
Abstract
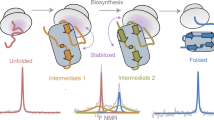
How do the key features of protein folding, elucidated from studies on native, isolated proteins, manifest in cotranslational folding on the ribosome? Using a well-characterized family of homologous α-helical proteins with a range of biophysical properties, we show that spectrin domains can fold vectorially on the ribosome and may do so via a pathway different from that of the isolated domain. We use cryo-EM to reveal a folded or partially folded structure, formed in the vestibule of the ribosome. Our results reveal that it is not possible to predict which domains will fold within the ribosome on the basis of the folding behavior of isolated domains; instead, we propose that a complex balance of the rate of folding, the rate of translation and the lifetime of folded or partly folded states will determine whether folding occurs cotranslationally on actively translating ribosomes.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

References
Braselmann, E., Chaney, J.L. & Clark, P.L. Folding the proteome. Trends Biochem. Sci. 38, 337–344 (2013).
Joh, N.H. et al. De novo design of a transmembrane Zn2+-transporting four-helix bundle. Science 346, 1520–1524 (2014).
Song, W.J. & Tezcan, F.A. A designed supramolecular protein assembly with in vivo enzymatic activity. Science 346, 1525–1528 (2014).
Koga, N. et al. Principles for designing ideal protein structures. Nature 491, 222–227 (2012).
Hingorani, K.S. & Gierasch, L.M. Comparing protein folding in vitro and in vivo: foldability meets the fitness challenge. Curr. Opin. Struct. Biol. 24, 81–90 (2014).
Frydman, J., Erdjument-Bromage, H., Tempst, P. & Hartl, F.U. Co-translational domain folding as the structural basis for the rapid de novo folding of firefly luciferase. Nat. Struct. Biol. 6, 697–705 (1999).
Kim, S.J. et al. Protein folding. Translational tuning optimizes nascent protein folding in cells. Science 348, 444–448 (2015).
Evans, M.S., Sander, I.M. & Clark, P.L. Cotranslational folding promotes β-helix formation and avoids aggregation in vivo. J. Mol. Biol. 383, 683–692 (2008).
Voss, N.R., Gerstein, M., Steitz, T.A. & Moore, P.B. The geometry of the ribosomal polypeptide exit tunnel. J. Mol. Biol. 360, 893–906 (2006).
Bhushan, S. et al. SecM-stalled ribosomes adopt an altered geometry at the peptidyl transferase center. PLoS Biol. 9, e1000581 (2011).
Bhushan, S. et al. α-Helical nascent polypeptide chains visualized within distinct regions of the ribosomal exit tunnel. Nat. Struct. Mol. Biol. 17, 313–317 (2010).
Nilsson, O.B. et al. Cotranslational protein folding inside the ribosome exit tunnel. Cell Reports 12, 1533–1540 (2015).
O'Brien, E.P., Hsu, S.T., Christodoulou, J., Vendruscolo, M. & Dobson, C.M. Transient tertiary structure formation within the ribosome exit port. J. Am. Chem. Soc. 132, 16928–16937 (2010).
Tu, L., Khanna, P. & Deutsch, C. Transmembrane segments form tertiary hairpins in the folding vestibule of the ribosome. J. Mol. Biol. 426, 185–198 (2014).
Kosolapov, A. & Deutsch, C. Tertiary interactions within the ribosomal exit tunnel. Nat. Struct. Mol. Biol. 16, 405–411 (2009).
Holtkamp, W. et al. Cotranslational protein folding on the ribosome monitored in real time. Science 350, 1104–1107 (2015).
O'Brien, E.P., Christodoulou, J., Vendruscolo, M. & Dobson, C.M. New scenarios of protein folding can occur on the ribosome. J. Am. Chem. Soc. 133, 513–526 (2011).
Ismail, N., Hedman, R., Lindén, M. & von Heijne, G. Charge-driven dynamics of nascent-chain movement through the SecYEG translocon. Nat. Struct. Mol. Biol. 22, 145–149 (2015).
Cymer, F. & von Heijne, G. Cotranslational folding of membrane proteins probed by arrest-peptide-mediated force measurements. Proc. Natl. Acad. Sci. USA 110, 14640–14645 (2013).
Ismail, N., Hedman, R., Schiller, N. & von Heijne, G. A biphasic pulling force acts on transmembrane helices during translocon-mediated membrane integration. Nat. Struct. Mol. Biol. 19, 1018–1022 (2012).
Goldman, D.H. et al. Mechanical force releases nascent chain-mediated ribosome arrest in vitro and in vivo. Science 348, 457–460 (2015).
Nilsson, O.B., Müller-Lucks, A., Kramer, G., Bukau, B. & von Heijne, G. Trigger factor reduces the force exerted on the nascent chain by a cotranslationally folding protein. J. Mol. Biol. 428, 1356–1364 (2016).
Nakatogawa, H. & Ito, K. Secretion monitor, SecM, undergoes self-translation arrest in the cytosol. Mol. Cell 7, 185–192 (2001).
Yap, M.N. & Bernstein, H.D. The plasticity of a translation arrest motif yields insights into nascent polypeptide recognition inside the ribosome tunnel. Mol. Cell 34, 201–211 (2009).
Tsai, A., Kornberg, G., Johansson, M., Chen, J. & Puglisi, J.D. The dynamics of SecM-induced translational stalling. Cell Reports 7, 1521–1533 (2014).
Gumbart, J., Schreiner, E., Wilson, D.N., Beckmann, R. & Schulten, K. Mechanisms of SecM-mediated stalling in the ribosome. Biophys. J. 103, 331–341 (2012).
Butkus, M.E., Prundeanu, L.B. & Oliver, D.B. Translocon “pulling” of nascent SecM controls the duration of its translational pause and secretion-responsive secA regulation. J. Bacteriol. 185, 6719–6722 (2003).
Shimizu, Y., Kanamori, T. & Ueda, T. Protein synthesis by pure translation systems. Methods 36, 299–304 (2005).
Batey, S. & Clarke, J. The folding pathway of a single domain in a multidomain protein is not affected by its neighbouring domain. J. Mol. Biol. 378, 297–301 (2008).
Batey, S. & Clarke, J. Apparent cooperativity in the folding of multidomain proteins depends on the relative rates of folding of the constituent domains. Proc. Natl. Acad. Sci. USA 103, 18113–18118 (2006).
Batey, S., Randles, L.G., Steward, A. & Clarke, J. Cooperative folding in a multi-domain protein. J. Mol. Biol. 349, 1045–1059 (2005).
Wensley, B.G., Kwa, L.G., Shammas, S.L., Rogers, J.M. & Clarke, J. Protein folding: adding a nucleus to guide helix docking reduces landscape roughness. J. Mol. Biol. 423, 273–283 (2012).
Wensley, B.G. et al. Experimental evidence for a frustrated energy landscape in a three-helix-bundle protein family. Nature 463, 685–688 (2010).
Cabrita, L.D. et al. A structural ensemble of a ribosome-nascent chain complex during cotranslational protein folding. Nat. Struct. Mol. Biol. 23, 278–285 (2016).
Cabrita, L.D., Dobson, C.M. & Christodoulou, J. Protein folding on the ribosome. Curr. Opin. Struct. Biol. 20, 33–45 (2010).
Kaiser, C.M., Goldman, D.H., Chodera, J.D., Tinoco, I. Jr. & Bustamante, C. The ribosome modulates nascent protein folding. Science 334, 1723–1727 (2011).
Scott, K.A., Randles, L.G. & Clarke, J. The folding of spectrin domains II: phi-value analysis of R16. J. Mol. Biol. 344, 207–221 (2004).
Wensley, B.G., Gärtner, M., Choo, W.X., Batey, S. & Clarke, J. Different members of a simple three-helix bundle protein family have very different folding rate constants and fold by different mechanisms. J. Mol. Biol. 390, 1074–1085 (2009).
Ingolia, N.T., Lareau, L.F. & Weissman, J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802 (2011).
Young, R. & Bremer, H. Polypeptide-chain-elongation rate in Escherichia coli B/r as a function of growth rate. Biochem. J. 160, 185–194 (1976).
Borgia, M.B. et al. Single-molecule fluorescence reveals sequence-specific misfolding in multidomain proteins. Nature 474, 662–665 (2011).
Borgia, A. et al. Localizing internal friction along the reaction coordinate of protein folding by combining ensemble and single-molecule fluorescence spectroscopy. Nat. Commun. 3, 1195 (2012).
Hill, S.A., Kwa, L.G., Shammas, S.L., Lee, J.C. & Clarke, J. Mechanism of assembly of the non-covalent spectrin tetramerization domain from intrinsically disordered partners. J. Mol. Biol. 426, 21–35 (2014).
Ke, S.H. & Madison, E.L. Rapid and efficient site-directed mutagenesis by single-tube 'megaprimer' PCR method. Nucleic Acids Res. 25, 3371–3372 (1997).
Zheng, L., Baumann, U. & Reymond, J.L. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115 (2004).
Bischoff, L., Berninghausen, O. & Beckmann, R. Molecular basis for the ribosome functioning as an L-tryptophan sensor. Cell Rep. 9, 469–475 (2014).
Chen, J.Z. & Grigorieff, N. SIGNATURE: a single-particle selection system for molecular electron microscopy. J. Struct. Biol. 157, 168–173 (2007).
Frank, J. et al. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 116, 190–199 (1996).
Penczek, P.A., Frank, J. & Spahn, C.M. A method of focused classification, based on the bootstrap 3D variance analysis, and its application to EF-G-dependent translocation. J. Struct. Biol. 154, 184–194 (2006).
Scheres, S.H. & Chen, S. Prevention of overfitting in cryo-EM structure determination. Nat. Methods 9, 853–854 (2012).
Pettersen, E.F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Acknowledgements
We thank A. Heuer for help preparing the figures. Supported by grants from the Swedish Cancer Foundation, the Swedish Research Council and the Knut and Alice Wallenberg Foundation (to G.v.H.); the Wellcome Trust (WT095195 to J.C.) and the European Research Council (ERC-2008-AdG 232648, to R.B.). J.C. is a Wellcome Trust Senior Research Fellow.
Author information
Authors and Affiliations
Contributions
O.B.N. and A.A.N. designed and carried out the experiments; J.J.H. and A.S. characterized the purified proteins; S.W. and R.B. were responsible for the cryo-EM experiments; A.S. wrote the manuscript; G.v.H. and J.C. conceived and planned the investigation and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 In vitro translation of the spectrin constructs.
(a) Schematic representation of the constructs used in the AP assays showing the Lep leader (blue), GSGS and SGSG linker sites (purple), the variable linker (green), the SecM stall site (grey) and the terminal Lep segment (orange). Constructs for β16, R16m5, R16m6, R16o15c and non-folding R15 and R16 all resemble R15. L, reported linker length (minimum L=21 amino acids, maximum L=61 amino acids). R15R16m5 and R153ProR16 resemble R15R16. Note that the R16 force profiles obtained with the LepB leader sequence are virtually identical (see Main text Fig 3 a,b). (b) R16 [L=27] and R16 [L=37] translated in the PURE system. Full-length (FL) and arrested (A) products are indicated. FLc, full-length control, where the crucial Pro at the C-terminal end of the SecM AP is mutated to Ala; Ac, arrest control, where the crucial Pro at the end of the AP is substituted with a stop codon.
Supplementary Figure 2 Reproducibility of the data.
Data for three separate repeats of wild-type R16 without the Lep leader are shown. Points in red are the full R16 trace (see Main text Fig 3b), points in green are repeats collected using an in vitro translation kit with a different lot number and points in blue are repeats collected in a different laboratory using an in vitro translation kit with a different lot number.
Supplementary Figure 3 Supplementary cryo-EM images.
(a) Resolution determination of the final reconstruction using Fourier shell correlation (FSC) indicating an average resolution of 4.8 Å. (b) Calculation of the local resolution using resmap (Kucukelbir, A. et al. Nat Methods 11, 63-65, 2014). (c) Superposition of the spectrin R16 domain (red) and the previously determined cryo-EM structure of the ADR1a zinc-finger domain (gold) in the ribosome exit tunnel12. Notably, the location in the exit tunnel of the C-terminus of the R16 domain is 25-30 Å away from the location of the C-terminus of ADR1a, consistent with the difference in linker lengths (L = 33 vs. 25 residues) used in the two constructs.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–3 and Supplementary Table 1 (PDF 460 kb)
Rigid body fit of the NMR structure of the R16 domain to the cryo-EM density map.
Rigid body fit of the NMR structure of the R16 domain colored according to r.m.s. deviation (blue, 0.5–1.9; white, 2–3.9; red, ≥4.0 Å) to the cryo-EM density map (Fig. 4c). (MP4 5049 kb)
Rights and permissions
About this article

Cite this article
Nilsson, O., Nickson, A., Hollins, J. et al. Cotranslational folding of spectrin domains via partially structured states. Nat Struct Mol Biol 24, 221–225 (2017). https://doi.org/10.1038/nsmb.3355
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nsmb.3355
