
Abstract
Development of breast cancer is linked to altered regulation of mammary gland developmental processes. A better understanding of normal mammary gland development can thus reveal possible mechanisms of how normal cells are re-programmed to become malignant. E2Fs 1-4 are part of the E2F transcription factor family with varied roles in mammary development, but little is known about the role of E2F5. A combination of scRNAseq and predictive signature tools demonstrated the presence of E2F5 in the mammary gland and showed changes in predicted activity during the various phases of mammary gland development. Testing the hypothesis that E2F5 regulates mammary function, we generated a mammary-specific E2F5 knockout mouse model, resulting in modest mammary gland development changes. However, after a prolonged latency the E2F5 conditional knockout mice developed highly metastatic mammary tumors. Whole genome sequencing revealed significant intertumor heterogeneity. RNAseq and protein analysis identified altered levels of Cyclin D1, with similarities to MMTV-Neu tumors, suggesting that E2F5 conditional knockout mammary glands and tumors may be dependent on Cyclin D1. Transplantation of the tumors revealed metastases to lymph nodes that were enriched through serial transplantation in immune competent recipients. Based on these findings, we propose that loss of E2F5 leads to altered regulation of Cyclin D1, which facilitates the development of metastatic mammary tumors after long latency. More importantly, this study demonstrates that conditional loss of E2F5 in the mammary gland leads to tumor formation, revealing its role as a transcription factor regulating a network of genes that normally result in a tumor suppressor function.
Similar content being viewed by others


The mammary gland is a complex organ that undergoes dynamic changes during different stages of development. Analysis of transcriptional profiles at each developmental stage, including pregnancy, lactation and involution, have revealed unique gene expression changes [1, 2]. One family of transcription factors that regulates these intricate transcriptional changes are the E2F transcription factors. The E2F transcription factor family consists of 8 members that are routinely divided to transcriptional activators and repressor [3,4,5,6] and are best known for their role in cell cycle progression [7,8,9,10]. However, they are a functionally diverse group of transcription factors with numerous studies highlighting their role in apoptosis [11], cell differentiation [12, 13], metabolism [14] and development [15,16,17,18,19]. The role of E2Fs in development was established through the characterization of single and compound E2F knockout mice. Following that, the role of E2Fs in mammary gland development was characterized in single knockouts of E2F1-4 [20]. Loss of E2F1, E2F3 or E2F4 resulted in mammary outgrowth delay and branching defects. However, these changes were not observed in E2F2 knockout mice. Given the high functional redundancy observed among E2Fs [21], the extent of compensation between the activator E2Fs in the mammary gland was explored. Using double knockout mice, E2F2 was found to partially compensate for the loss of E2F1 but not E2F3 in mammary ductal outgrowth, and transplant experiments demonstrated that these were cell autonomous effects [22].
E2F regulated functions are also integral in breast cancer development and progression to metastatic disease. For instance, specific activator E2Fs have unique roles in mediating metastasis, roles that are dependent upon the context of the activating oncogene with noted differences in tumors driven by Myc, Neu or PyMT [23,24,25,26,27]. E2Fs transcriptionally regulate a broad range of targets that are involved in many cancer associated pathways including apoptosis, genomic instability and DNA damage response [28]. Furthermore, E2Fs also have the potential to regulate oncogenes, with well characterized effects both up and downstream of Myc [6, 29,30,31]. The role of the E2Fs in development of the mammary gland and in altering tumor biology have been described for the activator E2Fs and repressor E2F4. However, the main E2F repressors include both E2F4 and E2F5 and the role of E2F5 in mammary development is not known. Similar to E2F4, E2F5 is considered to be a transcriptional repressor and canonically functions to repress cell cycle progression [32, 33]. Furthermore, E2F4 and E2F5 share the most structural similarities among all the E2F members and have demonstrated functional redundancy [32]. The role of E2F5 in tumorigenesis is conflicted and unclear as the literature for E2F5 in cancer biology reveals some studies suggesting an inhibition of transformation [34] while others note an oncogenic role [35,36,37]. Given the wide variety of target genes regulated by the E2Fs, it is likely that the role of E2F5 is largely tissue, cell type and context dependent, with roles that widely differ in tissue and tumor types. E2F5 has been understudied in part due to the early lethality associated with the knockout which results in hydrocephaly in prepubertal mice [19], making phenotypic examination and experimental manipulation more difficult. Interestingly, the hydrocephaly is distinct and is not observed in E2F4 knockout mice [18] but both E2Fs have functional overlap and combine to contribute to cell cycle regulation [32].
Here we have hypothesized that E2F5 plays an essential role in mammary gland development. To test this prediction, we generated mice with a conditional ablation of E2F5 in the mammary gland. In these mice we observed alterations to development and after a long latency, formation of metastatic tumors. Importantly, the transplantable tumors from these mice develop lymph node metastases, a trait not widely reported for other genetically engineered mouse models of breast cancer.
Results
To begin to investigate a potential role for E2F5 in mammary development and function, we examined a scRNAseq dataset [38] that was clustered into the various stages of mammary development, including nulliparious, pregnant, lactating and involuting (Fig. 1A). As a control, we first examined this scRNAseq data for E2F1 expression (Fig. 1B) and noted expected expression in the nulliparous and early pregnancy stages. We then examined this data for E2F5 expression (Fig. 1C), it revealed the highest level of expression of E2F5 in the lactating and involuting cells. Given the expression in different cell types we assessed E2F5 expression in a survey of various cell types within the mammary gland [39], using an involuting gland to have good representation of immune cells during remodeling (Fig. 1D). This revealed that E2F5 expression was present in several cell types, which Han et al. identified through various markers, including secretory alveoli as well as endothelial cells and fibroblasts (Fig. 1E and Supplemental Fig. 1). Interestingly, a comparison of differentiation states from the Bach et al. data revealed that E2F5 was expressed in progenitor cells as well as the differentiated secretory and hormone sensing lineages, a sharp contrast to the E2F1, a typical activator E2F (Fig. 1F). While expression of E2F5 leads to plausible hypotheses about function, expression is not linked to activity. To assess this, we generated a signature for E2F5 activation by overexpressing E2F5 using an adenoviral vector in Human Mammary Epithelial Cells (HMECs), with increasing multiplicity of infection tied to E2F5 levels (Fig. 1G). After infection, RNA was isolated after 18 h to generate a gene expression signature using microarrays. HMECs infected with the E2F5 construct were compared to controls with a GFP construct and a signature was generated using a Bayesian approach [40]. Despite being classically known as a transcriptional repressor, E2F5 expression resulted in both upregulated and downregulated genes, suggesting that E2F5 may also play a role as transcriptional activator [32, 33]. The up and down regulated genes in the signature were then tested for their predictive activity on a series of human breast cancer cell lines where predicted activity was compared to protein levels, validating the signature (Fig. 1H). A mammary gland developmental dataset [1] was then limited to the E2F5 signature genes and clustered, revealing that E2F5 regulated genes stratified mammary developmental stages (Fig. 1I). In addition, we characterized the subset of E2F5 regulated genes enriched in each developmental stage using gene ontology (Supplemental table 1). In the virgin and early pregnancy stages from Fig. 1I, the up and down regulated genes expressed after E2F5 overexpression were associated with various ontologies. For example, the most significant ontology in virgin and early pregnancy was organelle transport along microtubules (Supplemental Table 1). Later pregnancy timepoints were seen to have apoptotic processes enriched. However, as shown in data below, loss of E2F5 does not appreciably alter lactation, generating questions as to why apoptotic pathways were observed in the late pregnancy data. Next, we tested genes with a fold change in a terminal end bud/duct dataset [41] and found that nearly 10% of terminal end bud genes were also E2F5 regulated genes (Supplemental Fig. 2). Thus, we hypothesized that E2F5 played both developmental and functional roles in the mouse mammary gland.

Using a mouse mammary scRNAseq dataset that was split to various functional stages (A) including nulliparous (Null P) pregnancy day 14.5 (Preg D14.5), lactation day 6 (Lact D6) and involution day 11 (Inv D11). For each stage, the level of E2F1 (B) and E2F5 (C) expression was plotted. Using a separate scRNAseq dataset that was not sorted for epithelial cells (D), expression of E2F5 was examined across cell populations in the involuting mammary gland, revealing expression in endothelial cells, fibroblasts and alveoli with elevated signal in red and lower signaling in green (E). Examining lineage commitment (F), we overlaid E2F1 and E2F5 expression with elevated expression in green. To generate a signature for E2F5 activity, HMECs were infected with increasing multiplicity of infection (MOI) for an adenovirus expressing GFP or E2F5. A western blot demonstrated increasing levels of E2F5 with increasing MOI (G). Generation of a signature for E2F5 activation revealed genes up (orange/red) and down (blue) regulated. The activity of the signature was predicted in several human breast cancer cell lines and was plotted against the observed levels in a western blot for E2F5 revealing correlation between the two (H). A mammary gland developmental dataset (Stein et al., 2004) was limited to the E2F5 signature genes and was clustered, revealing that genes regulated by E2F5 stratified mammary developmental stages (I).
To directly test the role of E2F5 in mammary development we sought to use a knockout mouse model. With the lethality observed in the global knockout [19], we generated a strain of mice with loxP sites flanking exons 2 and 3 of E2F5 (Fig. 2A, Supplemental Fig. 3A, B). Through breeding to the MMTV-Cre strain [42] with expression limited to mammary epithelium [43], we generated a mammary specific knockout of E2F5 (Fig. 2A). Prior to interbreeding with MMTV-Cre transgenics, the E2F5flox/flox mice were backcrossed into the FVB background for 12 generations. Introduction of MMTV-Cre to the E2F5flox/flox strain resulted in the expected incomplete excision, which was not complete since whole mammary glands were tested and MMTV directed Cre expression is limited to mammary epithelium (Supplemental Fig. 3C). Testing for a developmental role, we examined mammary gland outgrowth at 4 weeks of age where the littermate controls had terminal end buds that had driven outgrowth past the lymph node in the fat pad (Fig. 2D). The E2F conditional knockouts (E2F5 CKO) had delayed development (Fig. 2E). Quantification of these results revealed a consistent delay in ductal extension (Fig. 2F, p = 0.0019). Heterozygous mice were not examined since the delay was slight. Other stages of mammary gland function were also assayed, with lactation occurring normally as assessed by histology and pup weight (Supplemental Fig. 4). To test for effects in older virgin mice, we allowed a cohort of controls and E2F5 CKO mice to age to 12 months. This revealed standard development in the controls and surprising alveolar overgrowth in the virgin E2F5 CKO mice (Fig. 2G, H).

A gene targeting strategy to flank exons 2 and 3 of E2F5 with loxP sites was employed with genotyping primers shown (A). With the introduction of Cre Recombinase under the mammary epithelial specific control of the MMTV promoter/enhancer, exons 2 and 3 are lost resulting in a tissue specific knockout. Examining ductal extension through wholemounts at 4 weeks we examined 20 control mice (E2F5flox/flox) (B) and 14 E2F5 CKO mice (C), revealing a delay in outgrowth. This delay was quantified revealing a consistent outgrowth delay, 0 = 0.0019 (D). After mice aged to 12 months, virgin mammary glands were assessed by both wholemount and histology. Relative to the MMTV-Cre controls (E) with their somewhat spiked ductal appearance, the E2F5 CKO mammary glands resembled a lactating mammary gland with alveoli engulfing the entire fat pad (F).
With the overgrowth of the virgin mammary gland, we carefully observed the E2F5 CKO mice for possible mammary tumor development. After a long latency, virgin E2F5 CKO mice developed focal mammary tumors while the MMTV-Cre control line did not (Fig. 3A). Multiparous E2F5 CKO mice were noted to have a slight reduction in tumor latency, potentially due to more widespread Cre expression during lactation. Given that MMTV is hormonally responsive, Cre is expressed at a much higher level [42], resulting in more widespread mammary epithelial E2F5 knockout, and tumors that develop slightly more rapidly. Time to 50% tumor burden for each genotype was as follows: Cre NA, Multiparous 596 days, Virgin 681 days, n = 16, 34 and 44 respectively with 16, 15 and 29 censored data points. Using a log rank test, the difference between virgin and multiparous was significant with p = 0.003. The heterozygous conditional deletion mice were not monitored for tumor development, but no MMTV-Cre E2F5 flox/wild type mice were noted to develop a tumor over the time they were used for breeding purposes.

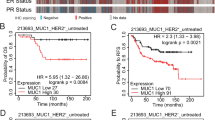
Regular palpation of the mammary glands revealed the onset of tumor formation in E2F5 CKO virgin and multiparous mice, but not in the control MMTV-Cre line (A). The resulting tumors were histologically diverse with numerous patterns noted, including squamous (B), solid with collagen tracks (C), papillary (D) and microacinar (E). 20-micron scale bars are included. The tumors were also metastatic, a pulmonary section reveals numerous metastatic lesions at both low and high power (F), this includes adenosquamous (left panel), normal lung (center panel) and EMT (right panel). Applying the E2F5 signature to human breast cancer stratified the patients to high/low quartiles. These results were consistent with the mouse model as low E2F5 activity was associated with worse survival (G).
The resulting tumors exhibited varied histological patterns, with a broad spectrum of subtypes reminiscent of those noted in other heterogenous strains such as MMTV-PyMT [25, 44]. Indeed, many of the same histopathologies previously noted in the PyMT and Myc strains were present, including adenosquamous (Fig. 3B), solid with collagen tracks (Fig. 3C), papillary (Fig. 3D and microacinar (Fig. 3E). No appreciable differences in pathology were noted in tumors from virgin and multiparous mice. Several tumors were tested by IHC for both ER and PR and were uniformly negative. CK8 and CK5 IHC staining revealed a mosaic pattern, suggesting both basal and luminal characteristics depending on the tumor (Supplemental Fig. 5). These tumors were highly metastatic with 74% of tumor bearing mice developing metastasis, with an average of 2.31 pulmonary metastases detected in a single histological section across the lobes of the lungs. An example of the pulmonary metastases is shown with several regions of the lungs magnified, illustrating that metastases in the lung can contain different pathologies for each metastatic lesion (Fig. 3F with adenosquamous (left panel), normal lung (center panel) and EMT (right panel)). In addition, liver metastasis was occasionally noted in the tumor bearing mice (4 metastases in 33 livers examined). Other metastatic lesions were infrequently noted on the thoracic wall, in the lymph nodes and in the intraperitoneal cavity as shown in Supplemental Fig. 6. No differences were noted in metastasis rates between virgin and multiparous tumors and it is unclear why only 74% of tumors were metastatic. With loss of E2F5 resulting in tumor formation in the mouse model, we hypothesized that E2F5 function may be related to human breast cancer development or clinical outcomes. Testing the E2F5 signature in a human breast cancer dataset allowed us to stratify patients to high/low quartiles of E2F5 activity. Survival analysis of these patients revealed that low E2F5 activity was associated with a reduction in survival rates (Fig. 3G). Moreover, at the single gene level, low levels of E2F5 expression in HER2 and triple negative subtypes of breast cancer was linked to worse outcomes (Supplemental Fig. 7A–D) [45]. Taken together, these data suggest that E2F5 expression and activity normally has a protective role in human breast cancer.
To explore the mechanisms by which E2F5 may regulate tumor development and progression we used an integrated approach by analyzing genomic and transcriptomic data. We examined whole genome sequence from five E2F5 CKO tumors with diverse histopathology (Supplemental Fig. 8). A representative circos plot for one of the tumors is shown with chromosomes around the periphery (Fig. 4A) with the remainder in supplemental data (Supplemental Fig. 9A–D, Supplemental Fig. 10A–E). Starting from the outermost region of the plot, an ideogram with labelled mouse chromosomes is provided. Single nucleotide variants (SNV) are shown as points with color reflecting putative impact, followed by copy number variants (CNV) with deletions and amplifications shown with color blocks. In the center, translocations and inversions are shown with lines (Fig. 4A). The differences between the circos plots were readily evident when comparing circos plots (Supplemental 9A-D). The combined SNV and CNV events impacting at least three tumors are shown (Fig. 4B), with some genes having more than one event per gene. To directly compare both shared and unique CNV events, we generated a graph demonstrating the CNV events found in the 5 tumors (Fig. 4C). All CNVs are listed in Supplemental Table 2. The CNV analysis revealed a number of regions that were partially conserved. A closer examination of chromosome 6 revealed that three of the five tumors shared an amplification event that encompasses Wnt2, Cav1 and Met (Fig. 4D). The boxed region in Fig. 4D is seen in expanded form in Fig. 4E, where the black line shows the diploid level and the amplification extent in three tumors is evident. Other regions of interest were noted on chromosome 3 and 8, where SNVs were enriched. Numerous SNVs in genes of potential interest were noted in multiple tumors, including in genes such as FBXO15 (5 of 5 tumors) and TSHZ1 (4 of 5 tumors). Other SNVs were highly impactful but were only noted in individual tumors, include p53, KRas, NUDT7 and MTRF1. SNVs that were visually inspected in the sequence data are included in Supplemental Table 2. Comparison to other WGS data for PyMT and Neu tumors revealed divergence between the tumor genotypes at the SNV level, with the most shared SNVs being between Neu and E2F5 CKO tumors (Supplemental Fig. 11). Examining the mutational spectrum of the tumors using the COSMIC SBS mutation signatures (Fig. 4F), revealed aging and HRR Deficient as the predominant signatures in the E2F5 CKO tumors. It should be noted that the aging signature may be present simply due to the age of the mice at which tumors developed.

Whole genome sequencing on five E2F5 CKO tumors were generated. Analysis of WGS data revealed CNVs, SNVs and translocation for each tumor. An example of a Circos plot generated for a single tumor provides a bird eye view of the genetic alteration identified (A). Starting from the outermost region of each plot, an ideogram with labelled mouse chromosomes is first, followed by SNVs at the next innermost ring. SNVs were marked as low (yellow), moderate (orange), and high (red) predicted impact as defined by Mutect2 annotation. The next innermost ring contains all predicted CNVs as defined by the consensus of Delly and Lumpy; deletions are colored blue and duplications are colored red. Within each tumor, the height of CNVs are scaled relative to the CNV with the largest amount of evidence supporting it as determined by Lumpy annotation (e.g. a duplication with 40 pieces of evidence will only be half the height of a deletion with 80 pieces of evidence). The width of each CNV is determined by the start and stop positions determined by the consensus of Delly and Lumpy on the length of the genome. Duplications point outward while deletions point inward starting from a shared midpoint. The innermost ring contains predicted high impact translocations and high to moderate impact inversions by the consensus of Delly and Lumpy calls. Inversions are colored black. Translocation color matches the ideogram color of one of the two chromosomes involved in the event. For all CNV and SNVs found in at least 3 tumors an oncoprint style plot was generated with SNV/CNV per tumor shown above and the SNV/CNV per gene shown at right. For each tumor sample in each column, only the high confidence calls are presented (B). Copy number alterations found in the tumors are illustrated with each column representing a chromosome location (C). Red indicates amplification while blue indicates deletion. A closer examination at the CNVs located on Chromosome 6 revealed some shared events between the tumors (D). The boxed region in (D) is expanded for the three tumors containing an amplification at that point in panel E. The COSMIC SBS mutation signatures enriched among the tumors were identified and are presented in panel F.
In addition to the WGS characterization, we also examined the transcriptional profiles of E2F5 CKO tumors through bulk RNAseq on samples derived from whole MMTV-Cre mammary glands (aged 635, 546 and 537 days), whole E2F5 CKO mammary glands (aged 470, 482 and 580 days), E2F5 CKO mammary tumors from all observed histological subtypes and tumor cell lines derived from the E2F5 CKO tumors (E2F5CKO tumor cell lines). First, we performed differential gene expression analysis between whole MMTV-Cre mammary glands and E2F5 CKO mammary glands as well as with E2F5 CKO tumors using the RNA-seq data. After filtering out differentially expressed genes with a fold change <2, the resulting lists of genes were further filtered based on its percent altered in human breast cancer (cut off >5%). The resulting upregulated and downregulated genes were analyzed using Enrichr, a gene set enrichment analysis tool [46]. The analysis revealed that the upregulated genesets are enriched for genes that are putative targets of E2F4 and E2F6 (Fig. 5A). Although this may not be surprising as there may be compensation, it does suggest that these differentially expressed genes are likely direct E2F targets and are dysregulated in E2F5CKO tumors. Given that this is a new model, we tested for relation to the known PAM50 subtypes, demonstrating that the tumors aligned with a mixture of Basal, Luminal A and HER2+ve tumors (Supplemental Fig. 12). Geneset enrichment analysis on the downregulated genes revealed enrichment for putative targets such as PPARG, SUZ12, MYDO1 and ESR1. (Supplemental Fig. 13)). Pathway analysis revealed that the upregulated geneset were enriched for pathways associated with cell cycling, Fanconi anemia, DNA repair, and DNA replication signaling pathways with loss of E2F5 (Fig. 5B). In contrast, the downregulated genesets were enriched for processes involved in normal metabolism (Supplemental Figure 13). We then examined the differentially expressed genes in StringDb to visualize which genes have known or predicted interactions with E2F5 (Fig. 5C). To further narrow down our target gene population, we examined the level of gene expression in both E2F5CKO mammary gland and tumor cell lines. Notably, we observed that Cyclin D1 transcripts were elevated in E2F5CKO mammary glands and significantly elevated in E2F5CKO tumor cell lines (Fig. 5D). Given that Cyclin D1 appears to be elevated in pre-tumor E2F5 CKO mammary glands, we hypothesize that alteration of Cyclin D1 expression may be critical for tumor formation in E2F5CKO tumors. In line with the differential gene expression analysis, Cyclin D1 and several other genes demonstrated increased expression in E2F5CKO mammary glands and tumors relative to MMTV-Cre mammary glands through qRT-PCR (Supplemental Fig. 14A–E). However, the most striking difference was seen in Cyclin D1 where there was a significant increase in E2F5CKO tumors relative to control in the RNAseq data (Fig. 5D), confirmed with a 15-fold increase in Cyclin D1 levels observed through qRT-PCR (Fig. 5E). To examine whether Cyclin D1 potentially has a prominent role in E2F5 tumors, we analyzed the levels of Cyclin D family members in E2F5CKO tumor in comparison to Wnt-1 and Neu tumors. This revealed that levels of Cyclin D1 were consistent in all three models. However, only cyclin D2 was detected in Wnt-1 tumors (Fig. 6F). The E2F5 CKO tumors resembled the MMTV-Neu tumors for expression of Cyclin D1, D2 and D3, suggesting that like MMTV-Neu the E2F5 CKO tumors were dependent on Cyclin D1. To further examine the potential inverse relationship between E2F5 and Cyclin D1, we examined Cyclin D1 levels in the setting of E2F5 overexpression in HMECs. Consistent with our previous findings, we found that Cyclin D1 is downregulated in E2F5 overexpressing HMECs that were used in the E2F5 signature creation (Supplemental Fig. 14F).

Analysis of differentially upregulated genes in E2F5KO using EnRichR on whole mammary glands lacking E2F5 relative to E2F5 CKO tumors revealed putative targets (A) and biological pathways (B). Further analysis with StringDb revealed the predicted interactions between E2F5 and the differentially upregulated genes (C). Using a filtering strategy to determine genes involved with E2F5 CKO tumor formation, we identified Cyclin D1. Comparison of mammary gland expression of cyclin D1 through RNAseq revealed an upregulation in whole mammary glands, tumors and more extensively in the cell lines derived from the tumors (D). Validation of these results through qRT-PCR for cyclin D1 in 3 MMTV-Cre and 3 E2F5 CKO whole mammary glands revealed a 15 fold upregulation of cyclin D1 in the pre-tumor mammary glands (t-test P-value 0.077) (E). Examination of Wnt1, Neu and E2F5 CKO tumors revealed that each strain had elevated Cyclin D1 but only Wnt1 tumors had upregulation of Cyclin D2 (F). Indeed, the E2F5 CKO tumors closely resembled the MMTV-Neu tumors with elevated Cyclin D1 and lower levels of both Cyclin D2 and D3 (quantification in Supplemental Table 4).

Implantation of E2F5 CKO tumors into FVB MMTV-Cre recipients resulted in mammary and axillary tumor formation. The strategy for serial transplantation of E2F5 CKO axillary tumors into the abdominal mammary gland to enrichlymphatic metastasis is shown (A). In an example of an enrichment necropsy, the primary tumor has been excised (abdominal gland, bottom of panel) but a tumor has also formed in the region of the axial lymph node (top) (B) Labels include ALNT (axial lymph node tumor), LV (enlarged lymphatic vessel) and the remnants of the excised primary tumor (PT) in the abdominal gland. Cross section of the lymph node reveals both lymph (left side) tissue and metastatic (right side) cells (C). Staining with a pan-cytokeratin antibody reveals nests of metastatic cells throughout the lymph node (D). Cross section and staining of the enlarged vessel from (B) for podoplanin reveals a positively staining lymphatic vessel containing counterstained tumor cells (E). A summary for four transplanted lines shows the number of rounds of transplantation and where the optimal lymph node enrichment point was observed with the percent of tumor bearing mice with lymph node metastasis included for each (F). Examining human breast cancer with known lymph node status for the E2F5 activation gene signature revealed that metastatic tumors had lower E2F5 activity (G). Splitting the samples into quartiles, the samples with the lowest levels of E2F5 were most likely to have lymph node metastasis (P < 0.0001, Fisher test) (H).
The ability to model metastases, a feature reflective of human breast cancer, is an attractive component of this new mouse model of breast cancer but one that is offset by the extended latency. To overcome this limitation, we aimed to generate a transplantable metastatic mammary tumor bank. During necropsy of tumor bearing mice, small portions of the tumor were viable frozen. These small tumor fragments were then thawed and directly implanted into mammary glands without passing through tissue culture. MMTV-Cre mice were used as transplant recipients to prevent immune effects associated with the MMTV promoter enhancer48. 18 frozen and one fresh tumor were implanted into a small pocket created in the abdominal mammary gland of recipient mice, rapidly resulting in overt tumor formation. Strikingly, it was repeatedly noted that these mice developed a mammary tumor accompanied by a secondary metastatic tumor in the axial lymph node in 12 of the 19 lines at low penetrance. These axial lymph metastases were then implanted into recipient mammary fat pads in repeated rounds of transplantation (Fig. 6A). The axial lymph node tumors were routinely nearly as large as the primary tumor and an example of the axial lymph node metastasis is shown (top left of the image) after the primary tumor in the abdominal gland was removed at 1 cm endpoint (bottom portion of image) (Fig. 6B). Tumors only developed where implanted and in the axial lymph nodes. Examining the histology of these metastatic tumors in the axial lymph node revealed the presence of both lymphatic and tumor tissue (Fig. 6C). Pan-cytokeratin staining for epithelial derived tumor cells revealed nests of tumor cells within this axial lymph node (Fig. 6D). This was further examined in additional samples where in comparison to the normal lymph node, we were readily able to observe the tumor cells invading the lymph node (Supplemental Figure 15). Hypothesizing that the metastasis was occurring through the lymphatic vasculature, we sectioned across the region between the primary tumor and axial lymph node from Fig. 6B and stained for podoplanin, a marker of lymphatic vessel walls. This revealed staining of the vasculature with unstained tumor cells lodged within (Fig. 6E). Given that blood vasculature does not stain for podoplanin, that the tumor cells were negative for podoplanin and that this mouse developed extensive lymphatic metastasis, the positively stained cells are likely part of the lymphatic vasculature. Control transplants of MMTV-Neu and MMTV-PyMT tumors did not result in lymph node metastasis. To increase the penetrance of lymph node metastasis, we utilized prior enrichment strategies for breast metastases [47,48,49] where a sample of lymph node metastases was implanted into the mammary gland of a new recipient mice and the resulting axillary lymph node tumor was re-implanted into new recipient mice. This serial implantation resulted in the enrichment of lymph node metastasis from a penetrance of 23%, 50% and 73% in three separate tumor lines (Fig. 6F). With each round of implantation, the latency of lymph node metastasis formation was reduced. A summary of the enrichment transplantation revealed the ability across several lines to enrich for this property (Fig. 6F and Supplemental Table 3).
After observing lymphatic metastasis in the E2F5 CKO mouse model, we examined the role of E2F5 in human lymphatic metastasis. We predicted E2F5 activity in human breast cancer with known nodal status using the E2F5 signature described in Fig. 1G. The stratification of breast cancers by lymph node metastasis status revealed that those tumors with lymph node metastases had lower levels of E2F5 (Fig. 6G). Examining the upper and lower quartiles for E2F5 activity revealed almost a four-fold increase in lymphatic metastasis in the lower quartile of E2F5 activity (Fig. 6H). Together these data indicate that loss of E2F5 is associated with tumors that metastasize to the lymph node in both our conditional knockout mouse model as well as in human breast cancer.
Discussion
The repressive transcription factor E2F5 has been hypothesized to have a role in mammary gland development and tumor formation. Testing this hypothesis required generation of a conditional deletion due to the early lethality of the traditional E2F5 knockout due to developmental abnormalities [19]. Surprisingly only minor mammary gland defects were noted with the mammary specific loss of E2F5 but, after an extensive latency, metastatic mammary tumors developed. Notably, these tumors metastasized to the lymph node and this property was enhanced with serial transplantation. Mechanistically, we demonstrated an upregulation of Cyclin D1, with similar expression pattern of Cyclin D family observed in MMTV-Neu, a model where tumorigenicity was dependent upon Cyclin D1 [50].
During generation of the E2F5 gene signature we overexpressed E2F5 in HMECs and collected RNA 18 h after infection, a standard procedure that allows immediate transcriptional events to be assayed [40, 51]. The conventional role for E2F5 is thought to be a transcriptional repressor [7, 32], although several more recent publications have demonstrated a role as an activator in diverse settings include zebrafish [52] and cervical cancer [53]. Our results demonstrate that E2F5 can function both as an activator and as a repressor in mammary epithelial cells, dependent upon the gene. This is consistent with prior work suggesting that the context of E2F binding is an essential component in determining repressor and activator function [54].
While the predictive bioinformatic data strongly suggested a role for E2F5 in several stages of mammary development, from regulation of genes expressed in the terminal end bud to stratification of the major stages of mammary development by E2F5 signature genes, the observed phenotype was a relatively minor delay in outgrowth. This was a transient delay with virgin adult mice being indistinguishable from controls and unlike the E2F4 knockouts, the E2F5 CKO mice retained full lactational function of the mammary gland. Prior work has shown a range of individualized roles for E2F1-4 in mammary development with E2F4 having the most severe developmental effects and a near inability to rear pups [20]. However, consistent with the notion that the E2Fs can compensate for each other [21], functional compensation during mammary gland development was noted in the E2F knockouts [22]. The idea of familial compensation was also reinforced by effects seen in E2F4/5 double knockouts that were more extensive than individual knockouts in cell cycle progression. Further, double knockouts suffered from neonatal lethality while the individual knockouts were viable at birth [32]. Together our data and the literature suggest that E2F5 functions to regulate development in a context dependent manner and other E2Fs may compensate for loss of E2F5 and mitigate the developmental abnormalities.
After an extended latency the E2F5 CKO mice spontaneously developed mammary tumors. Given this latency, we hypothesized that a number of genomic events would be required for transformation and progression to metastatic disease. Integrating our bioinformatic analysis and in vitro studies, we identified a number of potential target genes. While some were unique to individual tumors, including p53 and KRas mutations, other events were shared across the E2F5 conditional knockout tumors. One common event across all tumors was the alteration of Cyclin D1 levels. Cyclin D1 is one of the most commonly amplified and/or overexpressed genes in human breast cancer with overexpression noted in nearly 50% of tumors [55]. Overexpression of Cyclin D1 in the mouse mammary gland results in mammary tumor development after 18 month latency, supporting a key role in tumorigenesis [56]. The requirement for Cyclin D1 in various mammary tumor GEMs has previously been demonstrated, including Neu but not Wnt-1. This is largely driven by the effects of other Cyclin family members in the Wnt-1 model while Neu tumors are dependent upon Cyclin D1. Based on gene expression analysis, E2F5 expression is inversely correlated with Cyclin D1, suggesting that E2F5 may be negatively regulating Cyclin D1. In addition, the levels of Cyclin D1, D2 and D3 in E2F5 CKO tumors compared to MMTV-Wnt-1 [57] and MMTV-Neu [58] tumors strongly suggest that E2F5 CKO tumors are analogous to MMTV-Neu and are dependent upon Cyclin D1. Indeed, since E2F5 CKO tumors, like MMTV-Neu, mainly expresses Cyclin D1, the E2F5 tumors likely resemble the dependence of MMTV-Neu on Cyclin D1 [50, 59, 60]. Furthermore, Cyclin D1 expression in MMTV-Neu tumors is mediated by E2F1 [50]. Other studies have also demonstrated that E2F1 and E2F4 can directly bind to and regulate Cyclin D1 expression [61]. Given the functional redundancy and shared binding motif between E2F family members, it is likely that E2F5 can also regulate Cyclin D1 expression. Taken together, we propose that loss of E2F5 in the mammary gland leads to deregulation of Cyclin D1, contributing to tumor development and progression. Given the wide range of SNV and CNV alterations, we suggest that this occurs in a complex mutational environment with numerous other pathways. Although there is evidence suggesting that E2F5 may be directly Cyclin D1 expression, it is also possible that disruption of E2F5 leads to dysregulation of other targets that can result in Cyclin D1 expression. To confirm the role of Cyclin D1 in E2F5 CKO mice tumorigenesis, our future studies will characterize the effects of loss of Cyclin D1 in the E2F5 CKO model.
The role of Cyclin D1 in comparison to other tumor models also raised the issue of how similar these tumors were to other models. Clearly the tumors were divergent from models such as MMTV-PyMT and MMTV-Neu given the characteristics that included a much longer tumor latency and a propensity for lymphatic metastasis. While a gene expression comparison on tumor data is possible, we have previously shown that these patterns are dominated by the histological subtype [44]. Instead, we compared the WGS data we previously generated for PyMT and Neu tumors [62] and compared it with the E2F5 CKO tumors, which revealed a unique mutational spectrum. Despite this, the E2F5 CKO tumors share the Cyclin family pattern with Neu tumors. This suggests some similarity to the Neu tumors but the mutational analysis also provides key differences.
In the study of mammary tumors that spontaneously metastasize in genetically engineered mouse models, pulmonary metastasis is typically noted, a feature that was also observed in the E2F5 CKO mice. Interestingly, we discovered that E2F5 CKO mammary tumors transplanted into the abdominal mammary fat pad had a propensity to metastasize to the axillary lymph node. Given that axillary lymph nodes are most commonly the first site of metastasis in human breast cancer, we sought to enrich the ability of E2F5 CKO tumors to metastasize to the axillary lymph node. Using a serial transplantation technique to re-transplant the axillary tumor into the abdominal mammary fat pad, we generated a syngeneic transplantation model that developed lymph node metastasis within one month of transplant and with >80% penetrance. Generally, the current mouse models of breast cancer rarely metastasize to the lymph node [63]. Thus, this model of enriched lymph node tumors is unique and can be a tool to examine the mechanisms driving lymph node metastasis. However, it should be noted that this transplantation technique includes a 1 x 1x 1 mm fragment of the tumor which includes cells from the microenvironment, including other cell types such as fibroblasts, endothelial cells and a variety of immune cells and this may alter the properties and signaling of the cancer cells.
Taken together, we have identified a novel role of E2F5 as a function tumor suppressor utilizing a conditional knockout mouse model. This is consistent with prior literature suggesting E2F5 has varied roles, including as a tumor suppressor [33]. In contrast, there are studies suggesting E2F5 may behave as an oncogenes in various cancer types including breast cancer. For example, a previous study demonstrates that knocking down E2F5 in human breast cancer cell lines inhibited cell proliferation, migration and invasion [64]. It is unclear why there is conflicting data on the role of E2F5 as an oncogene or tumor suppressor but, as with many genes, context is key. Importantly, all previous studies describing the oncogenic role of E2F5 in breast cancer have been completed in cell line models, whereas this is the first study that characterizes loss of E2F5 in the mammary gland of a mouse model. Interestingly, a similar paradigm exists for E2F8, an atypical E2F family member, where a possible oncogenic role was uncovered when knocked down in transformed cells and a mouse model knockout revealed a tumor suppressor effect [65,66,67,68]. We theorize that like E2F8 and other E2F family members, E2F5 may behave as an oncogene or tumor suppressor depending on context. Importantly, this context may be dependent upon tumor type, and on when the dysregulation of E2F5 occurs, during precancerous vs transformed stage.
This study has resulted in the development of a novel mouse model of breast cancer with histologically diverse mammary tumors and metastatic lesions. A unique feature of this model is its prolonged tumor latency which is similar to human breast cancers, where the majority of cancers occur in older postmenopausal women. Although breast cancer is an age-related disease, the role of aging in tumorigenesis has not been well defined. Studying age-related impacts on tumorigenesis has been limited in part by the availability of transgenic mouse models with prolonged tumor latency [69]. Since the majority of available transgenic mouse models develop tumors rapidly, it does not allow time for age-related changes to occur. Thus, E2F5CKO mice, which has an average tumor latency of 18–21 months, may be a favorable model to elucidate the role of aging in breast cancer development and progression. Finally, the E2F5 CKO syngeneic transplantation model can be a significant resource to studying the mechanism of lymphatic metastasis.
Material and methods
E2F5 expression in mammary development stages and cell types
To analyze E2F5 expression in a single cell RNA-seq mammary development dataset, the following website was used: https://marionilab.cruk.cam.ac.uk/mammaryGland/. To analyze E2F5 expression in various mammary cell types, the following website was used: https://bis.zju.edu.cn/MCA/.
Mouse dataset usage
The following published microarray datasets were used for analysis: terminal end bud and duct (GSE2988) and mammary gland developmental stages (GSE12247).
Newly generated RNAseq and WGS data has been deposited at the NIH NLM under BioProject # PRJNA887715.
E2F5 regulated genes
Human Mammary Epithelial cells were infected with adenovirus expressing E2F5 or GFP. Cells were collected eighteen hours after infection. Total RNA was extracted using Qiagen RNeasy Mini kit and submitted to Michigan State University Genomics Core facility for gene expression analysis using Affymetrix Human Genome U133 chip. Robust Multichip Average (RMA) algorithm was used to normalized microarray dataset. Significance Analysis of Microarray was used to identify differentially expressed genes in HMEC-E2F5.
E2F5 activation signature generation
E2F5 activation signature was generated based on previously described bayesian approach [40]. The E2F5 activation signature was applied to publicly available human breast cancer cell line data set GSE3156. To validate the signature, E2F5 expression was evaluated in two cell lines with the highest predicted E2F5 activity and two cell lines with the lowest predicted E2F5 activity via Western blot.
Clustering
Cluster 3.0 was used to perform unsupervised hierarchical clustering. Heatmap was generated using MATLAB imagesc function and Broad institute’s Morpheus and Genepattern [70] interface.
Gene ontology
The subset of E2F5 regulated genes enriched in each mammary developmental stage were characterized by gene ontology using https://maayanlab.cloud/Enrichr/ (Kuleshov et al. 2016).
Animal generation
All animal husbandry and use was in compliance with local, national and institutional guidelines. Ethical approval for the study was approved by Michigan State University Animal Care & Use Committee (IACUC) under AUF 06/18-084-00. E2F5 CKO mice were generated by flanking exons 2 and 3 of the E2F5 gene with loxP sites. E2F5flox/flox mice were interbred with MMTV-Cre mice [42]. Mice were monitored weekly for tumor development. The endpoint for primary tumor was 2000 mm3.
Histology
For wholemount analysis, abdominal mammary fat pads were excised and placed on glass slides. The slides were incubated in acetone for 24 h, rehydrated through an ethanol progression and stained in Harris’ Modified Hematoxylin for 24 h. The slides were destained in acidified ethanol. Slides were dehydrated and mounted with permount. To evaluate mammary outgrowth, the distance from the nipple to the leading edge of the epithelium and the distance from the nipple to the midpoint of the thoracic lymph node were measured. Samples for histology were fixed in 10% formalin and submitted to Michigan State University Pathology lab.
Whole genome sequence generation and analysis
Whole genome sequencing (WGS) sample reads were concatenated and quality checked using FastQC/0.11.7. Sample reads had adapter ends trimmed off using Trimmomatic/0.38 [71] and again checked for quality using FastQC. Paired WGS reads were aligned to the GRCm38-mm10 reference genome using Burrows-Wheeler Aligner/0.7.17 [72]. Read groups were added using Picard/2.22.1 from the Broad Institute http://broadinstitute.github.io/picard. Reads were then indexed and sorted using SAMtools/1.11 [73]. Duplicate reads were removed using Picard.
Variant calling
Final bam files had single nucleotide variants (SNVs) called using the consensus of Mutect2 under GATK/4.0.5.1 [74] and VarScan/2.4.1 [75]. Translocations, inversions, and copy number variants (CNVs) were called using the consensus of Delly/0.7.8 [76] and Lumpy/0.2.13 [77] for each tumor sample. Annotations of all samples were done using SnpEff/4.1d [78]. Tabular data from tumor VCF files were sorted and processed using a custom Python script using Python 3.8.8, Pandas 1.2.4, and NumPy 1.20.1. All unaltered VCF files are available in supplementary.
Circos visualization
Circos plots were generated using Circos/0.69-6 [79].
CNVkit Plots
CNVkit plots were generated on CNVKit/0.9.9 [80]. The “cnvkit.py batch –method wgs” option was used to generate the copy number ratios and copy segments files for downstream visualization. The “cnvkit.py scatter” command was used to generate the scatter plots with all default setting maintained. Heatmaps were generated using the “cnvkit.py heatmap -d” options. Tumor calls were made against normal FVB tissue (ERR046395). To reduce false positives, we ran our same pipeline on the Sanger Mouse Genomes Project (https://www.sanger.ac.uk/data/mouse-genomes-project/) FVB/NJ background release 1604 bam file. All SNVs found in either FVB background were filtered out from the tumor data. Only SNVs with a somatic p-value <= 0.05 were included in all downstream analyses as determined by VarScan. CNVs and inversions found in the FVB/NJ background that are on the same chromosome and have a difference of less than 100 bps in length from either FVB background event are excluded from analysis. For translocations, all events on either FVB background that are within 1 kb of either the donor or receiver position on each chromosome are dropped. These steps were accomplished using a custom python script.
Mutation signature
The Catalogue of Somatic Mutations in Cancer (COSMIC) single base substitution (SBS) signatures for E2F5 flox/flox tumors were derived using the deconstructSigs R package [81]. The mouse GRCm38 (mm10) reference assembly was used for trinucleotide context when calling SBS signatures. The resulting weights were plotted using matplotlib in Python.
Cell culture
Human mammary epithelial cells were cultured in Mammary Epithelial Cell Basal Medium (ATCC PCS-600-030) supplemented with rH-insulin (5 ug/mL), L-glutamine (6 mM), epinephrine (1 µM), apo-transferrin (5 µg/ml), rH-TGF-α (5 ng/ml), extractP (0.4%) and hydrocortisone hemisuccinate (100 ng/ml).
E2F5KO tumor cell line generation
E2F5KO mammary tumor pieces were placed on a culture dish with Dulbecco’s Modified Eagle Medium (Millipore Sigma D5030) supplemented with 10% Fetal bovine serum and 1x Antibiotic/Antimycotic to allow for cells to dissociate. Cells were passaged several times to enrich for epithelial cell population. Western blot was using an E2F5 antibody to demonstrate loss of E2F5 in isolated cell lines (Supplemental Fig.16). Tumor cell lines were validated for tumorigenic potential by implantation into recipient (MMTV-Cre) lines where they were noted to form tumors.
RNA-sequencing
Flash frozen tumor pieces were homogenized using Fisher Homogenizer 150 (Thermo Scientific). Total RNA was isolated using QIAGEN RNeasy Midi Kit (Hilden, Germany #75142) with the manufacturer’s protocol. RNA concentration was measured by Qubit (Thermo Scientific) and Agilent 2100 Bioanalyzer. RNA was submitted to Novogene to generate gene expression data. RNA samples with RIN > 7 was used for library preparation using the Illumina Tru-Seq stranded total RNA kit. RNA library was sequenced to a depth of >20 M reads/sample with paired end 150 base paired reads on Illumina NovaSeq 6000. Adaptors were removed from reads using Trimmomatic v0.33. Quality control was performed using FastQC v0.11.5. Reads were aligned and mapped using STAR [82]. RSEM was used to quantify and normalize reads [83]. Differential gene expression analysis was performed using EdgeR [84]. Pathway analysis was performed using https://maayanlab.cloud/Enrichr/ [46]. Protein-protein interaction network was generated with https://string-db.org/ [85].
Quantitative RT-PCR
Flash frozen tumor pieces were homogenized using Fisher Homogenizer 150 (Thermo Fisher). Total RNA was isolated using QIAGEN RNeasy Midi Kit (Qiagen 75142) with the manufacturer’s protocol. Quantitative RT-PCR was performed using Luna Universal One-Step RT-qPCR kit (New England Biolabs E3005S) according to manufacturer’s protocol using Agilent Mx3000P instrument. Primers were designed using Primer Bank tool (https://pga.mgh.harvard.edu/primerbank/). The following primers were used (5’ to 3’): CCND1 forward, TGACTGCCGAGAAGTTGTGC; CCND1 reverse, CTCATCCGCCTCTGGCATT; Gapdh forward, AGGTCGGTGTGAACGGATTTG; Gapdh reverse, TGTAGACCATGTAGTTGAGGTCA. Primer efficiency was 90–110% for all primers used. Delta-delta CT method was used for fold change analysis. For mammary gland comparisons, 3 control and 3 conditional knockout samples were used and experiments were done in triplicate.
Immunoblotting
To extract RNA from tissue, samples were homogenized using mortar and pestle in liquid nitrogen. Sample were lysed in RIPA lysis buffer (Thermo Scientific 89900) with proteinase inhibitor (Thermo Scientific A32963) for 1 h on ice with constant agitation. Protein was quantitated using BCA Protein Assay Kit (Thermo Scientific 3225) and then boiled at 100 °C for 5 min. Samples were loaded onto an 8–12% polyacrylamide gel. Separated protein was transferred onto an Immobilon- FL PVDF membrane (Millipore Sigma PFL00010). Membranes were blocked in 5% milk in TBS with 0.1% Tween-20 (TBS-Tween) for 1 h and then incubated in primary antibody overnight at 4 °C. Following three washes in TBS-Tween the membrane was incubated in the appropriate antibody at a dilution 1:10,000 in 5% milk in TBS-Tween for 1 h at room temperature. Membranes were washed 3x in TBS-Tween and imaged on LI-COR Odyssey imaging system. The following antibodies were used: 1:100 E2F5 (Santa Cruz Biotech sc-374268), 1:1000 Grb2 (Cell Signaling Technology 3976), 1:4000 Vinculin (Cell Signaling Technology 13901), 1:2000 Cyclin D1 (Thermo Scientific 516356.), 1:2000 Cyclin D3 (Thermo Scientific PA5-97551) and 1:1000 Cyclin D2 (Proteintech 10934-1-AP). Image studio version 5.5 was used for analysis and quantification of the Western blot.
Mammary fat pad transplantation
E2F5 CKO mammary tumors were harvested and stored in DMEM with 20% FBS and 10% DMSO at −80 °C before long term storage in liquid nitrogen vapor phase. Tumors were thawed and orthotopically implanted into the abdominal mammary gland of 6-to-10-week old MMTV Cre female mice. Mice were palpated 2x a week for mammary tumor formation. When the tumor size reached 2000 mm3, samples were harvested for further analysis.
E2F5 activation in clinical samples
E2F5 activation signature was applied to a human breast cancer database using MATLAB [86]. Graphpad was used to create the survival plot in patients with high vs low E2F5 activation. Broad Institute’s Morpheus was used to generate the heatmap demonstrating E2F5 activation in human breast cancer patient and its lymph node metastasis status.
Statistical analysis
Except otherwise noted, all statistical comparisons are performed with an unpaired students two-tailed, unpaired t-test.
Data availability
Data is freely available and bioinformatic data has been deposited at BioProject # PRJNA887715.
References
Stein T, Morris JS, Davies CR, Weber-Hall SJ, Duffy MA, Heath VJ, et al. Involution of the mouse mammary gland is associated with an immune cascade and an acute-phase response, involving LBP, CD14 and STAT3. Breast Cancer Res. 2004;6:R75–91.
Clarkson RW, Wayland MT, Lee J, Freeman T, Watson CJ. Gene expression profiling of mammary gland development reveals putative roles for death receptors and immune mediators in post-lactational regression. Breast Cancer Res. 2004;6:R92–109.
Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–62.
Van Den Heuvel S, Dyson NJ. Conserved functions of the pRB and E2F families. Nature reviews. Mol cell Biol. 2008;9:713–24.
Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002;3:11–20.
Sears RC, Nevins JR. Signaling networks that link cell proliferation and cell fate. J Biol Chem. 2002;277:11617–20.
Attwooll C, Lazzerini Denchi E, Helin K. The E2F family: specific functions and overlapping interests. EMBO J. 2004;23:4709–16.
Johnson DG, Schwarz JK, Cress WD, Nevins JR. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature. 1993;365:349–52.
Laresgoiti U, Apraiz A, Olea M, Mitxelena J, Osinalde N, Rodriguez JA, et al. E2F2 and CREB cooperatively regulate transcriptional activity of cell cycle genes. Nucleic Acids Res. 2013;41:10185–98.
Mudryj M, Devoto SH, Hiebert SW, Hunter T, Pines J, Nevins JR. Cell cycle regulation of the E2F transcription factor involves an interaction with cyclin A. Cell. 1991;65:1243–53.
Hallstrom TC, Mori S, Nevins JR. An E2F1-dependent gene expression program that determines the balance between proliferation and cell death. Cancer Cell. 2008;13:11–22.
Asp P, Acosta-Alvear D, Tsikitis M, van Oevelen C, Dynlacht BD. E2f3b plays an essential role in myogenic differentiation through isoform-specific gene regulation. Genes Dev. 2009;23:37–53.
Chong JL, Wenzel PL, Saenz-Robles MT, Nair V, Ferrey A, Hagan JP, et al. E2f1-3 switch from activators in progenitor cells to repressors in differentiating cells. Nature. 2009;462:930–4.
Fajas L. Metabolic control in cancer cells. Ann Endocrinol (Paris). 2013;74:71–73.
Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG Jr., Livingston DM, et al. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell. 1996;85:549–61.
Murga M, Fernandez-Capetillo O, Field SJ, Moreno B, Borlado LR, Fujiwara Y, et al. Mutation of E2F2 in mice causes enhanced T lymphocyte proliferation, leading to the development of autoimmunity. Immunity. 2001;15:959–70.
Humbert PO, Verona R, Trimarchi JM, Rogers C, Dandapani S, Lees JA. E2f3 is critical for normal cellular proliferation. Genes Dev. 2000;14:690–703.
Rempel RE, Saenz-Robles MT, Storms R, Morham S, Ishida S, Engel A, et al. Loss of E2F4 activity leads to abnormal development of multiple cellular lineages. Mol Cell. 2000;6:293–306.
Lindeman GJ, Dagnino L, Gaubatz S, Xu Y, Bronson RT, Warren HB, et al. A specific, nonproliferative role for E2F-5 in choroid plexus function revealed by gene targeting. Genes Dev. 1998;12:1092–8.
Andrechek ER, Mori S, Rempel RE, Chang JT, Nevins JR. Patterns of cell signaling pathway activation that characterize mammary development. Development. 2008;135:2403–13.
Kong LJ, Chang JT, Bild AH, Nevins JR. Compensation and specificity of function within the E2F family. Oncogene. 2007;26:321–7.
To B, Andrechek ER. Transcription factor compensation during mammary gland development in E2F knockout mice. PLoS One. 2018;13:e0194937.
Fujiwara K, Yuwanita I, Hollern DP, Andrechek ER. Prediction and Genetic Demonstration of a Role for Activator E2Fs in Myc-Induced Tumors. Cancer Res. 2011;71:1924–32.
Yuwanita I, Barnes D. S, Andrechek ER. Increased metastasis with loss of E2F2 in Myc-driven tumors. Oncotarget. 2015;6:38210–24.
Hollern DP, Honeysett J, Cardiff RD, Andrechek ER. The E2F transcription factors regulate tumor development and metastasis in a mouse model of metastatic breast cancer. Mol Cell Biol. 2014;34:3229–43.
Andrechek ER. HER2/Neu tumorigenesis and metastasis is regulated by E2F activator transcription factors. Oncogene. 2015;34:217–25.
Rennhack J, Andrechek E. Conserved E2F mediated metastasis in mouse models of breast cancer and HER2 positive patients. Oncoscience. 2015;2:867–71.
Black EP, Hallstrom T, Dressman HK, West M, Nevins JR. Distinctions in the specificity of E2F function revealed by gene expression signatures. Proc Natl Acad Sci USA. 2005;102:15948–53.
Leone G, Sears R, Huang E, Rempel R, Nuckolls F, Park CH, et al. Myc requires distinct E2F activities to induce S phase and apoptosis. Mol Cell. 2001;8:105–13.
Pusapati RV, Weaks RL, Rounbehler RJ, McArthur MJ, Johnson DG. E2F2 suppresses Myc-induced proliferation and tumorigenesis. Mol Carcinog. 2010;49:152–6.
Rounbehler RJ, Rogers PM, Conti CJ, Johnson DG. Inactivation of E2f1 enhances tumorigenesis in a Myc transgenic model. Cancer Res. 2002;62:3276–81.
Gaubatz S, Lindeman GJ, Ishida S, Jakoi L, Nevins JR, Livingston DM, et al. E2F4 and E2F5 play an essential role in pocket protein-mediated G1 control. Mol Cell. 2000;6:729–35.
Pierce AM, Schneider-Broussard R, Philhower JL, Johnson DG. Differential activities of E2F family members: unique functions in regulating transcription. Mol Carcinog. 1998;22:190–8.
Chen C, Wells AD. Comparative analysis of E2F family member oncogenic activity. PLoS One. 2007;2:e912.
Jiang Y, Yim SH, Xu HD, Jung SH, Yang SY, Hu HJ, et al. A potential oncogenic role of the commonly observed E2F5 overexpression in hepatocellular carcinoma. World J Gastroenterol. 2011;17:470–7.
Polanowska J, Le Cam L, Orsetti B, Valles H, Fabbrizio E, Fajas L, et al. Human E2F5 gene is oncogenic in primary rodent cells and is amplified in human breast tumors. Genes Chromosomes Cancer. 2000;28:126–30.
Umemura S, Shirane M, Takekoshi S, Kusakabe T, Itoh J, Egashira N, et al. Overexpression of E2F-5 correlates with a pathological basal phenotype and a worse clinical outcome. Br J Cancer. 2009;100:764–71.
Bach K, Pensa S, Grzelak M, Hadfield J, Adams DJ, Marioni JC, et al. Differentiation dynamics of mammary epithelial cells revealed by single-cell RNA sequencing. Nat Commun. 2017;8:2128.
Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell. 2018;173:1307.
Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–7.
Kouros-Mehr H, Werb Z. Candidate regulators of mammary branching morphogenesis identified by genome-wide transcript analysis. Dev Dyn. 2006;235:3404–12.
Andrechek ER, Hardy WR, Siegel PM, Rudnicki MA, Cardiff RD, Muller WJ. Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proc Natl Acad Sci USA. 2000;97:3444–9.
Andrechek ER, White D, Muller WJ. Targeted disruption of ErbB2/Neu in the mammary epithelium results in impaired ductal outgrowth. Oncogene. 2005;24:932–7.
Hollern DP, Swiatnicki MR, Andrechek ER. Histological subtypes of mouse mammary tumors reveal conserved relationships to human cancers. PLoS Genet. 2018;14:e1007135.
Gyorffy B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput Struct Biotechnol J. 2021;19:4101–9.
Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–97.
Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–9.
Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–49.
Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–24.
Lee RJ, Albanese C, Fu M, D’Amico M, Lin B, Watanabe G, et al. Cyclin D1 is required for transformation by activated Neu and is induced through an E2F-dependent signaling pathway. Mol Cell Biol. 2000;20:672–83.
Gatza ML, Lucas JE, Barry WT, Kim JW, Wang Q, Crawford MD, et al. A pathway-based classification of human breast cancer. Proc Natl Acad Sci USA. 2010;107:6994–9.
Xie H, Kang Y, Wang S, Zheng P, Chen Z, Roy S, et al. E2f5 is a versatile transcriptional activator required for spermatogenesis and multiciliated cell differentiation in zebrafish. PLoS Genet. 2020;16:e1008655.
Teissier S, Pang CL, Thierry F. The E2F5 repressor is an activator of E6/E7 transcription and of the S-phase entry in HPV18-associated cells. Oncogene. 2010;29:5061–70.
Freedman JA, Chang JT, Jakoi L, Nevins JR. A combinatorial mechanism for determining the specificity of E2F activation and repression. Oncogene. 2009;28:2873–81.
Buckley MF, Sweeney KJ, Hamilton JA, Sini RL, Manning DL, Nicholson RI, et al. Expression and amplification of cyclin genes in human breast cancer. Oncogene. 1993;8:2127–33.
Wang TC, Cardiff RD, Zukerberg L, Lees E, Arnold A, Schmidt EV. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature. 1994;369:669–71.
Li Y, Hively WP, Varmus HE. Use of MMTV-Wnt-1 transgenic mice for studying the genetic basis of breast cancer. Oncogene. 2000;19:1002–9.
Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci USA. 1992;89:10578–82.
Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017–21.
Zhang Q, Sakamoto K, Liu C, Triplett AA, Lin WC, Rui H, et al. Cyclin D3 compensates for the loss of cyclin D1 during ErbB2-induced mammary tumor initiation and progression. Cancer Res. 2011;71:7513–24.
Watanabe G, Albanese C, Lee RJ, Reutens A, Vairo G, Henglein B, et al. Inhibition of cyclin D1 kinase activity is associated with E2F-mediated inhibition of cyclin D1 promoter activity through E2F and Sp1. Mol Cell Biol. 1998;18:3212–22.
Rennhack JP, To B, Swiatnicki M, Dulak C, Ogrodzinski MP, Zhang Y, et al. Integrated analyses of murine breast cancer models reveal critical parallels with human disease. Nat Commun. 2019;10:3261.
To B, Isaac D, Andrechek ER. Studying Lymphatic Metastasis in Breast Cancer: Current Models, Strategies, and Clinical Perspectives. J Mammary Gland Biol Neoplasia. 2020;25:191–203.
Xu H, Fei D, Zong S, Fan Z. MicroRNA-154 inhibits growth and invasion of breast cancer cells through targeting E2F5. Am J Transl Res. 2016;8:2620–30.
Thurlings I, Martinez-Lopez LM, Westendorp B, Zijp M, Kuiper R, Tooten P, et al. Synergistic functions of E2F7 and E2F8 are critical to suppress stress-induced skin cancer. Oncogene. 2017;36:829–39.
Kent LN, Rakijas JB, Pandit SK, Westendorp B, Chen HZ, Huntington JT, et al. E2f8 mediates tumor suppression in postnatal liver development. J Clin Invest. 2016;126:2955–69.
Park SA, Platt J, Lee JW, López-Giráldez F, Herbst RS, Koo JS. E2F8 as a Novel Therapeutic Target for Lung Cancer. J Natl Cancer Inst. 2015;107.https://doi.org/10.1093/jnci/djv151.
Deng Q, Wang Q, Zong WY, Zheng DL, Wen YX, Wang KS, et al. E2F8 contributes to human hepatocellular carcinoma via regulating cell proliferation. Cancer Res. 2010;70:782–91.
Rennhack J, To B, Wermuth H, Andrechek ER. Mouse Models of Breast Cancer Share Amplification and Deletion Events with Human Breast Cancer. J Mammary Gland Biol Neoplasia. 2017;22:71–84.
Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38:500–1.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76.
Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–i339.
Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15:R84.
Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6:80–92.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45.
Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol. 2016;12:e1004873.
Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 2011;12:323.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40.
von Mering C, Jensen LJ, Snel B, Hooper SD, Krupp M, Foglierini M, et al. STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005;33:D433–437.
Mihaly Z, Kormos M, Lanczky A, Dank M, Budczies J, Szasz MA, et al. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Res Treat. 2013;140:219–32.
Funding
This work was supported by a grant from the Elsa U Pardee Foundation and NICHD 1R01HD104606-01 to ERA, and Aitch Foundation scholarships to BT and JPR.
Author information
Authors and Affiliations
Contributions
BT was involved with conceptualization, methodology, investigation, and writing, CB analyzed data, J-RJ was involved in methodology and investigation, JG-L, JV, RR, JR, DH, LJ, DJ, MS, EV, RK and JH were involved in analyzing data and investigation, JN was involved in conceptualization and ERA was involved in conceptualization, methodology, validation, analysis, investigation, writing, supervision, administration and funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
To, B., Broeker, C., Jhan, JR. et al. Insight into mammary gland development and tumor progression in an E2F5 conditional knockout mouse model. Oncogene (2024). https://doi.org/10.1038/s41388-024-03172-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41388-024-03172-4
