, histone demethylases (HDMs). Methylation of histones occurs at arginine and lysine residues. Arginine and lysine both can be monomethylated or dimethylated, whereas lysine is also capable of being trimethylated. Histone methylation can either promote or inhibit gene expression, which depends on the specific situation. For example, lysine methylation at H3K9, H3K27, and H4K20 is generally associated with suppression of gene expression, whereas methylation of H3K4, H3K36, and H3K79 induces gene expression.360 Mutation of H3K27M (lysine 27 to methionine) and H3K36M are two important oncogenic events, and H3K27M and H3K36M serve as drivers of pediatric gliomas and sarcomas. H3K27M has been identified in more than 70% of diffuse intrinsic pontine gliomas (DIPGs) and 20% of pediatric glioblastomas, which results in a global reduction in the trimethylation of H3K27 (H3K27me3).361,362,363 However, the H3K36M mutation impairs the differentiation of mesenchymal progenitor cells and generates undifferentiated sarcoma, leading to increased levels of H3K27me3 and global loss of H3K36 (me2 and me).364,365 Meanwhile, depletion of H3K36 methyltransferases results in similar phenotypes to those seen with H3K36M mutation.364 To date, KMTs (lysine methyltransferases) have been better studied than arginine methyltransferases (PRMTs) due to their sequence of discovery, different prevalence and impact. Their targets are not limited to only histones, they also modify other key proteins, such as the tumor suppressor p53, TAF10, and Piwi proteins.366,367,368
Histone methyltransferases
All KMTs contain a 130-amino-acid conserved domain, the SET (suppressor of variegation, enhancer of Zeste, trithorax) domain, except for DOT1L. The SET domain is responsible for the enzymatic activity of SET-containing KMTs. Instead of methylating lysine residues in histone tails, DOT1L methylates lysine in the globular core of the histone, and its catalytic domain is more similar to that of PRMTs.369,370 The enzymatic activity of KMTs results in the transfer of a methyl group from S-adenosylmethionine (SAM) to a the ε-amino group of a lysine residue. The first identified KMT was SUV39H1, which targets H3K9.371 Sequentially, more than 50 SET-containing proteins have been identified with proven or predicted lysine methylation potential. Of note, KMTs are highly specific enzymes, meaning that they are highly selective for lysine residues they can methylate and the specific methylation degree they can achieve. For example, SUV39H1 and SUV39H2 specifically methylate histone 3 at lysine 9 (H3K9), and DOT1L only methylates H3K79.371 Based on their structure and sequence around the SET domain, generally, KMTs can be divided into six groups, SUV39, SET1, SET2, EZH, SMYD, and RIZ (PRDM) (reviewed by Volkel and Angrand372). The Pre-SET domain of the SUV39 family contains nine conserved cysteines that coordinate with three zinc ions to function. The SET1 family members share a similar Post-SET motif that contains three conserved cysteine residues. The SET2 family possesses an AWS motif that contains 7–9 cysteines. Their SET domain is located between the AWS motif and a Post-SET motif. The members of the enhancer of zeste homolog (EZH) family are the catalytic components of polycomb repressive complexes (PRCs), which are responsible for gene silencing. EZH proteins have no Post-SET motif but have 15 cysteines in front of the SET domain and show no methylated activity as isolated proteins.373 PRC2 shows lysine methylation activity through its catalytic components, EZH2 or its homolog EZH1.374 EZH2 can methylate not only histone H3 but also histone H1 at lysine 26.375 The SMYD family members, which are SET and MYND domain-containing proteins, possesses a MYND (myeloid-nervy-DEAF1) domain, a zinc-finger motif responsible for protein–protein interaction.376 The RIZ (PRDM) family is a large family containing a homolog of the SET domain, the PR domain. The PR and SET domains share 20–30% sequence identity and are both capable of inducing histone H3 methylation.377 However, most members of the RIZ family responsible for histone methylation are still unknown. So far, two of them have been proven to induce the methylation of histones: PRDM2 (RIZ1) is associated with H3K9 methylation; and Meisetz, the mouse homolog of PRDM9, trimethylates H3K4.378 Meanwhile, PRDM1 has been identified to interact with EHMT2, a member of the SUV39 family. PRDM6 acts as a transcription suppressor by interacting with class I HDACs and EHMT2 to induce cell proliferation and inhibit cell differentiation.379 Meanwhile, the recruitment of EHMT2 is based on the formation of a complex with PRDM1.380 Due to the lack of a characteristic sequence or structure flanking the SET domain, other SET-containing KMTs, such as SET7/9, SET8, SUV4-20H1, and SUV4-20H2, cannot be classified into these families. Notably, some KMTs contain more than one domain, which allows them to interact with other proteins, especially other epigenetic modifying proteins. SUV39H1 possesses a chromodomain that directly binds to nucleic acids and forms heterochromatin.381 MLL1 recognizes unmethylated DNA through its CpG-interacting CXXC domain. SETDB1 contains an MBD that interacts with methylated DNA.382 The Tudor domain in SETDB1 may potentially recognize the methylation of lysine residues.383 ASH1 is able to interact with CBP, a HAT, via a bromodomain within ADH1.384
Protein arginine methyltransferases (PRMTs) can be divided into two groups. Among the nine PRMTs, only PRMT5, PRMT7, and PRMT9 are type II PRMTs, and the other five PRMTs, except for PRMT2, are type I PRMTs. PRMT2 was identified by sequence homology385 but has not shown any catalytic activity during investigations, although PRMT2 acts as a strong coactivator for androgen receptor (AR), which is thought to be associated with arginine methylation.386 Both types of PRMTs first catalyze the formation of monomethylarginine as an intermediate. However, sequentially, type I PRMTs can form asymmetric dimethylarginine (ADMA, Rme2a), but type II PRMTs form symmetric dimethylarginine (SDMA, Rme2s). Rme2a means two methyl groups on one ω-amino group, whereas an Rme2s has one methyl group on each ω-amino group. PRMT1-PRMT8 were investigated by Herrmann and Fackelmayer,387 and FBXO11 was identified as PRMT9, which symmetrically dimethylates arginine residues.388
Most enzymes for histone methylation are substrate-specific proteins; therefore, alterations in the aberrant expression of enzymes are usually associated with specific histone residue mutations. One of the best-known examples of alterations in tumorigenesis is H3K4me3, which is associated with biphenotypic (mixed lineage) leukemia (MLL). The location of the MLL gene is where chromosomal translocations in AML and ALL usually occur.389 When the MLL gene is translocated, the catalytic SET domain is lost, which results in MLL translocation-generated fusion proteins, which recruit DOT1L.390 Maintenance of MLL-associated ALL depends on the methylation of H3K79 catalyzed by DOT1L.391 Therefore, DOT1L is usually associated with hematological malignancies rather than solid tumors. Alteration of the EZH2-induced methylation of H3K27 has been observed in multiple cancers, including various solid tumors (prostate, breast, kidney, bladder, and lung cancers) and hematological malignancies.392 Meanwhile, overexpression of EZH2 has been found in multiple cancers and is associated with poor prognosis.393 Different mechanisms have been proposed to describe the role of EZH2 in tumorigenesis (Table 3).
Methyl-histone recognition proteins
“Readers” of histone methylation contain several specific domains recognizing lysine or arginine methylation, such as a chromodomain,394 the WD40 repeat, the MBT (malignant brain tumor) domain, the Tudor domain395 and the PHD (plant homeodomain) finger motif.396 Representative chromodomain-containing proteins in humans are HP1 and Chd1, which can recognize H3K9me and H3K27me, respectively.394,397 WDR5 is a protein containing WD40 repeats. In addition to H3K4me, WDR5 prefers to bind to H3K4me2 via a histone-methylating complex and is required for maintaining H3K4me3.395 Later, WDR5 was shown to directly read H3R2, a “WIN” motif of MLL1, as well as symmetrical H3R2 dimethylation through the WD40 domain.398 L3MBTLs are a group of proteins containing three MBT repeat domains. L3MBTL1 represses gene expression via monomethylation or dimethylation of H4K20 or H1BK26.399 BPTF, RAG2, PYGO, and the tumor suppressor ING2 are representative proteins containing PHD finger motifs. They are all able to recognize and bind to H3K4me3.400 Intriguingly, DNMT3L and BHC80 also possess a PHD finger motif, but they selectively bind to unmethylated H3K4.401,402 There are a number of proteins containing Tudor domains, with a representative protein being JMJD2A. JMJD2A is a histone demethylase that equally binds to H3K4me3 and H4K20me3403 (Table 3).
Histone demethylases
The identification of histone demethylases (HDMs or KDMs) has lagged behind that of HMTs. Thus far, KDMs can be classified into two groups. The amine-oxidase type lysine-specific demethylases (LSDs) and the highly conserved JumonjiC (JMJC) domain-containing histone demethylases. LSD1 and LSD2, also known as KDM1A and B, are flavin adenine dinucleotide (FAD)-dependent amine oxidases that can only demethylate monomethylated and dimethylated lysine residues. LSD1 has been identified to specifically activate androgen receptor (AR) target genes along with AR by demethylating H3K9.404 The human genome codes more than 30 JMJC-containing KDMs that are able to remove methyl groups from all three methyl-lysine states. JHDM1A was the first characterized JMJC domain-containing HDM and specifically demethylates H3K36me2 and H3K36me1.405 Not all JMJC domain-containing proteins are able to demethylate histone proteins, such as HIF1AN and the transmembrane phosphatidylserine receptor PTDSR. JMJC-containing HDMs can be divided into six families:360 the JHDM1, JHDM2 (JMJD1), JHMD3 (JMJD2), JARID, PHF, and UT families. Notably, not all of these families possess the ability of histone demethylation. However, some JMJC-containing proteins, including those that are not included in these six families, contain one or more methylated-histone-binding domains. Their potential to demethylate methyl-lysine or methyl-arginine must be investigated. In addition to demethylases for lysine residues, JMJD6 is the first described arginine demethylase and lysine hydroxylase. It can remove methyl groups from H3R2 and H4R3.406 Another kind of protein is peptidylarginine deiminases (PADs or PADIs) or protein-arginine deiminases, which are able to convert arginine and monomethylated arginine to citrulline.407
LSD1 (KDM1A) is one of the best-studied KDMs and has been found to be increased in multiple cancers. Inhibition of LSD1 leads to global H3K4 methylation and promotes differentiation of neuroblastoma cells.408 Unlike KDM1A, KDM1B is mostly involved in growing oocytes with a restricted expression pattern.409 Similar to the dual roles of LSD1, members of the KDM2 family can either promote tumor formation or inhibit tumorigenesis.410 Through dimethylating H3K36 in DUSP3 (dual specific phosphatase 3), KDM2A activates ERK1/2 expression in lung cancer cells.411 Knockout of KDM2B in breast cancer downregulates the tumor stem cell markers ALDH and CD44 via the repression of polycomb complexes. KDM2B is also overexpressed in pancreatic ductal adenocarcinoma (PDAC) and cooperates with KrasG12D to promote PDAC formation in mouse models.412 The LSD1 and KDM2 family possesses context-dependent tumor-promoting and -inhibiting functions, which might depend on the different features of various cancers and the specific substrates of the enzymes. Therefore, further studies should take the dual roles of these enzymes into consideration. KDM3A, induced by hypoxia and nutrient starvation within the tumor microenvironment, shows carcinogenic effects via the promotion of tumor cell migration and invasion. Inhibition of KDM3A downregulates tumor-associated angiogenesis and macrophage infiltration.413,414 KDM3C is required for MLL-AF9 leukemia maintenance and is mutated in patients with intracranial germline tumors.415,416 KDM4A, KDM4B, and KDM4C have shown increased expression in prostate cancer with decreased levels of H3K9me2/3 and increased levels of H3K9me1.417 H3K9me3 is thought to be a hallmark of heterochromatic areas of the genome. In addition, KDM4 family members were the first identified demethylases targeting trimethylated lysines. Aberrant expression of KDM4 family members might lead to instability of the genome and become involved in tumorigenesis.410 Members of the KDM6 family usually act as tumor suppressors and are thought to cause cell growth arrest.418 For example, the tumor suppressor proteins p16INK4A and p14ARF, encoded by the INK4A-ARF locus, are repressed by H3K27me3. When stimulated by oncogenic factors, KDM6B is recruited to the INK4A-ARF locus and activates the transcription of these two tumor suppressors.419 In colorectal cancer, KDM7C is required for the efficacy of oxaliplatin and doxorubicin and for the activation of p53420 (Table 3).
Noncoding RNA
Epigenetic related noncoding RNAs (ncRNAs) include microRNAs (miRNAs), small interfering RNA (siRNAs), Piwi-interacting RNA (piRNAs), and long noncoding RNAs (lncRNAs). MiRNAs, one of the most studied ncRNAs, are small RNAs between 19 and 22 nucleotides in length that play important roles in the regulation of gene expression by controlling mRNA translation. Intriguingly, the regions that miRNAs usually target are frequently associated with carcinogenesis.567 Generally, they can be divided into tumor-promoting and tumor-suppressing miRNAs. During tumorigenesis, oncogenic miRNAs such as miR-155, miR-21 and miR-17-92 are usually overexpressed, and tumor-suppressive miRNAs such as miR-15-16 are downregulated.568 There is another type of miRNA, cellular context-dependent miRNAs, functioning in tumorigenesis. For example, miR-146 has been shown to be overexpressed in multiple cancers, whereas a recent study has proven that miR-146 can reduce the expression of BRCA1.568,569 Meanwhile, the expression of proteins and enzymes is also regulated by certain miRNAs. MiR-101 directly represses EZH2, and abnormal downregulation of miR-101 has been observed in cancers.570,571 The expression of the miR-29 family is inversely correlated with that of DNMT3A and -3B in lung cancer tissues. Forced expression of miR-29 inhibits tumorigenesis by inducing reexpression of methylation-silenced tumor suppressor genes.572 LncRNAs are another large group of noncoding RNAs that play a vital role in tumorigenesis. Some lncRNAs are cancer type-specific, such as PCGEM1 in prostate cancer and HEIH in hepatocellular carcinoma.573,574 Many aberrant lncRNAs have been discovered in various cancers. Dysregulation of HOTAIR has been found in lung, pancreatic, and colorectal cancer.575,576,577
Therefore, ncRNAs can either be directly involved in tumorigenesis or indirectly affect tumor development by participating in other epigenetic events.
Inhibitors and clinical trials
Unlike genetic mutations, epigenetic alterations are reversible. Given the importance of epigenetic marks in tumorigenesis, the availability of corresponding inhibitors has attracted extensive attention. Meanwhile, epigenetic regulation of a gene usually requires more than one epigenetic event. Currently, there are six epigenetic drugs approved for clinical use by the FDA (Table 4).
Targeting DNA methylation
Blockade of DNMTs is the most effective way to prevent aberrant DNA hypermethylation. However, until now, targeting of the methyltransferase enzymes still lacks specificity and even causes hypomethylation of the global genome.578 Complete deletion of DNMT1 in mice results in embryonic lethality.579 Knockout of DNMT1 in fibroblast cells causes aberrant expression of 10% of genes and p53-dependent death.580 Administration of DNA methylation inhibitors results in tumorigenesis in male Fischer rats.581 Regulation of DNA methylation is vital in cell survival and function, and in addition to the specificity needed and the side effect associated, it is hard to identify proper drugs.
DNA methylation inhibitors can be divided into two groups: nucleoside analogs and nonnucleoside analogs. Nucleoside analogs have a modified cytosine ring and can be turned into nucleotides and incorporated into newly synthesized DNA or RNA. DNA methyltransferases are bound by covalent complexes with the analogs, which inhibits DNA methylation. 5-Azacitidine (5-Aza-CR) and 5-aza-2′-deoxycytidine (5-Aza-CdR) are currently the two most studied and promising demethylation agents.582 5-Aza-CR and zebularine are ribonucleoside analogs that can be phosphorylated to be able to incorporate into RNA. However, they can also be incorporated into DNA via the ribonucleotide reductase pathway. 5-Azacitidine, an analog of cytidine, is an injectable suspension for the treatment of myelodysplastic syndromes (MDSs). It promotes cell differentiation, demethylation, and reexpression of inactivated genes.583 The 5-azacitidine side effects include fetal abnormalities584 and decreased male fertility, especially at high doses, but its analog, 6-azacytidine, does not show such effects.585 Notably, after treating the noninvasive breast cancer cell lines MCF-7 and ZR-75-1 with azacytidine, the cells gained invasive abilities due to the hypomethylation of several prometastasis genes.586 Decitabine (5-Aza-CdR) and 5-fluoro-2′-deoxycytidine (5-F-CdR) are deoxyribonucleoside analogs that are capable of incorporating into DNA following phosphorylation. Decitabine (5-aza-2′-deoxycytidine) inhibits DNA methylation in a dosage-dependent manner. It can reactivate silenced genes at low doses but gains cytotoxicity at high doses, while myelosuppression is the major side effect at all doses.587. Dihydro-5-azacytidine (DHAC) is a biologically active and chemically stable analog of 5-azacitidine with decreased toxicity.588,589 Because of its hydrolytic stability, it may be administrated via prolonged i.v. infusion, potentially eliminating the acute toxicities caused by administration of 5-azacytidine.590 Zebularine is a potential oral DNA-demethylating drug with stability in acidic environments and in aqueous solutions.591 However, the near millimolar dose requirements and the limited bioavailability in rodents (<7%) and primates (<1%) leave zebularine far from clinical translation.592
Among the drugs discussed, 5-Aza-CR593 and 5-Aza-CdR594 have already been approved by the US Food and Drug Administration (FDA) for the treatment of certain subtypes of MDS and chronic myelomonocytic leukemia. Because of their intrinsic preference for newly synthetic DNA, they tend to affect dividing cells, i.e., cancer cells.595 Ongoing preclinical experiments and clinical trials are exploring their efficacy in solid tumors. The common side effects of these nucleoside-like analogs are mutagenic risk and genomic instability. Nonnucleoside analogs are capable of avoiding these side effects.
Currently, many nonnucleoside analogs have been developed to prevent DNA from aberrant hypermethylation. These drugs are usually small molecular inhibitors and directly target catalytic sites rather than incorporating into DNA. Based on a three-dimensional model of DNMT1, RG108 was designed to block the activity of this enzyme and cause demethylation.596 Psammaplin is a group of natural extracts from the sponge Pseudoceratina purpurea and is capable of inhibiting both DNA methyltransferases and histone deacetylases with mild cytotoxicity.597 Similarly, EGCG ((-)-epigallocatechin-3-gallate) is the major polyphenol from green tea and reversibly demethylates methyl-DNA, resulting in the reactivation of multiple key genes, including hMLH1, P16, and RA, in colon, esophageal, and prostate cancer cell lines.598 Both hydralazine and procainamide, two drugs associated with lupus-like autoimmune diseases, can inhibit DNA methylation and induce self-reactivity in cloned T-cell lines.599 They have promising tumor suppressor-reactivating and antitumor actions in breast cancer.600,601 Another strategy is developing antisense oligonucleotides to inhibit DNMT transcription. MG98 is a second-generation phosphorothioate antisense oligodeoxynucleotide that prevents DNMT1 mRNA translation effects but has no obvious antitumor effect.602 It has been under investigation in preclinical experiments and phase I/II clinical trials, especially in solid tumors.603,604 Of note, in a systemic analysis comparing nonnucleoside inhibitors with 5-Aza-CdR, the latter showed better efficacy in DNA demethylation inhibition.605
To date, hundreds of clinical trials have investigated the effects of anti-DNA methylation therapy for various cancers (Table 5).
Inhibitors of histone modifications
Compared with DNA methylation, histone modifications have been investigated in broader areas of diseases, including solid tumors, hematological malignancies, and even many inflammatory diseases (such as viral infection, diabetes and inflammatory lung diseases). During the process of gene silencing, lysine deacetylation and demethylation of H3K4 rather than demethylation of H3K9 or cytosine methylation might be the primary causative event.606 Therefore, histone modification plays an essential role in the regulation of gene expression, which also makes it a promising target for disease treatment. Clinical trials targeting histone acetylation and histone methylation are listed in Table 6 and Table 7, respectively.
Inhibitors for HATs and BETs
Generally, there are two strategies for preventing aberrant histone acetylation, including altering interactions within the active sites within HATs or using mimetic products of enzymatic substrates. To date, many inhibitors targeting BRD proteins have been investigated in clinical trials, whereas there are no clinical trials investigating inhibitors for HATs.
Bisubstrate inhibitors are selective inhibitors for PCAF, p300, and TIP60. They mimic two substrates of HATs: the cofactor acetyl coenzyme A (Ac-CoA) and a peptide resembling the lysine substrate.607,608 However, due to their peptidic nature and size, they are not membrane-permeable and require the assistance of a delivery system. Based on inhibitory strategies for HATs, nonpeptide small molecular inhibitors have been developing as potential therapeutic agents. Several small molecule inhibitors are natural products, including garcinol, curcumin, and anacardic acid.609,610,611 These natural HAT inhibitors lack selectivity between HATs and often have other targets. Therefore, structurally modified and synthetic compounds have been reported. Α-Methylene-g-butyrolactones are small molecular inhibitors of HATs with selectivity for either GCN5L2 or PCAF.612 Isothiazolone is another HAT inhibitor targeting p300 and PCAF.613 However, high reactivity towards thiolates limits the application of HAT inhibitors in biological systems. Other inhibitors of HATs, such as thiazide sulfonamide and C646, have been gradually identified and show promising effects in multiple cancers. Another strategy to inhibit HAT activity is to target protein–protein interactions between HATs and their interaction partners. This method is dependent on the function of the interactions rather than the acetylation activity of HATs. ICG-001 and PRI-724 are representatives of this kind of inhibitor. Appropriately applying HAT agonists is also important to correct aberrant acetylation during diseases. CTPB is derived from anacardic acid and selectively activates p300, resulting in gene transcription.609 TTK21 and SPV106 are two other agonists based on anacardic acid.
Binding to BRDs and blocking acetylated lysine recognition is another mechanism that inhibits acetylation. JQ1 and I-BET762 are two representative inhibitors of the BET family. JQ1 is a cell-permeable small molecule and can competitively bind to BRD4 fusion oncoproteins, such as BRD4-NUT, resulting in cancer cell differentiation and apoptosis.614 Similarly, I-BET762 is also a synthetic mimic of and competes with BRD4.615 Other compounds, such as MS417, OTX-015, RVX-208, OXFBD, I-BET151, PFI-1, MS436, and XD14, are also BET inhibitors and have been well illustrated in other published papers.616 We will focus on the associations between these compounds and cancers. However, a number of non-BET proteins containing BRDs have attracted considerable attention. Many non-BET bromodomain inhibitors are based on a structure called the “WPF shelf” and a “gatekeeper” residue located at the start of the C helix.617 Several HATs have a BRD, such as Gcn5, PCFA, p300, and CBP. Inhibitors for CBP include MS2126, MS7972, ischemin, SGC-CBP30 and I-CBP112; optimized 1-(1H-indol-1-yl) ethanone derivatives have also shown promising results in inhibiting CBP and p300.618 BAZ2A/B bromodomain inhibitors include BAZ2-ICR and GSK2801. The quinolone-fused lactam LP99 was the first synthetic selective inhibitor for BRD7/9. I-BRD9 was identified by GlaxoSmithKline (GSK) and is a selective inhibitor of BRD9, which has more than 200-fold selectivity for BRD9 over BRD7 and 700-fold selectivity for BRD9 over BET family members.619 PFI-3 is a potential inhibitor of SMARCA4 and PB1 with a stronger affinity for the bromodomain of SMARCA4. However, Vangamudi et al. identified that the ATPase domain within SMARC4 bypassed the anticancer effects related to the bromodomain since PFI-3 did not inhibit cell proliferation.620 The BRPF1 (bromodomain and PHD finger-containing 1) protein is part of the BRPF family, which is a component of MYST family complexes. The inhibitors of BRPF1 include PFI-4, OF-1, and NI-57. 1,3-Dimethyl benzimidazolones were the first selective inhibitors of BRPF1. PFI-4 and OF-1 are two close analogs of 1,3-dimethyl benzimidazolone that have been identified by the Structural Genomics Consortium (SGC). Another BRPF1 inhibitor, NI-57, was discovered by the SGC based on a new quinolinone scaffold. Both NI-57 and OF-1 are thought to interact with BRPF1-3 as pan-BRPF bromodomain inhibitors. Based on the bromodomain contained within both TRIM24 (tripartite motif containing protein 24) and BRPF1, a dual inhibitor, IACS-9571, has been identified.621 Bromosporine is a panbromodomain inhibitor with good cellular activity, whereas in a recent study, researchers noticed that bromodomain inhibitors only targeted the BET family rather than other BRDs.622
Inhibition of HDACs
Given that multiple methods can regulate HDAC activity, the designation of HDAC inhibitors has its own advantages. In the 1970s, butyrate was found to induce the accumulation of acetylated histones in cancer cells, which is thought to be associated with the inhibition of deacetylation.623 Later, a natural extract, trichostatin A (TSA), was identified to inhibit the activity of partially purified HDACs and induce cancer cell differentiation and apoptosis.624 Gradually, more natural and synthetic compounds have been identified to inhibit histone deacetylation. A study reported that administration of HDAC inhibitors only regulates a small number of genes (1–2%) but induces an obvious and rapid decrease in c-Myc gene expression, which indicated that a restricted set of cellular genes was uniquely sensitive to regulation of histone acetylation.625 The combination of two HDAC inhibitors, SAHA and TSA, induced melanoma cell growth arrest by upregulating p21, p27 and NF-κB, and MG132 can enhance the effect of TSA.626 The inhibition of HDACs has been investigated in various cancers, with promising antitumor effects.627,628 Based on the characteristics of their chemical structures, HDAC inhibitors can be divided into five groups: short-chain fatty acids, hydroxamic acids, benzamides, cyclic peptides, and hybrid molecules. In addition to those included in the five groups, some new synthetic compounds also act as inhibitors of HDACs.
The short-chain fatty acid group contains sodium butyrate, valproic acid (VPA), sodium phenylbutyrate, and AN-9 (pivaloyloxymethyl butyrate). The effective concentration of butyrate is usually at the micromolar level. The group of hydroxamic acids includes more than ten members and is the best-studied class. Structural analyses of TSA and suberoylanilide hydroxamic acid (SAHA) show that they are noncompetitive inhibitors of HDACs since they share significant homology with class I and class II HDACs, which makes them mimics of the lysine substrates.629 In addition, they chelate the active zinc ion in a bidentate manner, which is crucial for enzymatic activity.624 Hexamethylene bisacetamide (HMBA) is a representative of the hybrid polar compounds (HPCs), whereas second-generation HPCs, such as oxamflatin, SAHA, suberic bishydroxamic acid (SBHA), and m-carboxycinnamic acid bishydroxamide (CBHA), have shown better inhibition of HDACs and anticancer effects than first-generation agents.630 Oxamflatin, scriptaid, and amide are analogs of TSA and show anticancer effects.631,632,633 Benzamide inhibitors (MS-275, MGCD0103, and CI-994) are well-studied and show promising effects in the treatment of diseases, especially cancers. They inhibit histone deacetylation via binding to catalytic zinc ions within HDACs through carbonyl and amino groups. Inhibition of HDACs by benzamide inhibitors is thought to be reversible, but the bond may become tight and pseudoirreversible in a time-dependent manner.634,635 However, benzamide inhibitors have less activity than members of the hydroxamate or cyclic peptide families, with an effective concentration around the micromolar range.636 Cyclic peptides can be further divided into two groups: cyclic tetrapeptide containing a 2-amino-8-oxo-9, 10-epoxy-decanoyl (AOE) moiety (HC-toxin, trapoxin) and cyclic peptides without the AOE moiety (apicidin and romidepsin). The epoxyketone group is essential for the inhibitors to bind to active zinc ions, but the epoxyketone-based bond is irreversible. Trapoxin is a fungal cyclic peptide and can irreversibly inhibit the activity of HDACs.637 Romidepsin, also known as FK228, most likely relies on one of the thiol groups to coordinate to the active site zinc ion.638 Garlic-associated derivatives, such as diallylsulfide and allylmercaptan, are capable of generating a thiol group that makes them potential inhibitors of HDACs.639 K-trap, an analog of trapoxin, and other derivatives, including 9-acyloxyapicidins and 9-hydroxy, have been under investigation. Depudecin is a natural epoxide derivative isolated from the fungus Alternaria brassicicola. Psammaplins is isolated from a marine sponge Pseudoceratina purpurea. These two natural extracts can inhibit the activity of HDACs.
Early HDAC inhibitors were nonselective because of the high homology of the structure and catalytic mechanism of HDACs within each group. The first selective HDAC inhibitor was tubacin, which targets HDAC6 with increased tubulin acetylation but not histone acetylation.640 PCI-34051, a specific inhibitor of HDAC8, can induce caspase-dependent apoptosis in T-cell lymphoma but does not increase histone acetylation.641 Another benzamide inhibitor, SHI-1:2, shows HDAC1/HDAC2-specific inhibitory activity that is >100-fold more selective than that of other HDACs.642 New synthetic chemicals, such as SK7041 and splitomicin, selectively target class I HDACs and sir2-like family members, respectively. The same efforts have been made to develop inhibitors for sirtuins, the class III HDACs. Nicotinamide, a byproduct of the sirtuin enzyme reaction, is a widely used inhibitor of all sirtuins. Other compounds, such as cambinol, salermide, tenovin, EX-527, suramin, and AGK2, have also been reported as sirtuin inhibitors. Sirtuin inhibitors (such as nicotinamide) function via interactions with the NAD+ within the active site of sirtuins or through binding to acetyl-lysine.
Of note, second-generation HDACs, including hydroxamic acids (vorinostat (SAHA), belinostat (PXD101), LAQ824, and panobinostat (LBH589)) and benzamides (entinostat (MS-275), tacedinaline (CI-994), and mocetinostat (MGCD0103)), are currently in clinical trials, and some of them have already been approved for disease treatment. The success of romidepsin in phase I clinical trials in cutaneous and peripheral T-cell lymphoma accelerated the development of HDAC inhibitors as anticancer drugs. In 2006, SAHA (vorinostat) was first approved by the US Food and Drug Administration (FDA) for the treatment of cancer, restricted to patients with cutaneous T-cell lymphoma (CTCL), as an HDAC inhibitor.643 Romidepsin (Istodax) was the second approved HDAC inhibitor, which was approved in 2009. Three members of the benzamide family have also shown clinical significance in anticancer drug development. Belinostat (Beleodaq, previously known as PXD101) was approved in 2014 by the US FDA and European Medicines Agency to treat peripheral T-cell lymphoma. Another HDAC inhibitor, panobinostat, is a nonselective HDAC (pan-HDAC). It has shown promising effects in anticancer treatments; therefore, the FDA accelerated its approval for the treatment of patients with multiple myeloma. Intriguing, as we mentioned before, truncating mutations in HDAC2 have been found in sporadic carcinomas and colorectal cancer and result in resistance to traditional HDAC inhibitors.644 Mutations in other HDACs also exist; therefore, screening of these mutations in cancer can improve the efficacy of HDAC inhibitors.
Inhibitor of HMTs and HDMTs
EPZ004777 was the first identified selective inhibitor of DOT1L and selectively kills MLL-translocated cells over those without MLL translocation.645 However, due to its poor pharmacokinetic properties, a second generation of EPZ004777, EPZ-5767, was developed with a cyclobutyl ring replacing the ribose moiety.646 EPZ-5767 also shows synergistic effects with cytarabine, daunorubicin, and the DNMT inhibitor azacitidine in treatments for ALL with MLL translocation. EPZ-5767, though still showing low oral bioavailability, has been investigated in clinical trials for the treatment of leukemia with MLL rearrangement.647 There are several inhibitors of EZH2. 3-Deazaneplanocin A (DZNep), a derivative of the antibiotic neplanocin-A, is one of the most studied compounds. In fact, DZNep is a SAH-hydrolase inhibitor and decreases EZH2 expression via upregulation of SAH, which leads to degradation of PRC2 in a feedback inhibition mechanism.648,649 Another kind of inhibitor is SAM competitive inhibitors. SAM is responsible for the methyl moiety of KMTs. EI1, a small molecular inhibitor of EZH2, inhibits EZH2 activity by directly binding to EZH2 and competing with SAM.650 GSK343 and GSK126 are two other SAM competitive inhibitors that have been investigated in clinical trials.651,652 EPZ005687, a potent inhibitor of EZH2, significantly reduces H3K27 methylation in lymphoma cells with point mutations at the Tyr641 and Ala677 residues of EZH2 without obvious effects on the proliferation of wild-type cells.653 EPZ-6438, which shows similar effects and superior oral bioavailability, was developed next.654 CPI-1205 is a novel inhibitor of EZH2 that belongs to the pyridone family.
Tranylcypromine (TCP) is an approved drug for depression due to its ability to inhibit monoamine oxidase (MAO) activity. The structures of LSD enzymes and MAOs share many similarities. Therefore, the side effects of TCP as an HDMT inhibitor, including orthostatic hypotension, dizziness, and drowsiness,655 are mostly caused by targeting of MAO. Administration of TCP in MLL-AF9 leukemia promotes tumor cell differentiation and apoptosis.656 TCP is also capable of resensitizing non-acute promyelocytic leukemia (APL) AML cells to all-trans retinoic acid (ATRA) treatment via increasing H3K4me2 and the expression of myeloid-differentiation-associated genes.657 Several derivatives of TCP have been developed to achieve better bioavailability and selectivity, including OG-002, RN-1, SP2509, and GSK690.658,659,660 Another LSD1 selective inhibitor, ORY-1001, can also promote the differentiation of leukemia cell lines, especially cells with translocations in MLL, and has good oral bioavailability.661 To date, three LSD1 inhibitors, including TCP, ORY-1001, and GSK2879552, have been under investigation in clinical trials for the treatment of cancer patients. Daminozide (N-(dimethylamino) succinamic acid, 160 Da), a plant growth regulator, selectively inhibits KDM2/7 by chelating the active site metal.662 Daminozide and siRNA can similarly downregulate KDM7 expression and then regulate tumor-repopulating cells via demethylation of H3K9.663 GSK-J1 was the first identified KDM6 inhibitor with restricted cellular permeability, which resulted from its highly polar structure. Its ethyl ester, GSK-J4, possesses an improved ability to enter cells.664 However, scientists have found that GSK-J1 shows compatible selectivity for the KDM6 and KDM5 families and that GSK-J4 is also a potential inhibitor for KDM5B and KDM4C.665 EPT-103182, a selective inhibitor of KDM5B, has shown promising results in terms of antiproliferative effects in hematological and solid cancer cells. KDM8 and JMJD6 share homology and can be inhibited by a broad spectrum inhibitor, NOG.661
Specific inhibitors usually have similar selectivity to closely related homologs within a group, and even across different groups, which needs to be taken into consideration when using compounds that are not highly selective.
Combined therapy
Epigenetic regulation during tumorigenesis is complicated and involves multiple steps. Therefore, the combination of two or more therapies targeting various epigenetic events seems helpful. This combination synergistically inhibits the expression of tumor-growth-promoting genes and promotes the reexpression of tumor suppressor genes. 4SC-202 is a small molecular drug with dual effects that can inhibit HDAC1/2/3 and LSD1 with similar low micromolar potency. This drug is under clinical investigation. Other studies have administered two or more kinds of epigenetic drugs for anticancer therapy. Relevant clinical trials are listed in Table 8.
Conclusion
Although more specific mechanisms need to be investigated, it is well accepted that epigenetic events are important in normal biological processes as well as in tumorigenesis and that the epigenetic status is usually widely altered during cancer initiation. This makes epigenome-targeted therapy a promising strategy for the treatment of cancer. Based on the complexity of cancer, epigenetic alterations have influenced multiple aspects in cancer, such as the expression of oncogenes and tumor suppressor genes and signal transduction, resulting in enhanced cancer growth, invasion and metastasis. Although epigenetic therapy has a rational and profound basis in theory, some problems remain to be discussed and solved. The first and most important is the problem of selectivity. Epigenetic events are ubiquitously distributed across normal and cancer cells. In fact, some cancers depend on certain epigenetic alterations and can be sensitive to this regulation, whereas under usual regulation, normal cells have the ability to compensate for these epigenetic changes. Therefore, the priority is to determine the most important epigenetic alterations for different cancers. The second problem extends from the first problem. Thus far, epigenetic therapy has obtained impressive results in hematological malignancies but not in solid tumors. The properties of hematological malignant cells and solid tumor cells are different. However, researchers have still investigated the appropriate strategies for solid tumors. Since epigenetic alterations have effects on the sensitivity of small molecule targeted therapy and chemotherapy or radiotherapy, epigenetic-targeted therapy seems to be an important adjunctive therapy. The combination of epigenetic therapy and immunotherapy has also been investigated in preclinical and clinical trials.
Based on the achievements obtained, epigenetic-targeted therapy is a promising strategy for anticancer treatment. Epigenomes in cancer are related to many aspects during cancer initiation. A better understanding of the specific mechanisms underlying those alterations in different cancers is necessary. Meanwhile, optimized treatment options, including a variety of combinations, still remain to be discovered.
References
Waddington, C. H. The epigenotype. Int J. Epidemiol. 41, 10–13 (2012).
Holliday, R. The inheritance of epigenetic defects. Science 238, 163–170 (1987).
Bird, A. Perceptions of epigenetics. Nature 447, 396–398 (2007).
Shen, H. & Laird, P. W. Interplay between the cancer genome and epigenome. Cell 153, 38–55 (2013).
Holliday, R. A new theory of carcinogenesis. Br. J. Cancer 40, 513–522 (1979).
Seligson, D. B. et al. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 435, 1262–1266 (2005).
Fahrner, J. A., Eguchi, S., Herman, J. G. & Baylin, S. B. Dependence of histone modifications and gene expression on DNA hypermethylation in cancer. Cancer Res. 62, 7213–7218 (2002).
Ben-Porath, I. & Cedar, H. Epigenetic crosstalk. Mol. Cell. 8, 933–935 (2001).
Richards, E. J. & Elgin, S. C. Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell 108, 489–500 (2002).
Cedar, H. & Bergman, Y. Programming of DNA methylation patterns. Annu Rev. Biochem. 81, 97–117 (2012).
Sun, D. et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell. Stem Cell. 14, 673–688 (2014).
Valentini, E. et al. Analysis of the machinery and intermediates of the 5hmC-mediated DNA demethylation pathway in aging on samples from the MARK-AGE Study. Aging 8, 1896–1922 (2016).
Kulis, M. & Esteller, M. DNA methylation and cancer. Adv. Genet. 70, 27–56 (2010).
Karatzas, P. S., Mantzaris, G. J., Safioleas, M. & Gazouli, M. DNA methylation profile of genes involved in inflammation and autoimmunity in inflammatory bowel disease. Medicine 93, e309 (2014).
Easwaran, H., Tsai, H. C. & Baylin, S. B. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell. 54, 716–727 (2014).
Klutstein, M., Nejman, D., Greenfield, R. & Cedar, H. DNA methylation in cancer and aging. Cancer Res. 76, 3446–3450 (2016).
Keshet, I. et al. Evidence for an instructive mechanism of de novo methylation in cancer cells. Nat. Genet. 38, 149–153 (2006).
Ito, S. et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466, 1129–1133 (2010).
Keshet, I., Lieman-Hurwitz, J. & Cedar, H. DNA methylation affects the formation of active chromatin. Cell 44, 535–543 (1986).
Baylin, S. B. & Jones, P. A. A decade of exploring the cancer epigenome—biological and translational implications. Nat. Rev. Cancer 11, 726–734 (2011).
Merlo, A. et al. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat. Med. 1, 686–692 (1995).
Rideout, W. M., Coetzee, G. A., Olumi, A. F. & Jones, P. A. 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science 249, 1288–1290 (1990).
Kleihues, P. et al. Tumors associated with p53 germline mutations: a synopsis of 91 families. Am. J. Pathol. 150, 1–13 (1997).
Caputo, S. et al. Description and analysis of genetic variants in French hereditary breast and ovarian cancer families recorded in the UMD-BRCA1/BRCA2 databases. Nucleic Acids Res. 40, D992–D1002 (2012).
Christensen, B. C. et al. Breast cancer DNA methylation profiles are associated with tumor size and alcohol and folate intake. PLoS Genet. 6, e1001043 (2010).
Mayor, R. et al. Long-range epigenetic silencing at 2q14.2 affects most human colorectal cancers and may have application as a non-invasive biomarker of disease. Br. J. Cancer 100, 1534–1539 (2009).
Devaney, J. et al. Epigenetic deregulation across chromosome 2q14.2 differentiates normal from prostate cancer and provides a regional panel of novel DNA methylation cancer biomarkers. Cancer Epidemiol. Biomark. Prev. 20, 148–159 (2011).
Bell, A., Bell, D., Weber, R. S. & El-Naggar, A. K. CpG island methylation profiling in human salivary gland adenoid cystic carcinoma. Cancer 117, 2898–2909 (2011).
He, S. et al. Expression of DNMT1 and DNMT3a are regulated by GLI1 in human pancreatic cancer. PLoS ONE 6, e27684 (2011).
Ibrahim, A. E. et al. Sequential DNA methylation changes are associated with DNMT3B overexpression in colorectal neoplastic progression. Gut 60, 499–508 (2011).
Baylin, S. B. & Ohm, J. E. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction. Nat. Rev. Cancer 6, 107–116 (2006).
Markl, I. D. et al. Global and gene-specific epigenetic patterns in human bladder cancer genomes are relatively stable in vivo and in vitro over time. Cancer Res. 61, 5875–5884 (2001).
Wolf, S. F. et al. Methylation of the hypoxanthine phosphoribosyltransferase locus on the human X chromosome: implications for X-chromosome inactivation. Proc. Natl Acad. Sci. USA 81, 2806–2810 (1984).
Jones, P. A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492 (2012).
Lee, W. H. et al. Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc. Natl Acad. Sci. USA 91, 11733–11737 (1994).
Wilting, R. H. & Dannenberg, J. H. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist Updat. 15, 21–38 (2012).
Esteller, M. et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 343, 1350–1354 (2000).
Hegi, M. E. et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 352, 997–1003 (2005).
Esteller, M. et al. Hypermethylation of the DNA repair gene O(6)-methylguanine DNA methyltransferase and survival of patients with diffuse large B-cell lymphoma. J. Natl Cancer Inst. 94, 26–32 (2002).
Ramsahoye, B. H. et al. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl Acad. Sci. USA 97, 5237–5242 (2000).
Saxonov, S., Berg, P. & Brutlag, D. L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl Acad. Sci. USA 103, 1412–1417 (2006).
Smith, Z. D. & Meissner, A. DNA methylation: roles in mammalian development. Nat. Rev. Genet. 14, 204–220 (2013).
Mitchell, A. R. et al. Epigenetic control of mammalian centromere protein binding: does DNA methylation have a role. J. Cell Sci. 109(Pt 9), 2199–2206 (1996).
Schübeler, D. Function and information content of DNA methylation. Nature 517, 321–326 (2015).
Goyal, R., Reinhardt, R. & Jeltsch, A. Accuracy of DNA methylation pattern preservation by the Dnmt1 methyltransferase. Nucleic Acids Res. 34, 1182–1188 (2006).
Li, Y. et al. Stella safeguards the oocyte methylome by preventing de novo methylation mediated by DNMT1. Nature 564, 136–140 (2018).
Wu, H. & Zhang, Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 156, 45–68 (2014).
Okano, M., Bell, D. W., Haber, D. A. & Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257 (1999).
Chedin, F., Lieber, M. R. & Hsieh, C. L. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc. Natl Acad. Sci. USA 99, 16916–16921 (2002).
Robertson, K. D. et al. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res. 27, 2291–2298 (1999).
Piyathilake, C. J. et al. A higher degree of expression of DNA methyl transferase 1 in cervical cancer is associated with poor survival outcome. Int J. Women’s Health 9, 413–420 (2017).
Mizuno, S. et al. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood 97, 1172–1179 (2001).
Mirza, S. et al. Expression of DNA methyltransferases in breast cancer patients and to analyze the effect of natural compounds on DNA methyltransferases and associated proteins. J. Breast Cancer 16, 23–31 (2013).
Klose, R. J. & Bird, A. P. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 31, 89–97 (2006).
Feng, Q. & Zhang, Y. The MeCP1 complex represses transcription through preferential binding, remodeling, and deacetylating methylated nucleosomes. Genes Dev. 15, 827–832 (2001).
Yildirim, O. et al. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell 147, 1498–1510 (2011).
Villa, R. et al. The methyl-CpG binding protein MBD1 is required for PML-RARalpha function. Proc. Natl Acad. Sci. USA 103, 1400–1405 (2006).
Watanabe, S. et al. Methylated DNA-binding domain 1 and methylpurine-DNA glycosylase link transcriptional repression and DNA repair in chromatin. Proc. Natl Acad. Sci. USA 100, 12859–12864 (2003).
Bader, S. et al. MBD1, MBD2 and CGBP genes at chromosome 18q21 are infrequently mutated in human colon and lung cancers. Oncogene 22, 3506–3510 (2003).
Zhao, X. et al. Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proc. Natl Acad. Sci. USA 100, 6777–6782 (2003).
Sansom, O. J. et al. Deficiency of Mbd2 suppresses intestinal tumorigenesis. Nat. Genet. 34, 145–147 (2003).
Suzuki, H. et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat. Genet. 36, 417–422 (2004).
Campbell, P. M., Bovenzi, V. & Szyf, M. Methylated DNA-binding protein 2 antisense inhibitors suppress tumourigenesis of human cancer cell lines in vitro and in vivo. Carcinogenesis 25, 499–507 (2004).
Noh, E. J. et al. Methyl CpG-binding domain protein 3 mediates cancer-selective cytotoxicity by histone deacetylase inhibitors via differential transcriptional reprogramming in lung cancer cells. Cancer Res. 65, 11400–11410 (2005).
Biswas, M. et al. MBD3/NuRD loss participates with KDM6A program to promote DOCK5/8 expression and Rac GTPase activation in human acute myeloid leukemia. FASEB J. 33, 5268–5286 (2019).
Riccio, A. et al. The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with microsatellite instability. Nat. Genet. 23, 266–268 (1999).
Bader, S. A., Walker, M. & Harrison, D. J. A human cancer-associated truncation of MBD4 causes dominant negative impairment of DNA repair in colon cancer cells. Br. J. Cancer 96, 660–666 (2007).
Millar, C. B. et al. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science 297, 403–405 (2002).
Sansom, O. J., Bishop, S. M., Bird, A. & Clarke, A. R. MBD4 deficiency does not increase mutation or accelerate tumorigenesis in mice lacking MMR. Oncogene 23, 5693–5696 (2004).
Bernard, D. et al. The methyl-CpG-binding protein MECP2 is required for prostate cancer cell growth. Oncogene 25, 1358–1366 (2006).
Müller, H. M. et al. MeCP2 and MBD2 expression in human neoplastic and non-neoplastic breast tissue and its association with oestrogen receptor status. Br. J. Cancer 89, 1934–1939 (2003).
Mudbhary, R. et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell. 25, 196–209 (2014).
Unoki, M. et al. UHRF1 is a novel molecular marker for diagnosis and the prognosis of bladder cancer. Br. J. Cancer 101, 98–105 (2009).
Unoki, M. et al. UHRF1 is a novel diagnostic marker of lung cancer. Br. J. Cancer 103, 217–222 (2010).
Prokhortchouk, A. et al. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 15, 1613–1618 (2001).
van Roy, F. M. & McCrea, P. D. A role for Kaiso-p120ctn complexes in cancer. Nat. Rev. Cancer 5, 956–964 (2005).
Kim, K. et al. Identification of oncogenic microRNA-17–92/ZBTB4/specificity protein axis in breast cancer. Oncogene 31, 1034–1044 (2012).
Law, J. A. & Jacobsen, S. E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220 (2010).
Feng, S., Jacobsen, S. E. & Reik, W. Epigenetic reprogramming in plant and animal development. Science 330, 622–627 (2010).
Hajkova, P. et al. Epigenetic reprogramming in mouse primordial germ cells. Mech. Dev. 117, 15–23 (2002).
Tahiliani, M. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 (2009).
Hu, L. et al. Structural insight into substrate preference for TET-mediated oxidation. Nature 527, 118–122 (2015).
Ito, S. et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303 (2011).
He, Y. F. et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307 (2011).
Lorsbach, R. B. et al. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22; q23). Leukemia 17, 637–641 (2003).
Delhommeau, F. et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 360, 2289–2301 (2009).
Kosmider, O. et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica 94, 1676–1681 (2009).
Abdel-Wahab, O. et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 70, 447–452 (2010).
Yang, H. et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 32, 663–669 (2013).
Hsu, C. H. et al. TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep. 2, 568–579 (2012).
Lian, C. G. et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 150, 1135–1146 (2012).
Cao, Y. L. et al. Exosomal DNMT1 mediates cisplatin resistance in ovarian cancer. Cell Biochem Funct. 35, 296–303 (2017).
Rajendran, G. et al. Epigenetic regulation of DNA methyltransferases: DNMT1 and DNMT3B in gliomas. J. Neurooncol. 104, 483–494 (2011).
Xing, J. et al. Expression of methylation-related genes is associated with overall survival in patients with non-small cell lung cancer. Br. J. Cancer 98, 1716–1722 (2008).
Hong, L. et al. The interaction between miR-148a and DNMT1 suppresses cell migration and invasion by reactivating tumor suppressor genes in pancreatic cancer. Oncol. Rep. 40, 2916–2925 (2018).
Nagai, M., Nakamura, A., Makino, R. & Mitamura, K. Expression of DNA (5-cytosin)-methyltransferases (DNMTs) in hepatocellular carcinomas. Hepatol. Res. 26, 186–191 (2003).
Wang, Y., Hu, Y., Guo, J. & Wang, L. miR-148a-3p suppresses the proliferation and invasion of esophageal cancer by targeting DNMT1. Genet Test. Mol. Biomark. 23, 98–104 (2019).
Hu, Y. et al. LncRNA-SNHG1 contributes to gastric cancer cell proliferation by regulating DNMT1. Biochem Biophys. Res Commun. 491, 926–931 (2017).
Chen, F. et al. MiR-137 suppresses triple-negative breast cancer stemness and tumorigenesis by perturbing BCL11A-DNMT1 interaction. Cell Physiol. Biochem. 47, 2147–2158 (2018).
Gao, X. et al. Calpain-2 triggers prostate cancer metastasis via enhancing CRMP4 promoter methylation through NF-κB/DNMT1 signaling pathway. Prostate 78, 682–690 (2018).
Zhang, Y. et al. Effects of DNMT1 silencing on malignant phenotype and methylated gene expression in cervical cancer cells. J. Exp. Clin. Cancer Res. 30, 98 (2011).
Lee, E. et al. DNMT1 regulates epithelial-mesenchymal transition and cancer stem cells, which promotes prostate cancer metastasis. Neoplasia 18, 553–566 (2016).
Chen, B. F. et al. microRNA-199a-3p, DNMT3A, and aberrant DNA methylation in testicular cancer. Epigenetics 9, 119–128 (2014).
Sun, J. et al. miR-182 induces cervical cancer cell apoptosis through inhibiting the expression of DNMT3a. Int J. Clin. Exp. Pathol. 8, 4755–4763 (2015).
Pang, Y. et al. MYC and DNMT3A-mediated DNA methylation represses microRNA-200b in triple negative breast cancer. J. Cell Mol. Med. 22, 6262–6274 (2018).
Zhou, Y. et al. Ginsenoside 20(S)-Rg3 inhibits the warburg effect via modulating DNMT3A/ MiR-532-3p/HK2 pathway in ovarian cancer cells. Cell Physiol. Biochem. 45, 2548–2559 (2018).
Zhang, Q., Feng, Y., Liu, P. & Yang, J. MiR-143 inhibits cell proliferation and invasion by targeting DNMT3A in gastric cancer. Tumour Biol. 39, 1010428317711312 (2017).
Gao, Q. et al. Deletion of the de novo DNA methyltransferase Dnmt3a promotes lung tumor progression. Proc. Natl Acad. Sci. USA 108, 18061–18066 (2011).
Garzon, R. et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 113, 6411–6418 (2009).
Deivendran, S. et al. Metastasis-associated protein 1 is an upstream regulator of DNMT3a and stimulator of insulin-growth factor binding protein-3 in breast cancer. Sci. Rep. 7, 44225 (2017).
Tang, X. et al. Autocrine TGF-β1/miR-200s/miR-221/DNMT3B regulatory loop maintains CAF status to fuel breast cancer cell proliferation. Cancer Lett. 452, 79–89 (2019).
Chen, Z. et al. miR-124 and miR-506 inhibit colorectal cancer progression by targeting DNMT3B and DNMT1. Oncotarget 6, 38139–38150 (2015).
Qiu, W. et al. Kaempferol modulates DNA methylation and downregulates DNMT3B in bladder cancer. Cell Physiol. Biochem. 41, 1325–1335 (2017).
Peres, R. et al. Angiogenin contributes to bladder cancer tumorigenesis by DNMT3b-mediated MMP2 activation. Oncotarget 7, 43109–43123 (2016).
Song, N. et al. Lentivirus‑mediated knockdown of MeCP2 inhibits the growth of colorectal cancer cells in vitro. Mol. Med Rep. 13, 860–866 (2016).
Ray, B. K. et al. Epigenetic regulation by Z-DNA silencer function controls cancer-associated ADAM-12 expression in breast cancer: cross-talk between MeCP2 and NF1 transcription factor family. Cancer Res. 73, 736–744 (2013).
Xu, M. et al. MeCP2 suppresses LIN28A expression via binding to its methylated-CpG islands in pancreatic cancer cells. Oncotarget 7, 14476–14485 (2016).
Yaqinuddin, A. et al. Silencing of MBD1 and MeCP2 in prostate-cancer-derived PC3 cells produces differential gene expression profiles and cellular phenotypes. Biosci. Rep. 28, 319–326 (2008).
Zhang, B. et al. MBD1 is an epigenetic regulator of KEAP1 in pancreatic cancer. Curr. Mol. Med. 16, 404–411 (2016).
Pulukuri, S. M. & Rao, J. S. CpG island promoter methylation and silencing of 14-3-3σ gene expression in LNCaP and Tramp-C1 prostate cancer cell lines is associated with methyl-CpG-binding protein MBD2. Oncogene 25, 4559–4572 (2006).
Martin, V. et al. MBD2-mediated transcriptional repression of the p14ARF tumor suppressor gene in human colon cancer cells. Pathobiology 75, 281–287 (2008).
Pontes, T. B. et al. Reduced mRNA expression levels of MBD2 and MBD3 in gastric carcinogenesis. Tumour Biol. 35, 3447–3453 (2014).
Zhang, Y. et al. Epigenetic silencing of RNF144A expression in breast cancer cells through promoter hypermethylation and MBD4. Cancer Med. 7, 1317–1325 (2018).
Abdel-Rahman, W. M. et al. Truncation of MBD4 predisposes to reciprocal chromosomal translocations and alters the response to therapeutic agents in colon cancer cells. DNA Repair 7, 321–328 (2008).
Pierre, C. C., Hercules, S. M., Yates, C. & Daniel, J. M. Dancing from bottoms up—roles of the POZ-ZF transcription factor Kaiso in cancer. Biochim Biophys. Acta Rev. Cancer 1871, 64–74 (2019).
Wang, H. et al. Kaiso, a transcriptional repressor, promotes cell migration and invasion of prostate cancer cells through regulation of miR-31 expression. Oncotarget 7, 5677–5689 (2016).
Dai, S. D. et al. Kaiso is expressed in lung cancer: its expression and localization is affected by p120ctn. Lung Cancer 67, 205–215 (2010).
Bassey-Archibong, B. I. et al. Kaiso depletion attenuates the growth and survival of triple negative breast cancer cells. Cell Death Dis. 8, e2689 (2017).
Roussel-Gervais, A. et al. Loss of the methyl-CpG-binding protein ZBTB4 alters mitotic checkpoint, increases aneuploidy, and promotes tumorigenesis. Cancer Res. 77, 62–73 (2017).
Yu, Y. et al. Tumor suppressive ZBTB4 inhibits cell growth by regulating cell cycle progression and apoptosis in Ewing sarcoma. Biomed. Pharmacother. 100, 108–115 (2018).
Kim, K. et al. Induction of the transcriptional repressor ZBTB4 in prostate cancer cells by drug-induced targeting of microRNA-17-92/106b-25 clusters. Mol. Cancer Ther. 11, 1852–1862 (2012).
Jing, J. et al. The role of ZBTB38 in promoting migration and invasive growth of bladder cancer cells. Oncol. Rep. 41, 1980–1990 (2019).
Saidi, S., Popov, Z., Janevska, V. & Panov, S. Overexpression of UHRF1 gene correlates with the major clinicopathological parameters in urinary bladder cancer. Int Braz. J. Urol. 43, 224–229 (2017).
Jiao, D. et al. UHRF1 promotes renal cell carcinoma progression through epigenetic regulation of TXNIP. Oncogene 38, 5686–5699 (2019).
Kong, X. et al. Defining UHRF1 domains that support maintenance of human colon cancer DNA methylation and oncogenic properties. Cancer Cell. 35, 633–648.e7 (2019).
Hu, Q. et al. UHRF1 promotes aerobic glycolysis and proliferation via suppression of SIRT4 in pancreatic cancer. Cancer Lett. 452, 226–236 (2019).
Wei, C. et al. Upregulation of UHRF1 promotes the progression of melanoma by inducing cell proliferation. Oncol. Rep. 39, 2553–2562 (2018).
Gao, S. P. et al. UHRF1 promotes breast cancer progression by suppressing KLF17 expression by hypermethylating its promoter. Am. J. Cancer Res. 7, 1554–1565 (2017).
Lu, S. et al. Ubiquitin-like with PHD and ring finger domains 2 is a predictor of survival and a potential therapeutic target in colon cancer. Oncol. Rep. 31, 1802–1810 (2014).
Zhang, T. et al. UHRF2 decreases H3K9ac expression by interacting with it through the PHD and SRA/YDG domain in HepG2 hepatocellular carcinoma cells. Int J. Mol. Med. 39, 126–134 (2017).
Iguchi, T. et al. Identification of UHRF2 as a negative regulator of epithelial-mesenchymal transition and its clinical significance in esophageal squamous cell carcinoma. Oncology 95, 179–187 (2018).
Jin, C. et al. Loss of UHRF2 is associated with non-small cell lung carcinoma progression. J. Cancer 9, 2994–3005 (2018).
Si, Y. et al. Fisetin decreases TET1 activity and CCNY/CDK16 promoter 5hmC levels to inhibit the proliferation and invasion of renal cancer stem cell. J. Cell Mol. Med. 23, 1095–1105 (2019).
Huang, H. et al. TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc. Natl Acad. Sci. USA 110, 11994–11999 (2013).
Jiang, X. et al. Targeted inhibition of STAT/TET1 axis as a therapeutic strategy for acute myeloid leukemia. Nat. Commun. 8, 2099 (2017).
Chen, L. Y. et al. TET1 reprograms the epithelial ovarian cancer epigenome and reveals casein kinase 2α as a therapeutic target. J. Pathol. 248, 363–376 (2019).
Filipczak, P. T. et al. p53-Suppressed oncogene TET1 prevents cellular aging in lung cancer. Cancer Res. 79, 1758–1768 (2019).
Liu, C. et al. Decrease of 5-hydroxymethylcytosine is associated with progression of hepatocellular carcinoma through downregulation of TET1. PLoS ONE 8, e62828 (2013).
Guo, H. et al. TET1 suppresses colon cancer proliferation by impairing á-catenin signal pathway. J. Cell Biochem. 120, 12559–12565 (2019).
Cimmino, L. et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 170, 1079–1095.e20 (2017).
Langemeijer, S. M. et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat. Genet. 41, 838–842 (2009).
Zhu, X. & Li, S. TET2 inhibits tumorigenesis of breast cancer cells by regulating caspase-4. Sci. Rep. 8, 16167 (2018).
Huang, Y. et al. Loss of nuclear localization of TET2 in colorectal cancer. Clin. Epigenetics. 8, 9 (2016).
Chen, D. et al. TOP2A, HELLS, ATAD2, and TET3 are novel prognostic markers in renal cell carcinoma. Urology 102, 265.e1–265.e7 (2017).
Yang, L. et al. Reduced expression of TET1, TET2, TET3 and TDG mRNAs are associated with poor prognosis of patients with early breast cancer. PLoS ONE 10, e0133896 (2015).
Ye, Z. et al. TET3 inhibits TGF-β1-induced epithelial-mesenchymal transition by demethylating miR-30d precursor gene in ovarian cancer cells. J. Exp. Clin. Cancer Res. 35, 72 (2016).
Kouzarides, T. Chromatin modifications and their function. Cell 128, 693–705 (2007).
Audia, J. E. & Campbell, R. M. Histone modifications and cancer. Cold Spring Harb. Perspect. Biol. 8, a019521 (2016).
Hawkins, R. D. et al. Dynamic chromatin states in human ES cells reveal potential regulatory sequences and genes involved in pluripotency. Cell Res. 21, 1393–1409 (2011).
Hon, G. C., Hawkins, R. D. & Ren, B. Predictive chromatin signatures in the mammalian genome. Hum. Mol. Genet. 18, R195–R201 (2009).
Heintzman, N. D. et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112 (2009).
Seligson, D. B. et al. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 174, 1619–1628 (2009).
Fraga, M. F. et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 37, 391–400 (2005).
Di, C. V. & Schneider, R. Cancers with wrong HATs: the impact of acetylation. Brief. Funct. Genomics. 12, 231–243 (2013).
Sadoul, K., Wang, J., Diagouraga, B. & Khochbin, S. The tale of protein lysine acetylation in the cytoplasm. J. Biomed. Biotechnol. 2011, 970382 (2011).
Choudhary, C. et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 (2009).
Kleff, S., Andrulis, E. D., Anderson, C. W. & Sternglanz, R. Identification of a gene encoding a yeast histone H4 acetyltransferase. J. Biol. Chem. 270, 24674–24677 (1995).
Brownell, J. E. et al. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 84, 843–851 (1996).
Marmorstein, R. & Zhou, M. M. Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 6, a018762 (2014).
Heery, D. M. & Fischer, P. M. Pharmacological targeting of lysine acetyltransferases in human disease: a progress report. Drug Disco. Today 12, 88–99 (2007).
Yao, T. P. et al. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 93, 361–372 (1998).
Oike, Y. et al. Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: implications for a dominant-negative mechanism. Hum. Mol. Genet. 8, 387–396 (1999).
Tanner, K. G. et al. Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. J. Biol. Chem. 274, 18157–18160 (1999).
Tanner, K. G., Langer, M. R. & Denu, J. M. Kinetic mechanism of human histone acetyltransferase P/CAF. Biochemistry 39, 15652 (2000).
Yan, Y., Harper, S., Speicher, D. W. & Marmorstein, R. The catalytic mechanism of the ESA1 histone acetyltransferase involves a self-acetylated intermediate. Nat. Struct. Biol. 9, 862–869 (2002).
Liu, X. et al. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature 451, 846–850 (2008).
Marmorstein, R. Biochemical and structural characterization of recombinant histone acetyltransferase proteins. Methods Enzymol. 376, 106–119 (2004).
Chen, Y. F. et al. High expression levels of COX-2 and P300 are associated with unfavorable survival in laryngeal squamous cell carcinoma. Eur. Arch. Otorhinolaryngol. 270, 1009–1017 (2013).
Korzus, E. Rubinstein-Taybi syndrome and epigenetic alterations. Adv. Exp. Med. Biol. 978, 39–62 (2017).
Miller, R. W. & Rubinstein, J. H. Tumors in Rubinstein-Taybi syndrome. Am. J. Med. Genet. 56, 112–115 (1995).
Borrow, J. et al. The translocation t(8;16)(p11; p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat. Genet. 14, 33–41 (1996).
Panagopoulos, I. et al. Fusion of the MORF and CBP genes in acute myeloid leukemia with the t(10;16)(q22; p13). Hum. Mol. Genet. 10, 395–404 (2001).
Chaffanet, M. et al. MOZ is fused to p300 in an acute monocytic leukemia with t(8;22). Genes Chromosomes Cancer 28, 138–144 (2000).
Ida, K. et al. Adenoviral E1A-associated protein p300 is involved in acute myeloid leukemia with t(11;22)(q23;q13). Blood 90, 4699–4704 (1997).
Taki, T., Sako, M., Tsuchida, M. & Hayashi, Y. The t(11;16)(q23;p13) translocation in myelodysplastic syndrome fuses the MLL gene to the CBP gene. Blood 89, 3945–3950 (1997).
Muraoka, M. et al. p300 gene alterations in colorectal and gastric carcinomas. Oncogene 12, 1565–1569 (1996).
Gayther, S. A. et al. Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 24, 300–303 (2000).
Ozdağ, H. et al. Mutation analysis of CBP and PCAF reveals rare inactivating mutations in cancer cell lines but not in primary tumours. Br. J. Cancer 87, 1162–1165 (2002).
Burwinkel, B. et al. Association of NCOA3 polymorphisms with breast cancer risk. Clin. Cancer Res. 11, 2169–2174 (2005).
Wachtel, M. et al. Gene expression signatures identify rhabdomyosarcoma subtypes and detect a novel t(2;2)(q35; p23) translocation fusing PAX3 to NCOA1. Cancer Res. 64, 5539–5545 (2004).
Dhalluin, C. et al. Structure and ligand of a histone acetyltransferase bromodomain. Nature 399, 491–496 (1999).
Thompson, M. Polybromo-1: the chromatin targeting subunit of the PBAF complex. Biochimie 91, 309–319 (2009).
Zhang, Q. et al. Biochemical profiling of histone binding selectivity of the yeast bromodomain family. PLoS ONE 5, e8903 (2010).
Sanchez, R. & Zhou, M. M. The role of human bromodomains in chromatin biology and gene transcription. Curr. Opin. Drug Disco. Devel. 12, 659–665 (2009).
Filippakopoulos, P. & Knapp, S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 13, 337–356 (2014).
Li, Y. et al. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell 159, 558–571 (2014).
Morin, R. D. et al. Genetic landscapes of relapsed and refractory diffuse large B-cell lymphomas. Clin. Cancer Res. 22, 2290–2300 (2016).
Perlman, E. J. et al. MLLT1 YEATS domain mutations in clinically distinctive Favourable Histology Wilms tumours. Nat. Commun. 6, 10013 (2015).
Chiang, C. M. Brd4 engagement from chromatin targeting to transcriptional regulation: selective contact with acetylated histone H3 and H4. F1000 Biol. Rep. 1, 98 (2009).
Zeng, L. & Zhou, M. M. Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 513, 124–128 (2002).
Pirrotta, V. The necessity of chromatin: a view in perspective. Cold Spring Harb. Perspect. Biol. 8, a019547 (2016).
Yang, Z. et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell. 19, 535–545 (2005).
Phelps, M. A. et al. Clinical response and pharmacokinetics from a phase 1 study of an active dosing schedule of flavopiridol in relapsed chronic lymphocytic leukemia. Blood 113, 2637–2645 (2009).
Rahl, P. B. et al. c-Myc regulates transcriptional pause release. Cell 141, 432–445 (2010).
French, C. A. et al. BRD-NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene 27, 2237–2242 (2008).
Zuber, J. et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528 (2011).
de Ruijter, A. J. et al. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 370, 737–749 (2003).
Verdin, E., Dequiedt, F. & Kasler, H. G. Class II histone deacetylases: versatile regulators. Trends Genet. 19, 286–293 (2003).
Seto, E. & Yoshida, M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 6, a018713 (2014).
Imai, S., Armstrong, C. M., Kaeberlein, M. & Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 (2000).
Liszt, G., Ford, E., Kurtev, M. & Guarente, L. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J. Biol. Chem. 280, 21313–21320 (2005).
Frye, R. A. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem Biophys. Res. Commun. 260, 273–279 (1999).
Du, J. et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 (2011).
Johnson, C. A. et al. Human class I histone deacetylase complexes show enhanced catalytic activity in the presence of ATP and co-immunoprecipitate with the ATP-dependent chaperone protein Hsp70. J. Biol. Chem. 277, 9590–9597 (2002).
Zhang, X. et al. Activation of the growth-differentiation factor 11 gene by the histone deacetylase (HDAC) inhibitor trichostatin A and repression by HDAC3. Mol. Cell Biol. 24, 5106–5118 (2004).
Hubbert, C. et al. HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458 (2002).
Luo, J. et al. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408, 377–381 (2000).
Vaziri, H. et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159 (2001).
Peng, L. et al. SIRT1 negatively regulates the activities, functions, and protein levels of hMOF and TIP60. Mol. Cell Biol. 32, 2823–2836 (2012).
Ayer, D. E. Histone deacetylases: transcriptional repression with SINers and NuRDs. Trends Cell Biol. 9, 193–198 (1999).
Wen, Y. D. et al. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc. Natl Acad. Sci. USA 97, 7202–7207 (2000).
Watson, P. J., Fairall, L., Santos, G. M. & Schwabe, J. W. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 481, 335–340 (2012).
Kang, H. et al. Peptide switch is essential for Sirt1 deacetylase activity. Mol. Cell. 44, 203–213 (2011).
Kim, E. J., Kho, J. H., Kang, M. R. & Um, S. J. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol. Cell. 28, 277–290 (2007).
Ocker, M. & Schneider-Stock, R. Histone deacetylase inhibitors: signalling towards p21cip1/waf1. Int J. Biochem Cell Biol. 39, 1367–1374 (2007).
Ozdağ, H. et al. Differential expression of selected histone modifier genes in human solid cancers. BMC Genomics. 7, 90 (2006).
Xue, L. et al. RNAi screening identifies HAT1 as a potential drug target in esophageal squamous cell carcinoma. Int J. Clin. Exp. Pathol. 7, 3898–3907 (2014).
Fan, P. et al. Overexpressed histone acetyltransferase 1 regulates cancer immunity by increasing programmed death-ligand 1 expression in pancreatic cancer. J. Exp. Clin. Cancer Res. 38, 47 (2019).
Miao, B. P. et al. Histone acetyltransferase 1 up regulates Bcl2L12 expression in nasopharyngeal cancer cells. Arch. Biochem Biophys. 646, 72–79 (2018).
Jin, X., Tian, S. & Li, P. Histone acetyltransferase 1 promotes cell proliferation and induces cisplatin resistance in hepatocellular carcinoma. Oncol. Res. 25, 939–946 (2017).
Zhang, J., Liu, M., Liu, W. & Wang, W. Ras-ERK1/2 signalling promotes the development of osteosarcoma through regulation of H4K12ac through HAT1. Artif. Cells Nanomed. Biotechnol. 47, 1207–1215 (2019).
Han, N. et al. HAT1 induces lung cancer cell apoptosis via up regulating Fas. Oncotarget 8, 89970–89977 (2017).
Yin, Y. W. et al. The histone acetyltransferase GCN5 expression is elevated and regulated by c-Myc and E2F1 transcription factors in human colon cancer. Gene Expr. 16, 187–196 (2015).
Zhao, L., Pang, A. & Li, Y. Function of GCN5 in the TGF-β1-induced epithelial-to-mesenchymal transition in breast cancer. Oncol. Lett. 16, 3955–3963 (2018).
Zhao, C. et al. C5a induces A549 cell proliferation of non-small cell lung cancer via GDF15 gene activation mediated by GCN5-dependent KLF5 acetylation. Oncogene 37, 4821–4837 (2018).
Watts, G. S. et al. The acetyltransferase p300/CBP-associated factor is a p53 target gene in breast tumor cells. Neoplasia 6, 187–194 (2004).
Brasacchio, D. et al. Down-regulation of a pro-apoptotic pathway regulated by PCAF/ADA3 in early stage gastric cancer. Cell Death Dis. 9, 442 (2018).
Yu, C. et al. Phenethyl isothiocyanate inhibits androgen receptor-regulated transcriptional activity in prostate cancer cells through suppressing PCAF. Mol. Nutr. Food Res. 57, 1825–1833 (2013).
Xie, X. et al. Tip60 is associated with resistance to X-ray irradiation in prostate cancer. FEBS Open Bio. 8, 271–278 (2018).
Wang, L. T. et al. TIP60-dependent acetylation of the SPZ1-TWIST complex promotes epithelial-mesenchymal transition and metastasis in liver cancer. Oncogene 38, 518–532 (2019).
Sakuraba, K. et al. Down-regulation of Tip60 gene as a potential marker for the malignancy of colorectal cancer. Anticancer Res. 29, 3953–3955 (2009).
McGuire, A. et al. Quantifying Tip60 (Kat5) stratifies breast cancer. Sci. Rep. 9, 3819 (2019).
Yang, Y. et al. Tat-interactive protein-60KDA (TIP60) regulates the tumorigenesis of lung cancer in vitro. J. Cancer 8, 2277–2281 (2017).
Jaganathan, A. et al. Coactivator MYST1 regulates nuclear factor-κB and androgen receptor functions during proliferation of prostate cancer cells. Mol. Endocrinol. 28, 872–885 (2014).
Hayashi, Y. et al. NUP98-HBO1-fusion generates phenotypically and genetically relevant chronic myelomonocytic leukemia pathogenesis. Blood Adv. 3, 1047–1060 (2019).
Quintela, M. et al. HBO1 directs histone H4 specific acetylation, potentiating mechano-transduction pathways and membrane elasticity in ovarian cancer cells. Nanomedicine 17, 254–265 (2019).
Chen, Z. et al. HBO1 promotes cell proliferation in bladder cancer via activation of Wnt/β-catenin signaling. Mol. Carcinog. 57, 12–21 (2018).
Deguchi, K. et al. MOZ-TIF2-induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer Cell. 3, 259–271 (2003).
Mohammadi, K. et al. Investigation of the changes in the expression levels of MOZ gene in colorectal cancer tissues. J. Gastrointest. Oncol. 10, 68–73 (2019).
Yu, L. et al. Identification of MYST3 as a novel epigenetic activator of ERα frequently amplified in breast cancer. Oncogene 36, 2910–2918 (2017).
Vizmanos, J. L. et al. t(10;16)(q22; p13) and MORF-CREBBP fusion is a recurrent event in acute myeloid leukemia. Genes Chromosomes Cancer 36, 402–405 (2003).
Ono, H., Basson, M. D. & Ito, H. P300 inhibition enhances gemcitabine-induced apoptosis of pancreatic cancer. Oncotarget 7, 51301–51310 (2016).
Pattabiraman, D. R. et al. Interaction of c-Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood 123, 2682–2690 (2014).
Hou, X. et al. p300 promotes proliferation, migration, and invasion via inducing epithelial-mesenchymal transition in non-small cell lung cancer cells. BMC Cancer 18, 641 (2018).
Xu, B. et al. The LIM protein Ajuba recruits DBC1 and CBP/p300 to acetylate ERα and enhances ERα target gene expression in breast cancer cells. Nucleic Acids Res. 47, 2322–2335 (2019).
Wang, Y. M. et al. Histone acetyltransferase p300/CBP inhibitor C646 blocks the survival and invasion pathways of gastric cancer cell lines. Int J. Oncol. 51, 1860–1868 (2017).
He, H. et al. Selective p300 inhibitor C646 inhibited HPV E6-E7 genes, altered glucose metabolism and induced apoptosis in cervical cancer cells. Eur. J. Pharmacol. 812, 206–215 (2017).
Huh, J. W. et al. Prognostic impact of p300 expression in patients with colorectal canceu. J. Surg. Oncol. 108, 374–377 (2013).
Takeuchi, A. et al. p300 mediates cellular resistance to doxorubicin in bladder cancer. Mol. Med Rep. 5, 173–176 (2012).
Inagaki, Y. et al. Epigenetic regulation of proliferation and invasion in hepatocellular carcinoma cells by CBP/p300 histone acetyltransferase activity. Int J. Oncol. 48, 533–540 (2016).
Díaz-Beyá, M. et al. Acute myeloid leukemia with translocation (8;16)(p11; p13) and MYST3-CREBBP rearrangement harbors a distinctive microRNA signature targeting RET proto-oncogene. Leukemia 27, 595–603 (2013).
Jin, L. et al. Therapeutic targeting of the CBP/p300 bromodomain blocks the growth of castration-resistant prostate cancer. Cancer Res. 77, 5564–5575 (2017).
Ding, L. et al. CBP loss cooperates with PTEN haploinsufficiency to drive prostate cancer: implications for epigenetic therapy. Cancer Res. 74, 2050–2061 (2014).
Jia, D. et al. Crebbp loss drives small cell lung cancer and increases sensitivity to HDAC inhibition. Cancer Discov. 8, 1422–1437 (2018).
Pavón, M. A. et al. CKMT1 and NCOA1 expression as a predictor of clinical outcome in patients with advanced-stage head and neck squamous cell carcinoma. Head. Neck. 38(Suppl 1), E1392–E1403 (2016).
Qin, L. et al. NCOA1 directly targets M-CSF1 expression to promote breast cancer metastasis. Cancer Res. 74, 3477–3488 (2014).
Ma, Y. S. et al. High expression of miR-105–1 positively correlates with clinical prognosis of hepatocellular carcinoma by targeting oncogene NCOA1. Oncotarget 8, 11896–11905 (2017).
Aikawa, Y. et al. PU.1-mediated upregulation of CSF1R is crucial for leukemia stem cell potential induced by MOZ-TIF2. Nat. Med. 16, 580–585 (2010). 1p following 585.
Fancher, A. T. et al. Reconfiguring the AR-TIF2 protein-protein interaction HCS Assay in Prostate Cancer Cells and Characterizing the Hits from a LOPAC Screen. Assay. Drug Dev. Technol. 14, 453–477 (2016).
O’Donnell, K. A. et al. A Sleeping Beauty mutagenesis screen reveals a tumor suppressor role for Ncoa2/Src-2 in liver cancer. Proc. Natl Acad. Sci. USA 109, E1377–E1386 (2012).
Yu, J. et al. Disruption of NCOA2 by recurrent fusion with LACTB2 in colorectal cancer. Oncogene 35, 187–195 (2016).
Kumar, S. et al. NCOA3-mediated upregulation of mucin expression via transcriptional and post-translational changes during the development of pancreatic cancer. Oncogene 34, 4879–4889 (2015).
Alkner, S. et al. Prognostic and predictive importance of the estrogen receptor coactivator AIB1 in a randomized trial comparing adjuvant letrozole and tamoxifen therapy in postmenopausal breast cancer: the Danish cohort of BIG 1–98. Breast Cancer Res. Treat. 166, 481–490 (2017).
Zou, J. X. et al. ACTR/AIB1/SRC-3 and androgen receptor control prostate cancer cell proliferation and tumor growth through direct control of cell cycle genes. Prostate 66, 1474–1486 (2006).
Xie, D. et al. Correlation of AIB1 overexpression with advanced clinical stage of human colorectal carcinoma. Hum. Pathol. 36, 777–783 (2005).
Liu, W. H. & Chang, L. S. Arachidonic acid induces Fas and FasL upregulation in human leukemia U937 cells via Ca2+/ROS-mediated suppression of ERK/c-Fos pathway and activation of p38 MAPK/ATF-2 pathway. Toxicol. Lett. 191, 140–148 (2009).
Desai, S., Laskar, S. & Pandey, B. N. Autocrine IL-8 and VEGF mediate epithelial-mesenchymal transition and invasiveness via p38/JNK-ATF-2 signalling in A549 lung cancer cells. Cell Signal. 25, 1780–1791 (2013).
Sun, X., Lou, L., Zhong, K. & Wan, L. MicroRNA-451 regulates chemoresistance in renal cell carcinoma by targeting ATF-2 gene. Exp. Biol. Med. 242, 1299–1305 (2017).
Winter, A. G. et al. RNA polymerase III transcription factor TFIIIC2 is overexpressed in ovarian tumors. Proc. Natl Acad. Sci. USA 97, 12619–12624 (2000).
Xiao, L. et al. Induction of the CLOCK gene by E2-ERα signaling promotes the proliferation of breast cancer cells. PLoS ONE 9, e95878 (2014).
Xu, H., Wang, Z., Mo, G. & Chen, H. Association between circadian gene CLOCK and cisplatin resistance in ovarian cancer cells: a preliminary study. Oncol. Lett. 15, 8945–8950 (2018).
Xiang, R. et al. Circadian clock gene Per2 downregulation in non‑small cell lung cancer is associated with tumour progression and metastasis. Oncol. Rep. 40, 3040–3048 (2018).
Xie, X. W. et al. Expression of CIITA-related MHCII molecules in tumors linked to prognosis in hepatocellular carcinoma. Int J. Oncol. 34, 681–688 (2009).
Truax, A. D., Thakkar, M. & Greer, S. F. Dysregulated recruitment of the histone methyltransferase EZH2 to the class II transactivator (CIITA) promoter IV in breast cancer cells. PLoS ONE 7, e36013 (2012).
Satoh, A. et al. Epigenetic inactivation of class II transactivator (CIITA) is associated with the absence of interferon-gamma-induced HLA-DR expression in colorectal and gastric cancer cells. Oncogene 23, 8876–8886 (2004).
Starska, K. et al. Gene and protein expression of O-GlcNAc-cycling enzymes in human laryngeal cancer. Clin. Exp. Med. 15, 455–468 (2015).
Krześlak, A. et al. Gene expression of O-GlcNAc cycling enzymes in human breast cancers. Clin. Exp. Med. 12, 61–65 (2012).
Fontanals-Cirera, B. et al. Harnessing BET inhibitor sensitivity reveals AMIGO2 as a melanoma survival gene. Mol. Cell. 68, 731–744.e9 (2017).
Pérez-Pena, J. et al. Mapping Bromodomains in breast cancer and association with clinical outcome. Sci. Rep. 9, 5734 (2019).
Handoko, L. et al. JQ1 affects BRD2-dependent and independent transcription regulation without disrupting H4-hyperacetylated chromatin states. Epigenetics 13, 410–431 (2018).
Sherman, M. H. et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc. Natl Acad. Sci. USA 114, 1129–1134 (2017).
Zhang, Q. et al. By recruiting HDAC1, MORC2 suppresses p21 Waf1/Cip1 in gastric cancer. Oncotarget 6, 16461–16470 (2015).
Dong, Z. Y., Zhou, Y. R. & Wang, L. X. HDAC1 is indirectly involved in the epigenetic regulation of p38 MAPK that drive the lung cancer progression. Eur. Rev. Med. Pharm. Sci. 22, 5980–5986 (2018).
Zhang, N. et al. SREBP1, targeted by miR-18a-5p, modulates epithelial-mesenchymal transition in breast cancer via forming a co-repressor complex with Snail and HDAC1/2. Cell Death Differ. 26, 843–859 (2019).
Li, S. Z. et al. Nemo-like kinase (NLK) primes colorectal cancer progression by releasing the E2F1 complex from HDAC1. Cancer Lett. 431, 43–53 (2018).
von, B. J. et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology 137, 361–371.e1–5 (2009).
Tang, W. et al. The p300/YY1/miR-500a-5p/HDAC2 signalling axis regulates cell proliferation in human colorectal cancer. Nat. Commun. 10, 663 (2019).
Zhu, W. et al. FKBP3 promotes proliferation of non-small cell lung cancer cells through regulating Sp1/HDAC2/p27. Theranostics 7, 3078–3089 (2017).
Roy, S. S. et al. Significance of PELP1/HDAC2/miR-200 regulatory network in EMT and metastasis of breast cancer. Oncogene 33, 3707–3716 (2014).
Hulsurkar, M. et al. Beta-adrenergic signaling promotes tumor angiogenesis and prostate cancer progression through HDAC2-mediated suppression of thrombospondin-1. Oncogene 36, 1525–1536 (2017).
Miao, L. J. et al. Stat3 inhibits Beclin 1 expression through recruitment of HDAC3 in nonsmall cell lung cancer cells. Tumour Biol. 35, 7097–7103 (2014).
Chen, G. et al. Long noncoding RNA SBF2-AS1 promotes colorectal cancer proliferation and invasion by inhibiting miR-619–5p activity and facilitating HDAC3 expression. J. Cell Physiol. 234, 18688–18696 (2019).
Bayat, S. et al. Downregulation of HDAC2 and HDAC3 via oleuropein as a potent prevention and therapeutic agent in MCF-7 breast cancer cells. J. Cell Biochem. 120, 9172–9180 (2019).
Lucas, J. et al. Upregulation of PD‑L1 expression by resveratrol and piceatannol in breast and colorectal cancer cells occurs via HDAC3/p300‑mediated NF‑κB signaling. Int J. Oncol. 53, 1469–1480 (2018).
Kang, Y. et al. HDAC8 and STAT3 repress BMF gene activity in colon cancer cells. Cell Death Dis. 5, e1476 (2014).
Vanaja, G. R., Ramulu, H. G. & Kalle, A. M. Overexpressed HDAC8 in cervical cancer cells shows functional redundancy of tubulin deacetylation with HDAC6. Cell Commun. Signal. 16, 20 (2018).
Wang, Z. T. et al. Histone deacetylase inhibitors suppress mutant p53 transcription via HDAC8/YY1 signals in triple negative breast cancer cells. Cell Signal. 28, 506–515 (2016).
Wu, S. et al. Suberoylanilide hydroxamic acid (SAHA) promotes the epithelial mesenchymal transition of triple negative breast cancer cells via HDAC8/FOXA1 signals. Biol. Chem. 397, 75–83 (2016).
Halkidou, K. et al. Nuclear accumulation of histone deacetylase 4 (HDAC4) coincides with the loss of androgen sensitivity in hormone refractory cancer of the prostate. Eur. Urol. 45, 382–389 (2004).
Wang, B. et al. A dual role of miR-22 modulated by RelA/p65 in resensitizing fulvestrant-resistant breast cancer cells to fulvestrant by targeting FOXP1 and HDAC4 and constitutive acetylation of p53 at Lys382. Oncogenesis 7, 54 (2018).
Wei, J. Y. et al. Melatonin induces apoptosis of colorectal cancer cells through HDAC4 nuclear import mediated by CaMKII inactivation. J. Pineal Res. 58, 429–438 (2015).
Cao, C. et al. HDAC5-LSD1 axis regulates antineoplastic effect of natural HDAC inhibitor sulforaphane in human breast cancer cells. Int. J. Cancer 143, 1388–1401 (2018).
Liu, C. et al. Hypermethylation of miRNA-589 promoter leads to upregulation of HDAC5 which promotes malignancy in non-small cell lung cancer. Int. J. Oncol. 50, 2079–2090 (2017).
Sun, S. et al. HDAC6 inhibitor TST strengthens the antiproliferative effects of PI3K/mTOR inhibitor BEZ235 in breast cancer cells via suppressing RTK activation. Cell Death Dis. 9, 929 (2018).
Won, H. R. et al. A452, an HDAC6-selective inhibitor, synergistically enhances the anticancer activity of chemotherapeutic agents in colorectal cancer cells. Mol. Carcinog. 57, 1383–1395 (2018).
Wang, Z., Hu, P., Tang, F. & Xie, C. HDAC6-mediated EGFR stabilization and activation restrict cell response to sorafenib in non-small cell lung cancer cells. Med. Oncol. 33, 50 (2016).
Seidel, C. et al. 4-Hydroxybenzoic acid derivatives as HDAC6-specific inhibitors modulating microtubular structure and HSP90α chaperone activity against prostate cancer. Biochem. Pharmacol. 99, 31–52 (2016).
Witt, A. E. et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene 36, 1707–1720 (2017).
Wu, M. Y. et al. MiR-34a regulates therapy resistance by targeting HDAC1 and HDAC7 in breast cancer. Cancer Lett. 354, 311–319 (2014).
Miremadi, A., Oestergaard, M. Z., Pharoah, P. D. & Caldas, C. Cancer genetics of epigenetic genes. Hum. Mol. Genet. 16, R28–R49 (2007).
Salgado, E. et al. HDAC9 overexpression confers invasive and angiogenic potential to triple negative breast cancer cells via modulating microRNA-206. Biochem Biophys. Res. Commun. 503, 1087–1091 (2018).
Okudela, K. et al. Expression of HDAC9 in lung cancer-potential role in lung carcinogenesis. Int. J. Clin. Exp. Pathol. 7, 213–220 (2014).
Yang, Y. et al. HDAC10 promotes lung cancer proliferation via AKT phosphorylation. Oncotarget 7, 59388–59401 (2016).
Islam, M. M. et al. HDAC10 as a potential therapeutic target in ovarian cancer. Gynecol. Oncol. 144, 613–620 (2017).
Qin, J. et al. Hypoxia-inducible factor 1 alpha promotes cancer stem cells-like properties in human ovarian cancer cells by upregulating SIRT1 expression. Sci. Rep. 7, 10592 (2017).
Santolla, M. F. et al. SIRT1 is involved in oncogenic signaling mediated by GPER in breast cancer. Cell Death Dis. 6, e1834 (2015).
Sun, L. et al. A SUMOylation-dependent pathway regulates SIRT1 transcription and lung cancer metastasis. J. Natl Cancer Inst. 105, 887–898 (2013).
Farooqi, A. S. et al. Novel lysine-based thioureas as mechanism-based inhibitors of Sirtuin 2 (SIRT2) with anticancer activity in a colorectal cancer murine model. J. Med Chem. 62, 4131–4141 (2019).
Hoffmann, G., Breitenbücher, F., Schuler, M. & Ehrenhofer-Murray, A. E. A novel sirtuin 2 (SIRT2) inhibitor with p53-dependent pro-apoptotic activity in non-small cell lung cancer. J. Biol. Chem. 289, 5208–5216 (2014).
Li, Y. et al. SIRT2 promotes the migration and invasion of gastric cancer through RAS/ERK/JNK/MMP-9 pathway by increasing PEPCK1-related metabolism. Neoplasia 20, 745–756 (2018).
Li, Z. et al. SIRT2 inhibits non-small cell lung cancer cell growth through impairing Skp2-mediated p27 degradation. Oncotarget 7, 18927–18939 (2016).
Fiskus, W. et al. SIRT2 deacetylates and inhibits the peroxidase activity of peroxiredoxin-1 to sensitize breast cancer cells to oxidant stress-inducing agents. Cancer Res. 76, 5467–5478 (2016).
Damodaran, S. et al. Dysregulation of Sirtuin 2 (SIRT2) and histone H3K18 acetylation pathways associates with adverse prostate cancer outcomes. BMC Cancer 17, 874 (2017).
Xiong, Y. et al. SIRT3 deacetylates and promotes degradation of P53 in PTEN-defective non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 144, 189–198 (2018).
Wang, S. et al. Programmed death ligand 1 promotes lymph node metastasis and glucose metabolism in cervical cancer by activating integrin β4/SNAI1/SIRT3 signaling pathway. Oncogene 37, 4164–4180 (2018).
Wu, Y. et al. SIRT3 aggravates metformin-induced energy stress and apoptosis in ovarian cancer cells. Exp. Cell Res. 367, 137–149 (2018).
Quan, Y. et al. SIRT3 inhibits prostate cancer by destabilizing oncoprotein c-MYC through regulation of the PI3K/Akt pathway. Oncotarget 6, 26494–26507 (2015).
Lee, D. Y. et al. Regulation of SIRT3 signal related metabolic reprogramming in gastric cancer by Helicobacter pylori oncoprotein CagA. Oncotarget 8, 78365–78378 (2017).
Miyo, M. et al. Tumour-suppressive function of SIRT4 in human colorectal cancer. Br. J. Cancer 113, 492–499 (2015).
Sun, H. et al. SIRT4 acts as a tumor suppressor in gastric cancer by inhibiting cell proliferation, migration, and invasion. Onco Targets Ther. 11, 3959–3968 (2018).
Xu, L. et al. SIRT5 as a biomarker for response to anthracycline-taxane-based neoadjuvant chemotherapy in triple-negative breast cancer. Oncol. Rep. 39, 2315–2323 (2018).
Shi, L. et al. SIRT5-mediated deacetylation of LDHB promotes autophagy and tumorigenesis in colorectal cancer. Mol. Oncol. 13, 358–375 (2019).
Lu, W., Zuo, Y., Feng, Y. & Zhang, M. SIRT5 facilitates cancer cell growth and drug resistance in non-small cell lung cancer. Tumour Biol. 35, 10699–10705 (2014).
Tang, Z. et al. CDK2 positively regulates aerobic glycolysis by suppressing SIRT5 in gastric cancer. Cancer Sci. 109, 2590–2598 (2018).
Chen, X. F. et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 19, e45124 (2018).
Liu, Y. et al. Inhibition of SIRT6 in prostate cancer reduces cell viability and increases sensitivity to chemotherapeutics. Protein Cell. 4, 702–710 (2013).
Bauer, I. et al. The NAD+-dependent histone deacetylase SIRT6 promotes cytokine production and migration in pancreatic cancer cells by regulating Ca2+ responses. J. Biol. Chem. 287, 40924–40937 (2012).
Bai, L. et al. Upregulation of SIRT6 predicts poor prognosis and promotes metastasis of non-small cell lung cancer via the ERK1/2/MMP9 pathway. Oncotarget 7, 40377–40386 (2016).
Min, L. et al. Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat. Cell Biol. 14, 1203–1211 (2012).
Thirumurthi, U. et al. MDM2-mediated degradation of SIRT6 phosphorylated by AKT1 promotes tumorigenesis and trastuzumab resistance in breast cancer. Sci. Signal. 7, ra71 (2014).
Han, Y. et al. Hsa-miR-125b suppresses bladder cancer development by down-regulating oncogene SIRT7 and oncogenic long noncoding RNA MALAT1. FEBS Lett. 23, 3875–3882 (2013).
Yu, H. et al. Overexpression of sirt7 exhibits oncogenic property and serves as a prognostic factor in colorectal cancer. Clin. Cancer Res. 20, 3434–3445 (2014).
Tang, M. et al. Downregulation of SIRT7 by 5-fluorouracil induces radiosensitivity in human colorectal cancer. Theranostics 7, 1346–1359 (2017).
Tang, X. et al. SIRT7 antagonizes TGF-β signaling and inhibits breast cancer metastasis. Nat. Commun. 8, 318 (2017).
Jiang, Y., Han, Z., Wang, Y. & Hao, W. Depletion of SIRT7 sensitizes human non-small cell lung cancer cells to gemcitabine therapy by inhibiting autophagy. Biochem Biophys. Res. Commun. 506, 266–271 (2018).
Deubzer, H. E. et al. HDAC11 is a novel drug target in carcinomas. Int J. Cancer 132, 2200–2208 (2013).
Gong, D., Zeng, Z., Yi, F. & Wu, J. Inhibition of histone deacetylase 11 promotes human liver cancer cell apoptosis. Am. J. Transl. Res. 11, 983–990 (2019).
Huang, R. et al. Clinicopathological features and prediction values of HDAC1, HDAC2, HDAC3, and HDAC11 in classical Hodgkin lymphoma. Anticancer Drugs 29, 364–370 (2018).
Thole, T. M. et al. Neuroblastoma cells depend on HDAC11 for mitotic cell cycle progression and survival. Cell Death Dis. 8, e2635 (2017).
Völkel, P. & Angrand, P. O. The control of histone lysine methylation in epigenetic regulation. Biochimie 89, 1–20 (2007).
Wu, G. et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 44, 251–253 (2012).
Fang, D. et al. H3.3K27M mutant proteins reprogram epigenome by sequestering the PRC2 complex to poised enhancers. Elife 7, e36696 (2018).
Chan, K. M. et al. A lesson learned from the H3.3K27M mutation found in pediatric glioma: a new approach to the study of the function of histone modifications in vivo. Cell Cycle 12, 2546–2552 (2013).
Lu, C. et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352, 844–849 (2016).
Fang, D. et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science 352, 1344–1348 (2016).
Chuikov, S. et al. Regulation of p53 activity through lysine methylation. Nature 432, 353–360 (2004).
Kouskouti, A. et al. Gene-specific modulation of TAF10 function by SET9-mediated methylation. Mol. Cell. 14, 175–182 (2004).
Vagin, V. V. et al. Proteomic analysis of murine Piwi proteins reveals a role for arginine methylation in specifying interaction with Tudor family members. Genes Dev. 23, 1749–1762 (2009).
Frederiks, F., Stulemeijer, I. J., Ovaa, H. & van Leeuwen, F. A modified epigenetics toolbox to study histone modifications on the nucleosome core. Chembiochem 12, 308–313 (2011).
Min, J. et al. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell 112, 711–723 (2003).
Rea, S. et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599 (2000).
Zhang, X. et al. Structure of the Neurospora SET domain protein DIM-5, a histone H3 lysine methyltransferase. Cell 111, 117–127 (2002).
Cao, R. & Zhang, Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell. 15, 57–67 (2004).
Margueron, R. & Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349 (2011).
Kuzmichev, A., Jenuwein, T., Tempst, P. & Reinberg, D. Different EZH2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol. Cell. 14, 183–193 (2004).
Spellmon, N. et al. Structure and function of SET and MYND domain-containing proteins. Int J. Mol. Sci. 16, 1406–1428 (2015).
Derunes, C. et al. Characterization of the PR domain of RIZ1 histone methyltransferase. Biochem Biophys. Res. Commun. 333, 925–934 (2005).
Hayashi, K., Yoshida, K. & Matsui, Y. A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature 438, 374–378 (2005).
Davis, C. A. et al. PRISM/PRDM6, a transcriptional repressor that promotes the proliferative gene program in smooth muscle cells. Mol. Cell Biol. 26, 2626–2636 (2006).
Gyory, I. et al. PRDI-BF1 recruits the histone H3 methyltransferase G9a in transcriptional silencing. Nat. Immunol. 5, 299–308 (2004).
Johnson, W. L. et al. RNA-dependent stabilization of SUV39H1 at constitutive heterochromatin. Elife 6, e25299 (2017).
Hashimoto, H., Vertino, P. M. & Cheng, X. Molecular coupling of DNA methylation and histone methylation. Epigenomics 2, 657–669 (2010).
Jurkowska, R. Z. et al. H3K14ac is linked to methylation of H3K9 by the triple Tudor domain of SETDB1. Nat. Commun. 8, 2057 (2017).
Bantignies, F., Goodman, R. H. & Smolik, S. M. Functional interaction between the coactivator Drosophila CREB-binding protein and ASH1, a member of the trithorax group of chromatin modifiers. Mol. Cell Biol. 20, 9317–9330 (2000).
Katsanis, N., Yaspo, M. L. & Fisher, E. M. Identification and mapping of a novel human gene, HRMT1L1, homologous to the rat protein arginine N-methyltransferase 1 (PRMT1) gene. Mamm. Genome 8, 526–529 (1997).
Meyer, R., Wolf, S. S. & Obendorf, M. PRMT2, a member of the protein arginine methyltransferase family, is a coactivator of the androgen receptor. J. Steroid Biochem. Mol. Biol. 107, 1–14 (2007).
Herrmann, F. et al. Human protein arginine methyltransferases in vivo-distinct properties of eight canonical members of the PRMT family. J. Cell Sci. 122, 667–677 (2009).
Cook, J. R. et al. FBXO11/PRMT9, a new protein arginine methyltransferase, symmetrically dimethylates arginine residues. Biochem Biophys. Res. Commun. 342, 472–481 (2006).
Slany, R. K. The molecular biology of mixed lineage leukemia. Haematologica 94, 984–993 (2009).
Okada, Y. et al. Leukaemic transformation by CALM-AF10 involves upregulation of Hoxa5 by hDOT1L. Nat. Cell Biol. 8, 1017–1024 (2006).
Bernt, K. M. et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 20, 66–78 (2011).
Chase, A. & Cross, N. C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 17, 2613–2618 (2011).
Takawa, M. et al. Validation of the histone methyltransferase EZH2 as a therapeutic target for various types of human cancer and as a prognostic marker. Cancer Sci. 102, 1298–1305 (2011).
Bannister, A. J. et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410, 120–124 (2001).
Wysocka, J. et al. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell 121, 859–872 (2005).
Shi, X. et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442, 96–99 (2006).
Flanagan, J. F. et al. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature 438, 1181–1185 (2005).
Migliori, V. et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat. Struct. Mol. Biol. 19, 136–144 (2012).
Trojer, P. et al. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell 129, 915–928 (2007).
Adams-Cioaba, M. A. & Min, J. Structure and function of histone methylation binding proteins. Biochem Cell Biol. 87, 93–105 (2009).
Lan, F. et al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature 448, 718–722 (2007).
Ooi, S. K. et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448, 714–717 (2007).
Lee, J., Thompson, J. R., Botuyan, M. V. & Mer, G. Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nat. Struct. Mol. Biol. 15, 109–111 (2008).
Metzger, E. et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437, 436–439 (2005).
Tsukada, Y. et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816 (2006).
Chang, B., Chen, Y., Zhao, Y. & Bruick, R. K. JMJD6 is a histone arginine demethylase. Science 318, 444–447 (2007).
Wang, Y. et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 306, 279–283 (2004).
Schulte, J. H. et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 69, 2065–2071 (2009).
Ciccone, D. N. et al. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 461, 415–418 (2009).
Pedersen, M. T. & Helin, K. Histone demethylases in development and disease. Trends Cell Biol. 20, 662–671 (2010).
Wagner, K. W. et al. KDM2A promotes lung tumorigenesis by epigenetically enhancing ERK1/2 signaling. J. Clin. Invest. 123, 5231–5246 (2013).
Tzatsos, A. et al. KDM2B promotes pancreatic cancer via Polycomb-dependent and -independent transcriptional programs. J. Clin. Invest. 123, 727–739 (2013).
Tee, A. E. et al. The histone demethylase JMJD1A induces cell migration and invasion by up-regulating the expression of the long noncoding RNA MALAT1. Oncotarget 5, 1793–1804 (2014).
Osawa, T. et al. Inhibition of histone demethylase JMJD1A improves anti-angiogenic therapy and reduces tumor-associated macrophages. Cancer Res. 73, 3019–3028 (2013).
Wang, J. et al. Epigenetic regulation of miR-302 by JMJD1C inhibits neural differentiation of human embryonic stem cells. J. Biol. Chem. 289, 2384–2395 (2014).
Wang, L. et al. Novel somatic and germline mutations in intracranial germ cell tumours. Nature 511, 241–245 (2014).
Cloos, P. A. et al. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 442, 307–311 (2006).
Wang, J. K. et al. The histone demethylase UTX enables RB-dependent cell fate control. Genes Dev. 24, 327–332 (2010).
Agger, K. et al. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 23, 1171–1176 (2009).
Lee, K. H. et al. PHF2 histone demethylase acts as a tumor suppressor in association with p53 in cancer. Oncogene 34, 2897–2909 (2015).
Cai, M. et al. Epigenetic silenced miR-125a-5p could be self-activated through targeting Suv39H1 in gastric cancer. J. Cell Mol. Med. 22, 4721–4731 (2018).
Yu, T. et al. Metformin inhibits SUV39H1-mediated migration of prostate cancer cells. Oncogenesis 6, e324 (2017).
Mo, W. et al. mTOR inhibitors suppress homologous recombination repair and synergize with PARP inhibitors via regulating SUV39H1 in BRCA-proficient triple-negative breast cancer. Clin. Cancer Res. 22, 1699–1712 (2016).
Pandey, M. et al. Involvement of EZH2, SUV39H1, G9a and associated molecules in pathogenesis of urethane induced mouse lung tumors: potential targets for cancer control. Toxicol. Appl. Pharmacol. 280, 296–304 (2014).
Kang, M. Y. et al. Association of the SUV39H1 histone methyltransferase with the DNA methyltransferase 1 at mRNA expression level in primary colorectal cancer. Int J. Cancer 121, 2192–2197 (2007).
Yang, Z. et al. The KMT1A-GATA3-STAT3 circuit is a novel self-renewal signaling of human bladder cancer stem cells. Clin. Cancer Res. 23, 6673–6685 (2017).
Khanal, P. et al. Prolyl isomerase Pin1 negatively regulates the stability of SUV39H1 to promote tumorigenesis in breast cancer. FASEB J. 27, 4606–4618 (2013).
Rodrigues, C. et al. A SUV39H1-low chromatin state characterises and promotes migratory properties of cervical cancer cells. Exp. Cell Res. 378, 206–216 (2019).
Shuai, W. et al. SUV39H2 promotes colorectal cancer proliferation and metastasis via tri-methylation of the SLIT1 promoter. Cancer Lett. 422, 56–69 (2018).
Yoon, K. A. et al. Novel polymorphisms in the SUV39H2 histone methyltransferase and the risk of lung cancer. Carcinogenesis 27, 2217–2222 (2006).
Nishikawaji, T. et al. Oncogenic roles of the SETDB2 histone methyltransferase in gastric cancer. Oncotarget 7, 67251–67265 (2016).
Wang, Y. F. et al. G9a regulates breast cancer growth by modulating iron homeostasis through the repression of ferroxidase hephaestin. Nat. Commun. 8, 274 (2017).
Tian, Y. F. et al. Preprogramming therapeutic response of PI3K/mTOR dual inhibitor via the regulation of EHMT2 and p27 in pancreatic cancer. Am. J. Cancer Res. 8, 1812–1822 (2018).
Cui, J. et al. EHMT2 inhibitor BIX-01294 induces apoptosis through PMAIP1-USP9X-MCL1 axis in human bladder cancer cells. Cancer Cell Int. 15, 4 (2015).
Ezponda, T. et al. The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene 32, 2882–2890 (2013).
Ryu, T. Y. et al. SETDB1 regulates SMAD7 expression for breast cancer metastasis. BMB Rep. 52, 139–144 (2019).
Chen, K. et al. Histone methyltransferase SETDB1 promotes the progression of colorectal cancer by inhibiting the expression of TP53. J. Cancer 8, 3318–3330 (2017).
Wong, C. M. et al. Up-regulation of histone methyltransferase SETDB1 by multiple mechanisms in hepatocellular carcinoma promotes cancer metastasis. Hepatology 63, 474–487 (2016).
Fei, Q. et al. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat. Commun. 6, 8651 (2015).
Wu, P. C. et al. H3K9 histone methyltransferase, KMT1E/SETDB1, cooperates with the SMAD2/3 pathway to suppress lung cancer metastasis. Cancer Res. 74, 7333–7343 (2014).
Zhu, Q. et al. The Wnt-driven Mll1 epigenome regulates salivary gland and head and neck cancer. Cell Rep. 26, 415–428.e5 (2019).
Lu, C. et al. The MLL1-H3K4me3 axis-mediated PD-L1 expression and pancreatic cancer immune evasion. J. Natl Cancer Inst. 109, djw283 (2017).
Yang, Z. et al. Single-cell sequencing reveals variants in ARID1A, GPRC5A and MLL2 driving self-renewal of human bladder cancer stem cells. Eur. Urol. 71, 8–12 (2017).
Augert, A. et al. Small cell lung cancer exhibits frequent inactivating mutations in the histone methyltransferase KMT2D/MLL2: CALGB 151111 (Alliance). J. Thorac. Oncol. 12, 704–713 (2017).
Chen, J. et al. Pygo2 associates with MLL2 histone methyltransferase and GCN5 histone acetyltransferase complexes to augment Wnt target gene expression and breast cancer stem-like cell expansion. Mol. Cell Biol. 30, 5621–5635 (2010).
Xia, M. et al. Downregulation of MLL3 in esophageal squamous cell carcinoma is required for the growth and metastasis of cancer cells. Tumour Biol. 36, 605–613 (2015).
Kim, J. H. et al. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 74, 1705–1717 (2014).
Gallo, M. et al. MLL5 orchestrates a cancer self-renewal state by repressing the histone variant H3.3 and globally reorganizing chromatin. Cancer Cell. 28, 715–729 (2015).
Li, T. et al. SET1A cooperates with CUDR to promote liver cancer growth and hepatocyte-like stem cell malignant transformation epigenetically. Mol. Ther. 24, 261–275 (2016).
Karakashev, S. et al. CARM1-expressing ovarian cancer depends on the histone methyltransferase EZH2 activity. Nat. Commun. 9, 631 (2018).
Crea, F. et al. An EZH2 polymorphism is associated with clinical outcome in metastatic colorectal cancer patients. Ann. Oncol. 23, 1207–1213 (2012).
Reijm, E. A. et al. High protein expression of EZH2 is related to unfavorable outcome to tamoxifen in metastatic breast cancer. Ann. Oncol. 25, 2185–2190 (2014).
Kanu, N. et al. SETD2 loss-of-function promotes renal cancer branched evolution through replication stress and impaired DNA repair. Oncogene 34, 5699–5708 (2015).
Kim, I. K. et al. Acquired SETD2 mutation and impaired CREB1 activation confer cisplatin resistance in metastatic non-small cell lung cancer. Oncogene 38, 180–193 (2019).
Li, N. et al. AKT-mediated stabilization of histone methyltransferase WHSC1 promotes prostate cancer metastasis. J. Clin. Invest. 127, 1284–1302 (2017).
Li, J. et al. MiR-2392 suppresses metastasis and epithelial-mesenchymal transition by targeting MAML3 and WHSC1 in gastric cancer. FASEB J. 31, 3774–3786 (2017).
Saloura, V. et al. WHSC1L1-mediated EGFR mono-methylation enhances the cytoplasmic and nuclear oncogenic activity of EGFR in head and neck cancer. Sci. Rep. 7, 40664 (2017).
Chiou, S. H. et al. BLIMP1 induces transient metastatic heterogeneity in pancreatic cancer. Cancer Discov. 7, 1184–1199 (2017).
Sciortino, M. et al. Dysregulation of Blimp1 transcriptional repressor unleashes p130Cas/ErbB2 breast cancer invasion. Sci. Rep. 7, 1145 (2017).
Zhu, Z. et al. Downregulation of PRDM1 promotes cellular invasion and lung cancer metastasis. Tumour Biol. 39, 1010428317695929 (2017).
Kang, H. B. et al. PRDM1, a tumor-suppressor gene, is induced by genkwadaphnin in human colon cancer SW620 cells. J. Cell Biochem. 117, 172–179 (2016).
Emterling, A., Wallin, A., Arbman, G. & Sun, X. F. Clinicopathological significance of microsatellite instability and mutated RIZ in colorectal cancer. Ann. Oncol. 15, 242–246 (2004).
Abbondanza, C. et al. Identification of a functional estrogen-responsive enhancer element in the promoter 2 of PRDM2 gene in breast cancer cell lines. J. Cell Physiol. 227, 964–975 (2012).
Nanjundan, M. et al. Amplification of MDS1/EVI1 and EVI1, located in the 3q26.2 amplicon, is associated with favorable patient prognosis in ovarian cancer. Cancer Res. 67, 3074–3084 (2007).
Lu, Y. et al. EVI1 promotes epithelial-to-mesenchymal transition, cancer stem cell features and chemo-/radioresistance in nasopharyngeal carcinoma. J. Exp. Clin. Cancer Res. 38, 82 (2019).
Taniguchi, H. et al. Silencing PRDM14 expression by an innovative RNAi therapy inhibits stemness, tumorigenicity, and metastasis of breast cancer. Oncotarget 8, 46856–46874 (2017).
Watanabe, Y. et al. PRDM5 identified as a target of epigenetic silencing in colorectal and gastric cancer. Clin. Cancer Res. 13, 4786–4794 (2007).
Houle, A. A. et al. Aberrant PRDM9 expression impacts the pan-cancer genomic landscape. Genome Res. 28, 1611–1620 (2018).
Ruark, E. et al. Identification of nine new susceptibility loci for testicular cancer, including variants near DAZL and PRDM14. Nat. Genet. 45, 686–689 (2013).
Moriya, C. et al. Inhibition of PRDM14 expression in pancreatic cancer suppresses cancer stem-like properties and liver metastasis in mice. Carcinogenesis 38, 638–648 (2017).
Takahata, M. et al. SKI and MEL1 cooperate to inhibit transforming growth factor-beta signal in gastric cancer cells. J. Biol. Chem. 284, 3334–3344 (2009).
Reynoird, N. et al. Coordination of stress signals by the lysine methyltransferase SMYD2 promotes pancreatic cancer. Genes Dev. 30, 772–785 (2016).
Komatsu, S. et al. Overexpression of SMYD2 contributes to malignant outcome in gastric cancer. Br. J. Cancer 112, 357–364 (2015).
Sarris, M. E. et al. Smyd3 is a transcriptional potentiator of multiple cancer-promoting genes and required for liver and colon cancer development. Cancer Cell. 29, 354–366 (2016).
Cock-Rada, A. M. et al. SMYD3 promotes cancer invasion by epigenetic upregulation of the metalloproteinase MMP-9. Cancer Res. 72, 810–820 (2012).
Fenizia, C. et al. SMYD3 promotes the epithelial-mesenchymal transition in breast cancer. Nucleic Acids Res. 47, 1278–1293 (2018).
Hu, L., Zhu, Y. T., Qi, C. & Zhu, Y. J. Identification of Smyd4 as a potential tumor suppressor gene involved in breast cancer development. Cancer Res. 69, 4067–4072 (2009).
Kryczek, I. et al. IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 40, 772–784 (2014).
Cho, M. H. et al. DOT1L cooperates with the c-Myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nat. Commun. 6, 7821 (2015).
Liu, B. et al. MiR-502/SET8 regulatory circuit in pathobiology of breast cancer. Cancer Lett. 376, 259–267 (2016).
Yang, S. et al. Association of miR-502-binding site single nucleotide polymorphism in the 3′-untranslated region of SET8 and TP53 codon 72 polymorphism with non-small cell lung cancer in Chinese population. Acta Biochim Biophys. Sin. 46, 149–154 (2014).
Tryndyak, V. P., Kovalchuk, O. & Pogribny, I. P. Loss of DNA methylation and histone H4 lysine 20 trimethylation in human breast cancer cells is associated with aberrant expression of DNA methyltransferase 1, Suv4–20h2 histone methyltransferase and methyl-binding proteins. Cancer Biol. Ther. 5, 65–70 (2006).
Martín-Morales, L. et al. SETD6 dominant negative mutation in familial colorectal cancer type X. Hum. Mol. Genet. 26, 4481–4493 (2017).
Yao, R. et al. Lysines 207 and 325 methylation of WDR5 catalyzed by SETD6 promotes breast cancer cell proliferation and migration. Oncol. Rep. 40, 3069–3077 (2018).
Shen, C. et al. SET7/9 regulates cancer cell proliferation by influencing β-catenin stability. FASEB J. 29, 4313–4323 (2015).
Gu, Y. et al. Opposite effects of SET7/9 on apoptosis of human acute myeloid leukemia cells and lung cancer cells. J. Cancer 8, 2069–2078 (2017).
Montenegro, M. F. et al. Tumor suppressor SET9 guides the epigenetic plasticity of breast cancer cells and serves as an early-stage biomarker for predicting metastasis. Oncogene 35, 6143–6152 (2016).
Liu, L. M. et al. Methylation of C/EBPα by PRMT1 inhibits its tumor suppressive function in breast cancer. Cancer Res. 79, 2865–2877 (2019).
Avasarala, S. et al. PRMT1 is a novel regulator of epithelial-mesenchymal-transition in non-small cell lung cancer. J. Biol. Chem. 290, 13479–13489 (2015).
Mathioudaki, K. et al. The PRMT1 gene expression pattern in colon cancer. Br. J. Cancer 99, 2094–2099 (2008).
Lin, Z. et al. Overexpressing PRMT1 inhibits proliferation and invasion in pancreatic cancer by inverse correlation of ZEB1. IUBMB Life. 70, 1032–1039 (2018).
Zhong, J. et al. PRMT2β, a C-terminal splice variant of PRMT2, inhibits the growth of breast cancer cells. Oncol. Rep. 38, 1303–1311 (2017).
Al-Dhaheri, M. et al. CARM1 is an important determinant of ERα-dependent breast cancer cell differentiation and proliferation in breast cancer cells. Cancer Res. 71, 2118–2128 (2011).
Zhong, X. Y. et al. CARM1 methylates GAPDH to regulate glucose metabolism and is suppressed in liver cancer. Cell Rep. 24, 3207–3223 (2018).
Wang, Y. P. et al. Arginine methylation of MDH1 by CARM1 inhibits glutamine metabolism and suppresses pancreatic cancer. Mol. Cell. 64, 673–687 (2016).
Rengasamy, M. et al. The PRMT5/WDR77 complex regulates alternative splicing through ZNF326 in breast cancer. Nucleic Acids Res. 45, 11106–11120 (2017).
Fu, T., Lv, X., Kong, Q. & Yuan, C. A novel SHARPIN-PRMT5-H3R2me1 axis is essential for lung cancer cell invasion. Oncotarget 8, 54809–54820 (2017).
Demetriadou, C. et al. NAA40 contributes to colorectal cancer growth by controlling PRMT5 expression. Cell Death Dis. 10, 236 (2019).
Huang, S. et al. CAPG enhances breast cancer metastasis by competing with PRMT5 to modulate STC-1 transcription. Theranostics 8, 2549–2564 (2018).
Almeida-Rios, D. et al. Histone methyltransferase PRMT6 plays an oncogenic role of in prostate cancer. Oncotarget 7, 53018–53028 (2016).
Okuno, K. et al. Asymmetric dimethylation at histone H3 arginine 2 by PRMT6 in gastric cancer progression. Carcinogenesis 40, 15–26 (2019).
Chan, L. H. et al. PRMT6 regulates RAS/RAF binding and MEK/ERK-mediated cancer stemness activities in hepatocellular carcinoma through CRAF methylation. Cell Rep. 25, 690–701.e8 (2018).
Cheng, D. et al. PRMT7 contributes to the metastasis phenotype in human non-small-cell lung cancer cells possibly through the interaction with HSPA5 and EEF2. Onco. Targets Ther. 11, 4869–4876 (2018).
Yao, R. et al. PRMT7 induces epithelial-to-mesenchymal transition and promotes metastasis in breast cancer. Cancer Res. 74, 5656–5667 (2014).
Hernandez, S. J., Dolivo, D. M. & Dominko, T. PRMT8 demonstrates variant-specific expression in cancer cells and correlates with patient survival in breast, ovarian and gastric cancer. Oncol. Lett. 13, 1983–1989 (2017).
Bagger, S. O. et al. Aggressiveness of non-EMT breast cancer cells relies on FBXO11 activity. Mol. Cancer 17, 171 (2018).
Lee, Y. H. & Ann, D. K. Bi-phasic expression of Heterochromatin Protein 1 (HP1) during breast cancer progression: potential roles of HP1 and chromatin structure in tumorigenesis. J. Nat. Sci. 1, e127 (2015).
Shenoy, T. R. et al. CHD1 loss sensitizes prostate cancer to DNA damaging therapy by promoting error-prone double-strand break repair. Ann. Oncol. 28, 1495–1507 (2017).
Rodrigues, L. U. et al. Coordinate loss of MAP3K7 and CHD1 promotes aggressive prostate cancer. Cancer Res. 75, 1021–1034 (2015).
Pan, L., Tang, Z., Pan, L. & Tang, R. miR-3666 inhibits lung cancer cell proliferation, migration and invasion by targeting BPTF. Biochem Cell Biol. 97, 415–422 (2018).
Zhao, X. et al. BPTF promotes hepatocellular carcinoma growth by modulating hTERT signaling and cancer stem cell traits. Redox Biol. 20, 427–441 (2019).
Zeng, H. et al. Physical activity and breast cancer survival: an epigenetic link through reduced methylation of a tumor suppressor gene L3MBTL1. Breast Cancer Res. Treat. 133, 127–135 (2012).
Kumamoto, K. et al. ING2 is upregulated in colon cancer and increases invasion by enhanced MMP13 expression. Int J. Cancer 125, 1306–1315 (2009).
Okano, T. et al. Alterations in novel candidate tumor suppressor genes, ING1 and ING2 in human lung cancer. Oncol. Rep. 15, 545–549 (2006).
Li, Y. et al. RNA splicing of the BHC80 gene contributes to neuroendocrine prostate cancer progression. Eur. Urol. 76, 157–166 (2019).
Li, L. L. et al. JMJD2A contributes to breast cancer progression through transcriptional repression of the tumor suppressor ARHI. Breast Cancer Res. 16, R56 (2014).
Kim, T. D. et al. The JMJD2A demethylase regulates apoptosis and proliferation in colon cancer cells. J. Cell Biochem. 113, 1368–1376 (2012).
Kauffman, E. C. et al. Role of androgen receptor and associated lysine-demethylase coregulators, LSD1 and JMJD2A, in localized and advanced human bladder cancer. Mol. Carcinog. 50, 931–944 (2011).
Cao, C. et al. Functional interaction of histone deacetylase 5 (HDAC5) and lysine-specific demethylase 1 (LSD1) promotes breast cancer progression. Oncogene 36, 133–145 (2017).
Stewart, C. A. & Byers, L. A. Altering the course of small cell lung cancer: targeting cancer stem cells via LSD1 inhibition. Cancer Cell. 28, 4–6 (2015).
Liang, Y. et al. LSD1-mediated epigenetic reprogramming drives CENPE expression and prostate cancer progression. Cancer Res. 77, 5479–5490 (2017).
Wang, Y. et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138, 660–672 (2009).
Chen, L. et al. Functional characterization of lysine-specific demethylase 2 (LSD2/KDM1B) in breast cancer progression. Oncotarget 8, 81737–81753 (2017).
Chen, J. Y. et al. Lysine demethylase KDM2A inhibits TET2 to promote DNA methylation and silencing of tumor suppressor genes in breast cancer. Oncogenesis 6, e369 (2017).
Kong, Y. et al. RUNX3-mediated up-regulation of miR-29b suppresses the proliferation and migration of gastric cancer cells by targeting KDM2A. Cancer Lett. 381, 138–148 (2016).
Dhar, S. S. et al. Transcriptional repression of histone deacetylase 3 by the histone demethylase KDM2A is coupled to tumorigenicity of lung cancer cells. J. Biol. Chem. 289, 7483–7496 (2014).
Zacharopoulou, N. et al. The epigenetic factor KDM2B regulates cell adhesion, small rho GTPases, actin cytoskeleton and migration in prostate cancer cells. Biochim Biophys. Acta Mol. Cell Res. 1865, 587–597 (2018).
Kottakis, F. et al. NDY1/KDM2B functions as a master regulator of polycomb complexes and controls self-renewal of breast cancer stem cells. Cancer Res. 74, 3935–3946 (2014).
Peng, K. et al. Histone demethylase JMJD1A promotes colorectal cancer growth and metastasis by enhancing Wnt/β-catenin signaling. J. Biol. Chem. 293, 10606–10619 (2018).
Ramadoss, S. et al. Lysine-specific demethylase KDM3A regulates ovarian cancer stemness and chemoresistance. Oncogene 36, 6508 (2017).
Wade, M. A. et al. The histone demethylase enzyme KDM3A is a key estrogen receptor regulator in breast cancer. Nucleic Acids Res. 43, 196–207 (2015).
Cai, Y., Fu, X. & Deng, Y. Histone demethylase JMJD1C regulates esophageal cancer proliferation Via YAP1 signaling. Am. J. Cancer Res. 7, 115–124 (2017).
Chen, C. et al. Downregulation of histone demethylase JMJD1C inhibits colorectal cancer metastasis through targeting ATF2. Am. J. Cancer Res. 8, 852–865 (2018).
An, J. et al. HistoneH3 demethylase JMJD2A promotes growth of liver cancer cells through up-regulating miR372. Oncotarget 8, 49093–49109 (2017).
Wang, W. et al. KDM4B-regulated unfolded protein response as a therapeutic vulnerability in PTEN-deficient breast cancer. J. Exp. Med. 215, 2833–2849 (2018).
Zhao, L. et al. JMJD2B promotes epithelial-mesenchymal transition by cooperating with β-catenin and enhances gastric cancer metastasis. Clin. Cancer Res. 19, 6419–6429 (2013).
Fu, L. N. et al. Role of JMJD2B in colon cancer cell survival under glucose-deprived conditions and the underlying mechanisms. Oncogene 37, 389–402 (2018).
Luo, W. et al. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc. Natl Acad. Sci. USA 109, E3367–E3376 (2012).
An, Y. et al. circZMYM2 competed endogenously with miR-335-5p to regulate JMJD2C in pancreatic cancer. Cell Physiol. Biochem. 51, 2224–2236 (2018).
Peng, K. et al. Histone demethylase JMJD2D interacts with β-catenin to induce transcription and activate colorectal cancer cell proliferation and tumor growth in mice. Gastroenterology 156, 1112–1126 (2019).
Yang, G. J. et al. Selective inhibition of lysine-specific demethylase 5A (KDM5A) using a Rhodium(III) complex for triple-negative breast cancer therapy. Angew. Chem. Int Ed. Engl. 57, 13091–13095 (2018).
Uemura, M. et al. Jumonji domain containing 1A is a novel prognostic marker for colorectal cancer: in vivo identification from hypoxic tumor cells. Clin. Cancer Res. 16, 4636–4646 (2010).
Hayami, S. et al. Overexpression of the JmjC histone demethylase KDM5B in human carcinogenesis: involvement in the proliferation of cancer cells through the E2F/RB pathway. Mol. Cancer 9, 59 (2010).
Li, N. & Jiang, D. Jumonji domain containing 2C promotes cell migration and invasion through modulating CUL4A expression in lung cancer. Biomed. Pharmacother. 89, 305–315 (2017).
Lin, H. et al. KDM5c inhibits multidrug resistance of colon cancer cell line by down-regulating ABCC1. Biomed. Pharmacother. 107, 1205–1209 (2018).
Shen, X. et al. KDM5D inhibit epithelial-mesenchymal transition of gastric cancer through demethylation in the promoter of Cul4A in male. J. Cell Biochem. 120, 12247–12258 (2019).
Komura, K. et al. Resistance to docetaxel in prostate cancer is associated with androgen receptor activation and loss of KDM5D expression. Proc. Natl Acad. Sci. USA 113, 6259–6264 (2016).
Zhu, X. X. et al. Jarid2 is essential for the maintenance of tumor initiating cells in bladder cancer. Oncotarget 8, 24483–24490 (2017).
Tange, S. et al. JARID2 is involved in transforming growth factor-beta-induced epithelial-mesenchymal transition of lung and colon cancer cell lines. PLoS ONE 9, e115684 (2014).
Paccez, J. D. et al. Dihydroartemisinin inhibits prostate cancer via JARID2/miR-7/miR-34a-dependent downregulation of Axl. Oncogenesis 8, 14 (2019).
Ler, L. D. et al. Loss of tumor suppressor KDM6A amplifies PRC2-regulated transcriptional repression in bladder cancer and can be targeted through inhibition of EZH2. Sci. Transl. Med 9, eaai8312 (2017).
Andricovich, J. et al. Loss of KDM6A activates super-enhancers to induce gender-specific squamous-like pancreatic cancer and confers sensitivity to BET inhibitors. Cancer Cell. 33, 512–526.e8 (2018).
Mo, J. et al. Multifunctional nanoparticles for co-delivery of paclitaxel and carboplatin against ovarian cancer by inactivating the JMJD3-HER2 axis. Nanoscale 9, 13142–13152 (2017).
Wang, W. et al. KDM6B counteracts EZH2-mediated suppression of IGFBP5 to confer resistance to PI3K/AKT inhibitor treatment in breast cancer. Mol. Cancer Ther. 17, 1973–1983 (2018).
Ahn, J. et al. Target sequencing and CRISPR/Cas editing reveal simultaneous loss of UTX and UTY in urothelial bladder cancer. Oncotarget 7, 63252–63260 (2016).
Lee, K. H. et al. Histone demethylase KDM7A controls androgen receptor activity and tumor growth in prostate cancer. Int J. Cancer 143, 2849–2861 (2018).
Björkman, M. et al. Systematic knockdown of epigenetic enzymes identifies a novel histone demethylase PHF8 overexpressed in prostate cancer with an impact on cell proliferation, migration and invasion. Oncogene 31, 3444–3456 (2012).
Li, S. et al. Histone demethylase PHF8 promotes progression and metastasis of gastric cancer. Am. J. Cancer Res. 7, 448–461 (2017).
Shen, Y., Pan, X. & Zhao, H. The histone demethylase PHF8 is an oncogenic protein in human non-small cell lung cancer. Biochem Biophys. Res. Commun. 451, 119–125 (2014).
Zhao, Z. et al. Overexpression of histone demethylase JMJD5 promotes metastasis and indicates a poor prognosis in breast cancer. Int J. Clin. Exp. Pathol. 8, 10325–10334 (2015).
Hsia, D. A. et al. KDM8, a H3K36me2 histone demethylase that acts in the cyclin A1 coding region to regulate cancer cell proliferation. Proc. Natl Acad. Sci. USA 107, 9671–9676 (2010).
Abu-Jamous, B., Buffa, F. M., Harris, A. L. & Nandi, A. K. In vitro downregulated hypoxia transcriptome is associated with poor prognosis in breast cancer. Mol. Cancer 16, 105 (2017).
Liu, Y. et al. JMJD6 regulates histone H2A.X phosphorylation and promotes autophagy in triple-negative breast cancer cells via a novel tyrosine kinase activity. Oncogene 38, 980–997 (2019).
Lee, Y. F. et al. JMJD6 is a driver of cellular proliferation and motility and a marker of poor prognosis in breast cancer. Breast Cancer Res. 14, R85 (2012).
Zhang, Z., Yang, Y. & Zhang, X. MiR-770 inhibits tumorigenesis and EMT by targeting JMJD6 and regulating WNT/β-catenin pathway in non-small cell lung cancer. Life Sci. 188, 163–171 (2017).
Chang, X. et al. Investigating the pathogenic role of PADI4 in oesophageal cancer. Int J. Biol. Sci. 7, 769–781 (2011).
Calin, G. A. et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl Acad. Sci. USA 101, 2999–3004 (2004).
Kasinski, A. L. & Slack, F. J. Epigenetics and genetics. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer 11, 849–864 (2011).
Garcia, A. I. et al. Down-regulation of BRCA1 expression by miR-146a and miR-146b-5p in triple negative sporadic breast cancers. EMBO Mol. Med. 3, 279–290 (2011).
Friedman, J. M. et al. The putative tumor suppressor microRNA-101 modulates the cancer epigenome by repressing the polycomb group protein EZH2. Cancer Res. 69, 2623–2629 (2009).
Varambally, S. et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 322, 1695–1699 (2008).
Fabbri, M. et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3 A and 3B. Proc. Natl Acad. Sci. USA 104, 15805–15810 (2007).
Yang, L. et al. lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature 500, 598–602 (2013).
He, Y. et al. Long noncoding RNAs: Novel insights into hepatocelluar carcinoma. Cancer Lett. 344, 20–27 (2014).
Kim, K. et al. HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene 32, 1616–1625 (2013).
Kogo, R. et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 71, 6320–6326 (2011).
Nakagawa, T. et al. Large noncoding RNA HOTAIR enhances aggressive biological behavior and is associated with short disease-free survival in human non-small cell lung cancer. Biochem Biophys. Res. Commun. 436, 319–324 (2013).
Gius, D. et al. Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach. Cancer Cell. 6, 361–371 (2004).
Li, E., Bestor, T. H. & Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926 (1992).
Jackson-Grusby, L. et al. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat. Genet. 27, 31–39 (2001).
Carr, B. I. et al. The tumorigenicity of 5-azacytidine in the male Fischer rat. Carcinogenesis 5, 1583–1590 (1984).
Christman, J. K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 21, 5483–5495 (2002).
Jones, P. A. & Laird, P. W. Cancer epigenetics comes of age. Nat. Genet. 21, 163–167 (1999).
Rosen, M. B., House, H. S., Francis, B. M. & Chernoff, N. Teratogenicity of 5-azacytidine in the Sprague-Dawley rat. J. Toxicol. Environ. Health 29, 201–210 (1990).
Doerksen, T. & Trasler, J. M. Developmental exposure of male germ cells to 5-azacytidine results in abnormal preimplantation development in rats. Biol. Reprod. 55, 1155–1162 (1996).
Chik, F. & Szyf, M. Effects of specific DNMT gene depletion on cancer cell transformation and breast cancer cell invasion; toward selective DNMT inhibitors. Carcinogenesis 32, 224–232 (2011).
Issa, J. P. et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood 103, 1635–1640 (2004).
Beisler, J. A., Abbasi, M. M., Kelley, J. A. & Driscoll, J. S. Synthesis and antitumor activity of dihydro-5-azacytidine, a hydrolytically stable analogue of 5-azacytidine. J. Med. Chem. 20, 806–812 (1977).
Presant, C. A., Coulter, D., Valeriote, F. & Vietti, T. J. Contrasting cytotoxicity kinetics of 5-azacytidine and dihydro-5-azacytidine hydrochloride in L1210 leukemia in mice. J. Natl Cancer Inst. 66, 1151–1154 (1981).
Curt, G. A. et al. A phase I and pharmacokinetic study of dihydro-5-azacytidine (NSC 264880). Cancer Res. 45, 3359–3363 (1985).
Cheng, J. C. et al. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J. Natl Cancer Inst. 95, 399–409 (2003).
Holleran, J. L. et al. Plasma pharmacokinetics, oral bioavailability, and interspecies scaling of the DNA methyltransferase inhibitor, zebularine. Clin. Cancer Res. 11, 3862–3868 (2005).
Kaminskas, E. et al. Approval summary: azacitidine for treatment of myelodysplastic syndrome subtypes. Clin. Cancer Res. 11, 3604–3608 (2005).
Steensma, D. P. Decitabine treatment of patients with higher-risk myelodysplastic syndromes. Leuk. Res. 33(Suppl 2), S12–S17 (2009).
Liang, G. et al. Analysis of gene induction in human fibroblasts and bladder cancer cells exposed to the methylation inhibitor 5-aza-2′-deoxycytidine. Cancer Res. 62, 961–966 (2002).
Brueckner, B. et al. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 65, 6305–6311 (2005).
Piña, I. C. et al. Psammaplins from the sponge Pseudoceratina purpurea: inhibition of both histone deacetylase and DNA methyltransferase. J. Org. Chem. 68, 3866–3873 (2003).
Fang, M. Z. et al. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 63, 7563–7570 (2003).
Cornacchia, E. et al. Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J. Immunol. 140, 2197–2200 (1988).
Segura-Pacheco, B. et al. Reactivation of tumor suppressor genes by the cardiovascular drugs hydralazine and procainamide and their potential use in cancer therapy. Clin. Cancer Res. 9, 1596–1603 (2003).
Villar-Garea, A., Fraga, M. F., Espada, J. & Esteller, M. Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells. Cancer Res. 63, 4984–4989 (2003).
Davis, A. J. et al. Phase I and pharmacologic study of the human DNA methyltransferase antisense oligodeoxynucleotide MG98 given as a 21-day continuous infusion every 4 weeks. Invest N. Drugs 21, 85–97 (2003).
Amato, R. J. Inhibition of DNA methylation by antisense oligonucleotide MG98 as cancer therapy. Clin. Genitourin. Cancer 5, 422–426 (2007).
Plummer, R. et al. Phase I study of MG98, an oligonucleotide antisense inhibitor of human DNA methyltransferase 1, given as a 7-day infusion in patients with advanced solid tumors. Clin. Cancer Res. 15, 3177–3183 (2009).
Chuang, J. C. et al. Comparison of biological effects of non-nucleoside DNA methylation inhibitors versus 5-aza-2′-deoxycytidine. Mol. Cancer Ther. 4, 1515–1520 (2005).
Mutskov, V. & Felsenfeld, G. Silencing of transgene transcription precedes methylation of promoter DNA and histone H3 lysine 9. EMBO J. 23, 138–149 (2004).
Lau, O. D. et al. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol. Cell. 5, 589–595 (2000).
Yang, C., Ngo, L. & Zheng, Y. G. Rational design of substrate-based multivalent inhibitors of the histone acetyltransferase Tip60. ChemMedChem 9, 537–541 (2014).
Balasubramanyam, K., Swaminathan, V., Ranganathan, A. & Kundu, T. K. Small molecule modulators of histone acetyltransferase p300. J. Biol. Chem. 278, 19134–19140 (2003).
Balasubramanyam, K. et al. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J. Biol. Chem. 279, 51163–51171 (2004).
Balasubramanyam, K. et al. Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J. Biol. Chem. 279, 33716–33726 (2004).
Biel, M. et al. Design, synthesis, and biological evaluation of a small-molecule inhibitor of the histone acetyltransferase Gcn5. Angew. Chem. Int. Ed. Engl. 43, 3974–3976 (2004).
Stimson, L. et al. Isothiazolones as inhibitors of PCAF and p300 histone acetyltransferase activity. Mol. Cancer Ther. 4, 1521–1532 (2005).
Filippakopoulos, P. et al. Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2010).
Nicodeme, E. et al. Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123 (2010).
Brand, M. et al. Small molecule inhibitors of bromodomain-acetyl-lysine interactions. ACS Chem. Biol. 10, 22–39 (2015).
Romero, F. A. et al. Disrupting acetyl-lysine recognition: progress in the development of bromodomain inhibitors. J. Med. Chem. 59, 1271–1298 (2016).
Xiang, Q. et al. Discovery and optimization of 1-(1H-indol-1-yl)ethanone derivatives as CBP/EP300 bromodomain inhibitors for the treatment of castration-resistant prostate cancer. Eur. J. Med. Chem. 147, 238–252 (2018).
Theodoulou, N. H. et al. Discovery of I-BRD9, a selective cell active chemical probe for bromodomain containing protein 9 inhibition. J. Med. Chem. 59, 1425–1439 (2016).
Vangamudi, B. et al. The SMARCA2/4 ATPase domain surpasses the bromodomain as a drug target in SWI/SNF-mutant cancers: insights from cDNA rescue and PFI-3 inhibitor studies. Cancer Res. 75, 3865–3878 (2015).
Palmer, W. S. et al. Structure-guided design of IACS-9571, a selective high-affinity dual TRIM24-BRPF1 bromodomain inhibitor. J. Med. Chem. 59, 1440–1454 (2016).
Picaud, S. et al. Promiscuous targeting of bromodomains by bromosporine identifies BET proteins as master regulators of primary transcription response in leukemia. Sci. Adv. 2, e1600760 (2016).
Riggs, M. G., Whittaker, R. G., Neumann, J. R. & Ingram, V. M. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature 268, 462–464 (1977).
Yoshida, M., Kijima, M., Akita, M. & Beppu, T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 265, 17174–17179 (1990).
Van Lint, C., Emiliani, S. & Verdin, E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 5, 245–253 (1996).
Fandy, T. E. et al. Interactive effects of HDAC inhibitors and TRAIL on apoptosis are associated with changes in mitochondrial functions and expressions of cell cycle regulatory genes in multiple myeloma. Neoplasia 7, 646–657 (2005).
Fang, J. Y. Histone deacetylase inhibitors, anticancerous mechanism and therapy for gastrointestinal cancers. J. Gastroenterol. Hepatol. 20, 988–994 (2005).
Singh, T. R., Shankar, S. & Srivastava, R. K. HDAC inhibitors enhance the apoptosis-inducing potential of TRAIL in breast carcinoma. Oncogene 24, 4609–4623 (2005).
Finnin, M. S. et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401, 188–193 (1999).
Richon, V. M. et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl Acad. Sci. USA 95, 3003–3007 (1998).
Jung, M. et al. Amide analogues of trichostatin A as inhibitors of histone deacetylase and inducers of terminal cell differentiation. J. Med. Chem. 42, 4669–4679 (1999).
Kim, Y. B. et al. Oxamflatin is a novel antitumor compound that inhibits mammalian histone deacetylase. Oncogene 18, 2461–2470 (1999).
Su, G. H., Sohn, T. A., Ryu, B. & Kern, S. E. A novel histone deacetylase inhibitor identified by high-throughput transcriptional screening of a compound library. Cancer Res. 60, 3137–3142 (2000).
Bressi, J. C. et al. Benzimidazole and imidazole inhibitors of histone deacetylases: synthesis and biological activity. Bioorg. Med. Chem. Lett. 20, 3138–3141 (2010).
Ito, T. et al. Real-time imaging of histone H4K12-specific acetylation determines the modes of action of histone deacetylase and bromodomain inhibitors. Chem. Biol. 18, 495–507 (2011).
Suzuki, T. et al. Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives. J. Med. Chem. 42, 3001–3003 (1999).
Kijima, M. et al. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J. Biol. Chem. 268, 22429–22435 (1993).
Furumai, R. et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 62, 4916–4921 (2002).
Lea, M. A., Randolph, V. M. & Patel, M. Increased acetylation of histones induced by diallyl disulfide and structurally related molecules. Int J. Oncol. 15, 347–352 (1999).
Haggarty, S. J. et al. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl Acad. Sci. USA 100, 4389–4394 (2003).
Balasubramanian, S. et al. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 22, 1026–1034 (2008).
Methot, J. L. et al. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 18, 973–978 (2008).
Mann, B. S. et al. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 12, 1247–1252 (2007).
Ropero, S. et al. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat. Genet. 38, 566–569 (2006).
Daigle, S. R. et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 20, 53–65 (2011).
Daigle, S. R. et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 122, 1017–1025 (2013).
Basavapathruni, A. et al. Nonclinical pharmacokinetics and metabolism of EPZ-5676, a novel DOT1L histone methyltransferase inhibitor. Biopharm. Drug Dispos. 35, 237–252 (2014).
Miranda, T. B. et al. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 8, 1579–1588 (2009).
Glazer, R. I. et al. 3-Deazaneplanocin: a new and potent inhibitor of S-adenosylhomocysteine hydrolase and its effects on human promyelocytic leukemia cell line HL-60. Biochem Biophys. Res. Commun. 135, 688–694 (1986).
Qi, W. et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl Acad. Sci. USA 109, 21360–21365 (2012).
Verma, S. K. et al. Identification of Potent, Selective, Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS Med. Chem. Lett. 3, 1091–1096 (2012).
McCabe, M. T. et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112 (2012).
Knutson, S. K. et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 8, 890–896 (2012).
Knutson, S. K. et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol. Cancer Ther. 13, 842–854 (2014).
Fiedorowicz, J. G. & Swartz, K. L. The role of monoamine oxidase inhibitors in current psychiatric practice. J. Psychiatr. Pract. 10, 239–248 (2004).
Harris, W. J. et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 21, 473–487 (2012).
Schenk, T. et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 18, 605–611 (2012).
Liang, Y. et al. A novel selective LSD1/KDM1A inhibitor epigenetically blocks herpes simplex virus lytic replication and reactivation from latency. MBio 4, e00558–00512 (2013).
Neelamegam, R. et al. Brain-penetrant LSD1 inhibitors can block memory consolidation. ACS Chem. Neurosci. 3, 120–128 (2012).
Fiskus, W. et al. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia 28, 2155–2164 (2014).
Maes, T. et al. Advances in the development of histone lysine demethylase inhibitors. Curr. Opin. Pharmacol. 23, 52–60 (2015).
Rose, N. R. et al. Plant growth regulator daminozide is a selective inhibitor of human KDM2/7 histone demethylases. J. Med Chem. 55, 6639–6643 (2012).
Tan, Y. et al. Matrix softness regulates plasticity of tumour-repopulating cells via H3K9 demethylation and Sox2 expression. Nat. Commun. 5, 4619 (2014).
Kruidenier, L. et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 488, 404–408 (2012).
Heinemann, B. et al. Inhibition of demethylases by GSK-J1/J4. Nature 514, E1–E2 (2014).
Acknowledgements
This work is supported by the Excellent Youth Foundation of Sichuan Scientific Committee Grant in China (No. 2019JDJQ0008), the National Major Scientific and Technological Special Project for “Significant New Drugs Development” of China (No. 2018ZX09733001) , and the National Key Research and Development Program of China (No. 2016YFA0201402). This research did not receive any specific grants from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cheng, Y., He, C., Wang, M. et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Sig Transduct Target Ther 4, 62 (2019). https://doi.org/10.1038/s41392-019-0095-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-019-0095-0
This article is cited by
-
Epigenetic marvels: exploring the landscape of colorectal cancer treatment through cutting-edge epigenetic-based drug strategies
Clinical Epigenetics (2025)
-
Bridging tissue repair and epithelial carcinogenesis: epigenetic memory and field cancerization
Cell Death & Differentiation (2025)
-
Neuroblastoma susceptibility and association of N7-methylguanosine modification gene polymorphisms: multi-center case-control study
Pediatric Research (2025)
-
PRMT5 inhibition has a potent anti-tumor activity against adenoid cystic carcinoma of salivary glands
Journal of Experimental & Clinical Cancer Research (2025)
-
An Update and Translational Perspective in Genetics and Genomics of Breast Cancer
Current Breast Cancer Reports (2025)
