
Key Points
-
At least two forms of cell-autonomous immunity operate in higher organisms such as vertebrates: constitutive and inducible. The interferon (IFN) family of cytokines stimulates the inducible gene programme for mobilizing effector functions inside individual host cells.
-
IFN-induced effector proteins operate against most pathogen classes, especially bacteria, protozoan parasites and viruses.
-
Individual bacteria, protozoa and viruses occupy only a tiny fraction of the interior volume of a vertebrate cell. Hence, many IFN-inducible proteins are directly targeted to the site of microbial replication or generate toxic products capable of diffusing large intracellular distances to reach these microorganisms.
-
IFN-induced proteins inhibit intracellular bacteria and protozoa through a variety of mechanisms. These include: oxidative and nitrosative damage caused by cytotoxic gases (generated via IFN-inducible oxidoreductases); the recruitment of the autophagic machinery to deliver microorganisms to lysosomes (by IFN-inducible GTPases and cytosolic receptors); and the depletion of essential amino acids and divalent cations needed for microbial growth (by IFN-induced catabolic enzymes and efflux pumps, respectively).
-
IFN-induced antiviral mechanisms operate across most nucleated cells and at all stages of the viral life cycle, including entry, replication, capsid assembly and release. Several new proteins have recently been discovered that fulfil these different functions.
-
Much scientific effort over the last two decades has focused on how the innate immune system recognizes microbial pathogens. Attention is now beginning to turn towards understanding the effector mechanisms needed to sterilize these infections.
Abstract
Interferons (IFNs) induce the expression of hundreds of genes as part of an elaborate antimicrobial programme designed to combat infection in all nucleated cells — a process termed cell-autonomous immunity. As described in this Review, recent genomic and subgenomic analyses have begun to assign functional properties to novel IFN-inducible effector proteins that restrict bacteria, protozoa and viruses in different subcellular compartments and at different stages of the pathogen life cycle. Several newly described host defence factors also participate in canonical oxidative and autophagic pathways by spatially coordinating their activities to enhance microbial killing. Together, these IFN-induced effector networks help to confer vertebrate host resistance to a vast and complex microbial world.
Similar content being viewed by others

Main
Host effector mechanisms are essential for the survival of all multicellular organisms. This is exemplified by cell-autonomous immunity in plants, worms, flies and mammals. In Arabidopsis spp., for example, a definable set of resistance genes is mobilized during this programmed cell-intrinsic response to protect against diverse phytopathogens; this inherited response is sometimes referred to as the 'resistome'1,2. In higher species, however, the assembly of an antimicrobial arsenal or resistome takes on multiple forms, because the burden posed by infection in these organisms is considerable3. Indeed, as many as 1,400 phylogenetically distinct microorganisms can infect a single chordate host4.
To cope with this increased microbial challenge, vertebrates have evolved additional levels of cell-autonomous control beyond the pre-existing repertoire of constitutive host defence factors. These additional factors include hundreds of gene products that are transcribed in response to signals originating from the interferon (IFN), tumour necrosis factor (TNF), interleukin-1 (IL-1) and Toll-like receptor (TLR) families5,6. Many of the induced proteins confer direct microbicidal immunity in all nucleated cells7,8,9.
IFNs are among the most potent vertebrate-derived signals for mobilizing antimicrobial effector functions against intracellular pathogens8,10,11. Nearly 2,000 human and mouse IFN-stimulated genes (ISGs) have been identified to date, most of which remain uncharacterized (see the Interferome database)12 (Fig. 1). The recent large-scale examination of newly described ISGs reveals a highly diverse but integrated host defence programme dedicated to protecting the interior of a vertebrate cell13,14,15,16.

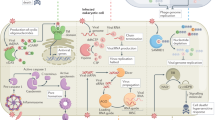
a | The evolution of cell-autonomous immunity and the emergence of interferon (IFN)-induced effector mechanisms around the protochordate–vertebrate split (∼530 million years ago). b | Cell-autonomous host defence proteins are canonically induced by IFNs via three receptor complexes with high affinities for their ligands (Ka < 10 nM−1)8. The first receptor complex is a tetramer — composed of two chains of IFNγ receptor 1 (IFNGR1) and two chains of IFNGR2 — that engages type II IFN (that is, IFNγ) dimers. The second is a heterodimer of IFNα/β receptor 1 (IFNAR1) and IFNAR2 that binds to the type I IFNs: a family consisting of 13 different IFNα subtypes and one IFNβ subtype in humans. In the third receptor complex, interleukin-10 receptor 2 (IL-10R2) associates with IFNλ receptor 1 (IFNLR1; also known as IL-28Rα) to bind to three different type III IFN (that is, IFNλ) ligands (see Ref. 8). Following receptor–ligand engagement, signals are transduced through signal transducer and activator of transcription 1 (STAT1) homodimers in response to IFNγ or through STAT1–STAT2 heterodimers in response to type I IFNs or IFNλ. Following their recruitment to the receptor complexes, these STAT molecules are phosphorylated by receptor-bound tyrosine kinases (namely, Janus kinases (JAKs) and tyrosine kinase 2 (TYK2)). Phosphorylated STAT1 homodimers (also known as GAF) translocate to the nucleus to bind to IFNγ-activated site (GAS) promoter elements to promote the IFN-induced expression of antimicrobial effector genes, some of which also require transactivation by IFN-regulatory factor 1 (IRF1) and IRF8. In the case of type I and III IFN signalling, phosphorylated STAT1–STAT2 dimers form a complex with IRF9 to yield IFN-stimulated gene factor 3 (ISGF3); this complex also translocates to the nucleus, where it binds to IFN-stimulated response elements (ISREs) in the promoters of different or overlapping IFN-stimulated effector genes.
When viewed on a microscopic scale, the cell interior represents an immense 'subterranean landscape' to patrol and defend. A single human macrophage, for example, occupies ∼5,000 μm3 (Ref. 17). Contrast this with a mature HIV-1 particle (∼200 nm3) or tubercle bacillus (∼5–10 μm3) and it quickly becomes apparent that most IFN-induced proteins will need to be dispatched to the site of pathogen replication to be effective18,19. Likewise, the ability of compartmentalized pathogens to remain largely sequestered in vesicles suggests that many IFN-induced effectors also need methods to detect these membrane-bound sanctuaries to eliminate the resident pathogens18,19,20.
Several ISGs fulfil both criteria. Members of an emerging superfamily of GTPases with immune functions recognize specific host lipid molecules on the pathogen vacuole to mark it for disruption or delivery to lysosomes21,22,23. Other recently identified IFN-induced proteins detect ubiquitylated bacteria in the cytosol24 or exposed glycans on host membranes that have been damaged by bacteria25, and these markers stimulate the removal of the infecting organism through autophagy. In addition, new antiviral factors distinguish the cellular entry, replication and exit points of HIV-1 and influenza A viruses13,15. Less discriminating effector mechanisms are also deployed; for example, diatomic radical gases such as superoxide (O2−) and nitric oxide (NO) circumvent the need for recognition of the membranes surrounding sequestered bacteria and protozoa inside host cells7,26. Because such gases can diffuse large distances (several micrometres), they can also enter adjacent cells to confer trans-acting immunity, a property first noted for NO against herpes simplex virus, ectromelia virus and vaccinia virus27. Both of these strategies rely on an expanded family of oxidoreductases and peroxidases that is now known to be present in essentially all phyla28.
It is the purpose of this Review to provide a broad conceptual framework for understanding IFN-induced cell-autonomous host defence and to highlight the growing list of effectors that combat internalized bacteria, protozoa and viruses at the level of the infected mammalian cell. It focuses principally on the downstream killing mechanisms, rather than on the well-known upstream microbial recognition and signalling events that elicit IFN production.
Cell-autonomous defence against bacteria
Bacteria infect host cells either through active invasion or via engulfment by professional phagocytes. Following their uptake, some bacterial species — such as Mycobacterium tuberculosis and Salmonella enterica serovars — inhabit membrane-bound compartments termed phagosomes, which they modify to limit their exposure to microbicidal factors20. By contrast, Chlamydia spp. reside in reticulate structures called inclusion bodies, which intercept Golgi-derived exocytic traffic as a source of nutrition29. Other bacterial species, including Listeria and Shigella spp., escape their vacuoles to replicate in the cytosol. In each subcellular locale, IFN-induced effector mechanisms are mobilized to defend the interior of the host cell against bacterial infection. These mechanisms rely on oxidative, nitrosative and protonative chemistries, as well as nutriprive (nutrient-restrictive) and membranolytic activities.
IFN-induced oxidative and nitrosative defence. Cytotoxic gases are one of the most ancient and important forms of cell-autonomous defence. These gases — collectively termed reactive oxygen species (ROS) and reactive nitrogen species (RNS) — are generated by oxidoreductases to confer microbicidal activity and regulate intracellular signalling7,26,28,30. The targets of ROS and RNS include bacterial DNA (which is damaged via guanine base oxidation), lipids (which are damaged via peroxidation), and haem groups or iron–sulphur clusters within bacterial enzymes7,26. Much of the redox damage caused by these gases can be traced to derivatives of O2− and NO. For example, the sequential addition of single electrons to O2− yields hydrogen peroxide (H2O2) and then the hydroxyl radical (·OH), both of which are more powerful oxidants than their predecessor26. Likewise, the reaction of NO with O2−, other ROS or thiols yields intermediates with potent bactericidal properties: dinitrogen oxides (N2O3 and N2O4), compound peroxides (ONOO−) and nitrosothiol adducts (RSNO)7,26. Within phagolysosomes, O2− undergoes spontaneous dismutation to H2O2, and stable nitrogenous end products such as nitrite (NO2−) are converted back at low pH to the volatile NO gas; both mechanisms aid bacterial killing7,9.
Given the toxicity of these molecules, it is not surprising that the production of ROS and RNS is tightly controlled and often compartmentalized to limit self-injury. This has the added benefit of maximizing microbicidal activity when production is localized to phagosomes and phagolysosomes that contain bacteria9. In mammals, three classes of cytokine-inducible oxidoreductases control ROS and RNS production. NADPH oxidases (NOXs) directly catalyse the production of O2−, whereas dual oxidases (DUOXs) produce H2O2 (Table 1; Supplementary information S1 (figure)). In addition, nitric oxide synthases (NOSs) synthesize NO, and the immunologically inducible isoform NOS2 (also known as iNOS) synthesizes large amounts of NO under infectious conditions. All three classes of oxidoreductases may act simultaneously, sometimes even within the same host cell, depending on the physiological setting and the activating stimuli7,28. Non-enzymatic sources of ROS and RNS can also contribute to host defence. For example, O2− can originate from mitochondrial leakage and NO can be generated by the action of gastric acid on NO2− that is produced from dietary nitrates (NO3−) by the oral microbiota7,28,31,32.
The NOX family of enzymes (NOX1 to NOX5) are the major ROS producers during infection28. NOX2 (also known as phagocyte oxidase) is responsible for the respiratory burst in neutrophils, monocytes, macrophages and eosinophils. Genetic evidence underscores its importance for host defence; indeed, congenital mutations in genes encoding NOX2 subunits give rise to a collective syndrome termed chronic granulomatous disease. Affected individuals suffer from recurrent infections with catalase-positive organisms such as Staphylococcus aureus, Serratia marcescens, Burkholderia cepacia, non-typhoidal Salmonella spp. and M. tuberculosis28,33 (Table 2).
NOX2 is a multisubunit enzyme comprising a transmembrane heterodimer — composed of gp91phox (also known as CYBB) and p22phox (also known as CYBA) — and three cytosolic subunits, namely p67phox (also known as NCF2), p47phox (also known as NCF1) and p40phox (also known as NCF4). The cytosolic subunits have SH3 domains that mediate intersubunit contacts and PX domains for binding membrane lipids once they translocate to the gp91phox–p22phox complexes at the plasma membrane or on plasma membrane-derived phagosomes28 (Table 1; Supplementary information S1 (figure)). The assembly and activation of NOX2 holoenzymes also requires several GTPases. RAC1 and RAC2 facilitate this process under basal conditions28, whereas the recently described GTPase guanylate-binding protein 7 (GBP7) operates after IFNγ stimulation16. IFNγ-induced GBP7 specifically recruits cytosolic p67phox–p47phox heterodimers to gp91phox–p22phox complexes on bacterial phagosomes containing Listeria monocytogenes or Mycobacterium bovis bacillus Calmette–Guérin (BCG)16 (Fig. 2). The proximity of phagosomal NOX2 to intraluminal bacteria may heighten IFN-induced killing, as subsequent fusion with lysosomes favours dismutation of O2− to the more-damaging oxidant H2O2 (Ref. 9). In addition to GBP7, the IFNγ-activated GTPase leucine-rich repeat kinase 2 (LRRK2) has recently been reported to promote NOX2 activity against S. Typhimurium34. How LRRK2 exerts its effects and whether it works in tandem with GBP7 on phagosomal membranes is currently unknown.

Interferon (IFN)-inducible proteins are required for host resistance to intracellular bacteria. a | Specific immunity-related GTPases (IRGs), guanylate-binding proteins (GBPs) and other GTPases translocate to compartmentalized bacteria in phagosomes or inclusion bodies. Here, different membrane regulatory complexes — IRGM1–snapin, GBP1–sequestosome 1 (SQSTM1) and GBP7–ATG4B — are assembled. These complexes initiate autophagic capture and SNARE-mediated fusion of the bacterial compartments with lysosomes16,21,22,23. In addition, IRGM3–IRGA6 (or IRGB10) mediate vacuole disruption19,52,53, and GBP7 (and possibly leucine-rich repeat kinase 2 (LRRK2)) help to assemble NADPH oxidase 2 (NOX2) on bacterial phagosomes, which mediates bacterial killing. Using this pathway, these GTPases can also deliver antimicrobial peptides (AMPs) to the autophagolysosome and, in the case of human IRGM, may instigate mitochondrial fission before autophagy59. Other IFN-inducible components, such as natural resistance-associated macrophage protein 1 (NRAMP1), help to exclude Mn2+ and Fe2+ from the bacterial phagosome, while importing protons (H+) into this compartment. Nitric oxide synthase 2 (NOS2), which synthesizes NO, works in concert with NOX2, which produces reactive oxygen species such as superoxide (O2−) and hydrogen peroxide (H2O2), to produce compound intermediates like peroxynitrite (not shown) that are highly bactericidal. b | An emerging signature for the recognition of some escaped bacteria in the cytosol is ubiquitylation (either single or multiple modifications with monoubiquitin and/or polyubiquitin chains) (see Ref. 47). SQSTM1, NDP52 and optineurin bind to ubiquitylated bacteria to initiate innate immune signalling and to recruit the autophagic machinery via LC3 family members. In addition, GBP1 and GBP2 polymerize around cytosolic bacteria in a ubiquitin-independent process that may recruit specific antimicrobial partners, while galectin 3 and galectin 8 bind to exposed glycans on the bacteria and, in the case of galectin 8, recruit NDP52 and downstream autophagic effectors25. SQSTM1 also activates a second antibacterial pathway involving diacylglycerol (DAG) and protein kinase Cδ (PKCδ) to induce NOX2 complex assembly. Dashed lines indicate possible routes and consequences. PtdIns(4,5)P2, phosphatidylinositol-4,5-bisphosphate; PtdIns(3,4,5)P3, phosphatidylinositol-3,4,5-trisphosphate; TBK1, TANK-binding kinase 1.
Other IFNγ-induced enzymes provide oxidative defence in non-phagocytic cells, such as epithelial cells lining the airways, oral cavity and gastrointestinal tract. The IFNγ-inducible enzymes NOX1 and DUOX2 generate O2− and H2O2, respectively, in these cells28 (Supplementary information S1 (figure)). At the plasma membrane, H2O2 can form hypothiocyanite (OSCN−), which acts as a potent chemorepellent against bacterial invasion and kills Listeria and Salmonella spp.35,36,37. Indeed, recent reports show that impaired clearance of Salmonella spp. follows the silencing of DUOX expression in zebrafish intestinal epithelium, indicating that such mechanisms operate during vertebrate immunity in vivo38. Thus, IFN-inducible NOXs and DUOXs restrict bacterial colonization not only of immune cells but also of stromal cells.
NOS2 is expressed in a variety of immune and non-immune cell types following stimulation by type I IFNs (that is, IFNα and IFNβ) and by IFNγ. Signals from other cytokines (notably, TNF, lymphotoxin-α and IL-1β) and from microbial products (such as lipopolysaccharides and lipopeptides) also synergize with IFNs for NOS2 induction7,39. To date, most work has focused on NOS2 activities in mouse macrophages, as human mononuclear phagocytes produce lower NO levels7. Experiments using NOS2 inhibitors that are relatively selective for this NOS isoform have implicated a role for NO and its derivatives in the early cell-autonomous immune response to intracellular bacteria7. This role was further delineated in mice and macrophages deficient for NOS2 and/or gp91phox39,40,41. M. tuberculosis is sensitive to NO-mediated killing but relatively resistant to O2− and H2O2, in part owing to its expression of the H2O2-detoxifying enzyme KatG42. NO exhibits molar potencies comparable to the current antibiotics used to treat tuberculosis, and the tuberculocidal activity of some new drugs (such as bicyclic nitroimidazoles) has been attributed to their release of NO43. By contrast, L. monocytogenes is sensitive to O2− and H2O2 but less vulnerable to NO, and S. enterica serovars are inhibited by both classes of chemicals39,41. Such differences reflect the metabolic pathways and microbial DNA repair processes targeted by ROS and RNS, as well as the detoxifying systems expressed by the bacteria7 (see Table 2). They may also reflect compartmentalization; for example, L. monocytogenes becomes sensitive to NO when trapped inside phagosomes, owing to synergism with other bactericidal insults or the heightened RNS concentrations that accumulate in a confined volume44. Therefore, phagosomal escape of L. monocytogenes before NOS2 recruitment could provide a survival benefit for the pathogen. For this reason, vertebrates have evolved other IFN-induced mechanisms to deal with bacterial escapees, as discussed below.
Lysosomal killing: phagosome maturation and autophagy. Acidified lysosomes are inimical for the growth of most bacteria. Here, a low pH (∼4.5–5.0) — which is generated via the action of proton-pumping vacuolar ATPases and maintained with the assistance of antiporters such as sodium/hydrogen exchanger 1 (NHE1) — enhances the bactericidal activity of both ROS and RNS7,9. In addition, an abundance of luminal proteases, lipases, glycosidases and antimicrobial peptides contributes to the sterilizing power of lysosomes45,46. This has resulted in some bacterial pathogens (such as M. tuberculosis) evolving strategies to avoid these degradative organelles, whereas other bacteria (such as L. monocytogenes) try to escape into the cytosol. Stimulation of the infected cell with IFNγ prevents both of these evasion strategies18,22,44.
At least two newly described families of IFN-inducible GTPases — the 21–47 kDa immunity-related GTPases (IRGs) and the 65–73 kDa GBPs — traffic to vacuolar and cytosolic bacteria, where they assemble membrane complexes to facilitate bacterial transfer to lysosomes or disruption of the pathogen compartment16,18,19,21,22,23. IFN-inducible GTPases function together with three ubiquitin-binding receptors — sequestosome 1 (SQSTM1; also known as p62), NDP52 and optineurin — that detect ubiquitylated structures on bacteria, as well as with galectins that detect glycans that are exposed during bacterial escape into the cytosol. These receptors recruit the autophagic machinery that engulfs bacteria for lysosomal delivery24,25,47,48,49,50. The resultant (auto)lysosomes kill and degrade the entrapped cargo.
IRGs were first shown to target phagosomes and direct lysosomal membrane traffic in IFNγ-activated macrophages infected with M. tuberculosis22. It is now known that IRGs also exert membrane regulatory functions on other bacterial compartments, and their action has also been observed in human and mouse fibroblasts and epithelial cells51,52,53,54. IRGs promote cell-autonomous immunity to vacuolar bacteria as diverse as M. tuberculosis, M. bovis, S. Typhimurium, Chlamydia trachomatis, Chlamydia psittaci, Legionella pneumophila and Crohn's disease-associated adherent invasive Escherichia coli (AIEC)22,23,51,52,53,54,55,56,57,58,59,60,61. Individual IRGs confer pathogen-specific immunity in vitro and in vivo, indicating that they have non-redundant functions during host defence (Table 2). Such specificity probably arises from the host-derived interacting partners and trafficking pathways used by a given IRG and the type of intracellular niche occupied by a given bacterial species18,19,51.
Recent studies have contributed to a conceptual framework for how IRGs orchestrate immunity to different compartmentalized pathogens21,23,53,54,59,60,62,63. This model posits cooperative interactions between IRG subclasses, as well as with SNARE proteins and autophagic effectors that may disrupt the pathogen-containing compartment before lysosomal delivery (Fig. 2).
IRGs are divided into two groups — GKS-containing IRGs and GMS-containing IRGs — based on their canonical (lysine-containing) and non-canonical (methionine-containing) G1 motifs within the conserved amino-terminal catalytic GTPase domain18,19,51 (Table 1). IRGs in the GMS-containing subclass (IRGM1, IRGM2 and IRGM3 in mice; IRGM in humans) appear to be intrinsic regulators that control the activities of their respective effectors, which can also include other IRGs21,23,53,54,59,60,62,63. For example, IRGM3 in the endoplasmic reticulum (ER) helps to maintain membranolytic GKS-containing IRGs (such as IRGA6 and possibly IRGB10) in the 'off' state by acting as a non-canonical guanine nucleotide dissociation inhibitor (GDI)62,63. When released from IRGM3, IRGA6 and IRGB10 directly target Chlamydia-containing inclusion bodies or disrupt the trafficking of sphingomyelin-containing exocytic vesicles to these organelles55,56. Such disruption probably results in autophagic engulfment of the pathogen53 and explains the susceptibility of Irgm3−/−, Irga6−/− and Irgb10−/− fibroblasts to infection with C. trachomatis or C. psittaci52,56,58.
IRGM1 and its smaller constitutive human orthologue, IRGM, engage their effectors when targeting M. tuberculosis, M. bovis, S. Typhimurium, AIEC or early L. monocytogenes phagosomes as part of the IFNγ-induced response to these bacteria21,22,23,51,54,57,59,60. The translocation of IRGM1 to mycobacterial phagosomes involves the recognition of specific host phosphoinositide lipids (namely, phosphatidylinositol-3,4,5-trisphosphate and, to a lesser extent, phosphatidylinositol-3,4-bisphosphate) on the nascent phagocytic cup21 (Fig. 2). Once recruited, IRGM1 interacts with and may regulate the assembly activity or phosphorylation status of snapin, a SNARE adaptor protein that recruits dynein motor complexes to traffic phagosomes and endosomesa long microtubules towards maturing autolysosomes21,64. Likewise, different human IRGM splice isoforms bind to the core autophagy proteins ATG5 and LC3B as well as the inner mitochondrial membrane lipid cardiolipin to induce mitochondrial fission and autophagy59,65; these functions of IRGM may underlie its protective response to mycobacterial, Salmonella spp. and AIEC infections23,54,59,60,65,66. Thus, a single GMS-containing IRG can act as a hub for coordinating membranolytic, fusogenic and fission events in an individual cell. This accounts in part for why deficiencies in GMS-containing IRGs cause such pronounced infectious phenotypes compared with those of GKS-containing IRGs18,20,52,56,57,58 (Table 2). It may also explain why human IRGM polymorphisms share genetic linkages with susceptibility to tuberculosis and Crohn's disease across so many geographically diverse populations66,67,68,69.
In contrast to the IFN-inducible IRGs, GBPs target escaped bacteria in addition to those residing within vacuoles16,70,71. Nucleotide-dependent self-assembly of some but not all GBPs — in which G domain dimers pair with the carboxy-terminal helical domain (CTHD) to form GBP tetramers — helps to partition GBPs between the cytosol and endomembranes of the cell16,72,73 (P. Kumar and J.D.M., unpublished observations) Table 1). A C-terminal CaaX motif used for isoprenylation also contributes to this membrane attachment16,72,73,74. These structural features — along with an ability to oligomerize with other GBPs or even interact with GMS-containing IRGs — dictate which endomembranes individual GBPs occupy16,72,73,74,75,76 and aid GBP targeting to both vacuolar and cytosolic bacteria (as the latter may retain remnants of damaged host membrane on the surface of their capsular coat following escape from the vacuole)16 (C. J. Bradfield, P. Kumar and J.D.M., unpublished observations). This translocation of GBPs to bacteria promotes intracellular defence against L. monocytogenes, S. Typhimurium, Chlamydia spp. and Mycobacterium spp. in both macrophages and epithelial cells16,70,71. The lack of such cell-autonomous defence probably contributes to the susceptibility of Gbp1−/− and Gbp5−/− mice to bacteria16 (Table 2).
The recent identification of interacting partners for GBP1 and GBP7 has begun to reveal the molecular mechanisms used by some of these GTPases to promote bacterial killing16. GBP1 interacts with the ubiquitin-binding protein SQSTM1, which delivers ubiquitylated protein cargo to autolysosomes, resulting in the generation of antimicrobial peptides that kill engulfed M. bovis and L. monocytogenes16,77,78. GBP7 recruits the autophagy protein ATG4B, which drives the extension of autophagic membranes around bacteria within damaged bacterial compartments and assembles NOX2 on these compartments16 (Fig. 2). GBP5, by contrast, binds NLRP3 (NOD-, LRR- and pyrin domain-containing 3) to promote specific inflammasome responses during the infection of IFNγ-activated macrophages by Listeria or Salmonella spp., whereas in non-phagocytic cells heterotypic interactions between GBPs may help to target cytosolic escaped bacteria to autolysosomes (A. R. Shenoy, C. J. Bradfield and J.D.M., unpublished observations). Thus, GBPs act in concert — both temporally and physically — to confer their antibacterial effects. Moreover, they integrate oxidative, lysosomal and possibly inflammasome-related killing as part of their host defence activities.
In addition to being targeted by GBPs, cytosolic bacteria have recently been shown to encounter a second line of cell-autonomous defence orchestrated by SQSTM1, NDP52, optineurin and galectins in macrophages and epithelial cells24,25,47,48,49,50. The IFN-inducible proteins SQSTM1 and NDP52, along with basally expressed optineurin, recognize bacteria coated with ubiquitin, whereas IFN-regulated galectins detect the β-galactoside moiety of polysaccharide sugars (host glycans and microbial carbohydrates) that become exposed on damaged membranes when bacteria escape their phagosome to reach the cytosol25,79,80,81. SQSTM1, NDP52 and optineurin all possess a C-terminal domain for binding ubiquitin and an internal or N-terminal region that interacts with LC3 autophagy proteins for delivering bacterial cargo to autophagic vacuoles (Fig. 2; Table 1). Galactin 3 and galactin 8 contain carbohydrate-recognition domains, and galectin 8 binds to NDP52, which links the recognition of sugar moieties on bacteria with the autophagic machinery further downstream25 (Fig. 2; Table 1).
NDP52 also recruits the IκB kinase (IKK) family kinase TBK1 (TANK-binding kinase 1) to ubiquitin-coated bacteria via the adaptor proteins SINTBAD (also known as TBKBP1) and/or NAP1 (also known as AZI2)24. TBK1 in turn phosphorylates optineurin to increase its affinity for ubiquitin; in this way, NDP52 and optineurin may cooperate to protect against infection24,47. Furthermore, NDP52 and SQSTM1 use septin- and actin-dependent autophagic pathways to target cytosolic Shigella spp. and the small percentage of S. Typhimurium that escape their vacuole48,49. By contrast, autophagic delivery of non-motile L. monocytogenes mutants occurs via a different, as yet unspecified, route49,50. Because SQSTM1 activates a second antibacterial pathway involving diacylglycerol to induce the assembly of NOX2 complexes82, parallels may be drawn with the GBPs, which induce both oxidative and autophagic pathways to confer cell-autonomous host defence.
Competing for intracellular cations. Facultative and obligate intracellular bacteria often have stringent metal cation requirements for growth inside mammalian host cells, which serve as a rich natural source of these chemical elements. As a result, IFN-induced mechanisms have evolved to restrict the intraphagosomal and cytosolic availability of Mn2+, Fe2+ and Zn2+, and to enhance the transport of Cu+ into the phagosome, as Cu+ helps to drive the formation of microbicidal ROS83,84,85. Indeed, the activation of macrophages by IFNγ lowers Mn2+, Fe2+ and Zn2+ concentrations by ∼2–6-fold and increases Cu+ levels by ∼5-fold within mycobacterial phagosomes86.
Part of the reduction in metal cation concentrations depends on a proton-dependent Mn2+ and Fe2+ efflux pump called natural resistance-associated macrophage protein 1 (NRAMP1; encoded by Slc11a1), which is upregulated by IFNγ83,87. NRAMP1 prevents ion sequestration specifically by phagosomal pathogens and competes with bacterial ion transporters for access to these nutritional metals83 (Fig. 2). For example, the growth of S. Typhimurium mutants that lack mntH (which encodes an NRAMP1 homologue with a high affinity for Mn2+ and Fe2+) or sitABCD (which encodes a second Mn2+-binding transport system) is attenuated in IFNγ-activated macrophages from mice that express the wild-type NRAMP1 efflux pump, but not in macrophages from congenic mice with a non-functional NRAMP1 efflux pump (derived from a defective Nramp1G169D allele)88. Similarly, infection of macrophages by an M. tuberculosis strain lacking Mramp (another bacterial NRAMP1 homologue) leads to increased Mn2+ and Fe2+ concentrations within the phagosome, and this may reduce bacterial viability89.
IFNγ stimulation also regulates other cation transport mechanisms, for example by inducing the relocation of the P-type ATPase Cu+ pump ATP7A to the phagosome, where it can transport Cu+ across the membrane to promote the generation of intraluminal hydroxyl radicals85. This again leads to intraphagosomal killing of bacteria. IFNγ stimulation concomitantly increases the expression of the Fe2+ exporter ferroportin 1 (also known as SLC40A1) at the plasma membrane, while decreasing transferrin receptor expression to limit Fe2+ uptake; both mechanisms further restrict the growth of S. Typhimurium in macrophages84.
In sum, synergistic IFN-inducible effector mechanisms are deployed in the cytosol and in diverse intracellular compartments to control bacterial infection. For example, IRGs, GBPs and recognition receptors help to direct vacuolar bacteria as well as 'marked' cytosolic bacteria to acidified autophagolysosomes. Low lysosomal pH, in turn, accelerates the dismutation of O2− to the more powerful oxidant H2O2, converts NO2− back to the toxic radical NO and drives hydroxyl radical formation with the aid of imported Cu+. Together, these IFN-regulated proteins help to maximize oxidative, nitrosative, protonative and membranolytic damage to bacterial targets in the lysosome.
Cell-autonomous defence against protozoa
In vertebrates, many protozoa are obligate intracellular pathogens that depend on the host cell for specific amino acids and metal ions. The nutritional and safety needs of different parasites often dictate the type of compartment they inhabit (reviewed in Ref. 90). For example, the apicomplexan parasite Toxoplasma gondii (which causes human toxoplasmosis) occupies a non-fusogenic vacuole that excludes most host-derived proteins, whereas the kinetoplastid parasites Trypanosoma cruzi (which is responsible for Chagas disease) and Leishmania spp. (which trigger cutaneous, mucocutaneous and visceral leishmaniasis) reside in the cytosol and in modified lysosomes, respectively90. These strategies operate effectively in resting cells by allowing the parasites access to nutrients while helping them to avoid contact with many host microbicidal proteins. However, once cells become stimulated with IFNs, new host defence pathways are transcriptionally induced to help limit parasite infection.
Parasiticidal activities. Previous studies have highlighted the role of NOS2-mediated killing in cell-autonomous defence against a variety of protozoa (reviewed in Ref. 7). The parasiticidal effects of NO are most evident in IFNγ-activated macrophages infected with Leishmania major amastigotes or T. cruzi trypomastigotes and in human and mouse hepatocytes infected with Plasmodium falciparum and Plasmodium yoelli sporozoites, respectively91,92,93 (Fig. 3). Furthermore, Nos2−/− mice were highly susceptible to these pathogens91,92,93 (Table 2). In the case of less virulent type II T. gondii tachyzoites, IFN-inducible NOS2 plays a more limited part, functioning at later time points94 after the IFN-inducible GTPases have contained parasite growth during the early stages of infection95. For virulent type I T. gondii strains, however, NOS2 is essential, because these parasites have evolved mechanisms to escape IRG-mediated inhibition in IFNγ-activated macrophages96. Here, NO does not appear to eliminate virulent T. gondii but instead imposes static, non-lethal control96. How NO inhibits Toxoplasma parasites, along with malaria, Leishmania and Trypanosoma parasites, remains incompletely understood, but haem-containing compounds (such as haemozoin) and protozoal cysteine proteases appear to be likely targets for S-nitrosylation, which can inactivate these enzymes7.

Different intracellular strategies are used by interferon (IFN)-inducible proteins against protozoa. Nitric oxide synthase 2 (NOS2) exerts potent parasiticidal activity, while GKS-containing immunity-related GTPases (IRGs) appear to be directly involved in parasite vacuole disruption once they reach the parisitophorous compartment. This proceeds via autophagy-independent trafficking after release from IRGM1–IRGM3 or ATG5 and is mediated by cooperative IRG loading62,63,103. Guanylate-binding proteins (GBPs) — specifically GBP1–GBP2 and GBP1–GBP5 complexes — also traffic to the parasitophorous vacuole, with uncharacterized effects on parasite control76. Natural resistance-associated macrophage protein 1 (NRAMP1) is important for restricting the uptake of Mn2+ and Fe2+ by this compartment, whereas indoleamine 2,3-dioxygenase 1 (IDO1) and/or IDO2 limit amino acid acquisition via the depletion of L-tryptophan. Dashed lines indicate possible routes or consequences.
Targeting the parasitophorous vacuole. As in the case of bacteria, IFN-inducible IRGs and GBPs defend the interior of the host cell against protozoa. IRGM1, IRGM3 and IRGA6 promote IFNγ-induced control (but not TNF- or CD40-dependent control) of avirulent T. gondii in macrophages and astrocytes96,97,98,99,100,101. IRGM1 also contributes to macrophage trypanocidal activity102 (Table 2). Inhibition of avirulent T. gondii appears to rely on several IRGs, with IRGM proteins providing a regulatory function by acting as GDIs that release GKS-containing IRGs to target the parasitophorous vacuole (Fig. 3). Recent studies invoke a hierarchical model in which IRGB6 and possibly IRGB10 act as forerunners to IRGA6 and then IRGD during their loading onto the parasitophorous vacuole some 90 minutes after parasite entry. The recruitment of these molecules is followed by vesiculation, membrane disruption and sometimes necroptosis62,63. What remains unknown are the structural and biochemical cues for targeting these molecules to the parasitophorous vacuole and whether membrane deformation is directly due to IRG activity or a result of some intermediary protein. These are topics of future investigation.
Other proteins assist the relocation of IRGs to the parasitophorous vacuole. For example, ATG5 facilitates the release and transit of IRGA6 from its bound state103. Heterotypic interactions between different GBPs have also recently been shown to underlie the vacuolar targeting of GBPs76 (Fig. 3). Hence, multiple parasitophorous vacuole-damaging mechanisms are likely to ensue as the IRGs and GBPs converge on this organelle. Because virulent T. gondii strains (but not avirulent strains) exclude IRGs and GBPs from the parasitophorous vacuole76,104,105, it is likely that these IFN-inducible GTPases exert a strong selective pressure via their membrane regulatory activities. Such pressure appears to be specific for different protozoa, as GBP1 is not recruited to T. cruzi compartments76.
Restricting nutrient acquisition. Nutriprive mechanisms are particularly effective against parasites. NRAMP1 prevents ion assimilation by Leishmania spp. (L. major and L. donovani)83 and indoleamine 2,3-dioxygenases (IDOs) hamper amino acid acquisition106. IDO1 and IDO2 are both IFN-inducible, haem-containing oxidoreductases that are responsible for the initial rate-limiting step of the kynurenine pathway, in which they degrade L-tryptophan to generate N-formylkynurenine (Fig. 3; Table 1). Removal of L-tryptophan restricts the growth of Leishmania spp. and T. gondii (as well as that of C. psittaci, Francisella spp., Rickettsia spp., herpes simplex virus 1 and hepatitis B virus) in IFNγ-activated macrophages, dendritic cells, fibroblasts, epithelial cells, astrocytes, endothelial cells and mesenchymal stem cells107,108,109,110,111. IDOs also inhibit T. cruzi via the downstream L-kynurenine catabolites 3-hydroxykynurenine and 3-hydroxyanthranilic acid, which are likely to be toxic for T. cruzi amastigotes and trypomastigotes112. Furthermore, in vivo blockade of IDOs using 1-methyltryptophan results in profound host susceptibility to T. gondii113. This host-protective role of IDOs against T. gondii, Leishmania spp. and Chlamydia spp. in humans is often superseded by the NOS2 pathway in other species (such as mice and rats), in which NOS2 may represent a more robust front-line defence mechanism7,108,111.
Overall, the relative potencies of NOS2, IDOs, IRGs and GBPs against protozoa reflect not only the species-specific pathways available in vertebrates but also the co-evolutionary adaptations used by different parasites to survive within vertebrate host cells.
Cell-autonomous defence against viruses
Viruses were the first reported targets of IFN-mediated immunity, and they are the one taxonomic group that can infect all nucleated cells (reviewed in Ref. 8). Less complex than eukaryotic protozoa and considerably smaller than most bacteria in terms of size and genome content, viruses nonetheless represent a major challenge to the host owing to their high mutation rates (up to 10−8 mutations per base per generation), their diverse cell tropisms and their ability to co-opt the replication machinery of the cell8. For these reasons, IFN-inducible proteins operate in multiple cell types and at all successive stages of the viral life cycle, including entry, replication, capsid assembly and release.
Blocking viral entry and uncoating. At least two IFN-inducible protein families have recently been shown to interfere with viral entry and uncoating: the IFN-inducible transmembrane (IFITM) proteins and the tripartite motif (TRIM) proteins.
IFITM1, IFITM2 and IFITM3 restrict the entry and endosomal fusion of influenza A virus and flaviviruses (such as West Nile virus and dengue virus) in both IFNγ- and IFNα-treated human cells13. Recent studies also extend the antiviral profile of these three IFITMs to include HIV-1, coronaviruses and the Marburg and Ebola filoviruses114,115,116. In the case of IFITM3, a C-terminal transmembrane region and S-palmitoylation contribute to its antiviral activity in membrane-bound compartments such as late endosomes and lysosomes115,116,117,118,119 (Table 3). IFITM3 is thought to deny cytosolic access to influenza A virus by preventing viral genomes from leaving the endocytic pathway119 (Fig. 4).

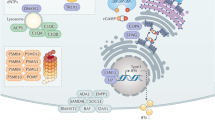
Multiple strategies are used by interferon (IFN)-inducible proteins to combat viruses. IFN-inducible effectors function at nearly every stage of the pathogen life cycle. For example, interferon-inducible transmembrane proteins (IFITMs) and tripartite motif proteins (TRIMs) act during viral entry and uncoating, and myxoma resistance proteins (MXs) block nucleocapsid transport. Inhibition of RNA reverse transcription, protein translation and stability is mediated by APOBEC3 (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide 3), SAMHD1 (SAM-domain- and HD-domain-containing protein 1), ADAR1 (adenosine deaminase, RNA-specific 1), NOS2 (nitric oxide synthase 2), OASs (2′-5′ oligoadenylate synthases), RNase L, ISG20 (IFN-stimulated gene 20 kDa protein), PKR (IFN-induced, RNA-activated protein kinase) and ISG15. Finally, viperin and tetherin help to prevent viral assembly and release, respectively. Some of the effectors (such as MX proteins) appear to operate in both the nucleus and the cytosol (not shown).
TRIMs also serve as viral restriction factors, particularly against retroviruses such as HIV-1. In vertebrates, many TRIMs are induced by IFNs (primarily by type I IFNs) in macrophages, myeloid dendritic cells, peripheral blood lymphocytes and fibroblasts120. TRIM-dependent antiviral activity relies on a shared N-terminal RING domain that functions as an E3 ligase and/or on a C-terminal SPRY domain that enables protein–protein interactions120,121 (Table 3). TRIM5α can restrict HIV-1 entry by binding to the retroviral capsid to accelerate its cytoplasmic uncoating and, as demonstrated more recently, by activating innate immune signalling through associations with the E2 ubiquitin-conjugating enzyme complex UBC13–UEV1A (also known as UBE2N–UBE2V1), which activates TGFβ-activated kinase 1 (TAK1) to induce immune genes122,123. Which of these two mechanisms predominates is as yet unresolved. In addition, TRIM22 combats hepatitis B virus and encephalomyocarditis virus by interfering with pre-genomic RNA synthesis and protease activity, whereas IFNβ-inducible TRIM79α restricts tick-borne encephalitis virus by mediating the lysosomal degradation of the viral RNA-dependent RNA polymerase NS5 (Refs 124, 125, 126). Furthermore, IFNα-inducible TRIM21 delivers incoming IgG-bound adenovirus to the proteasome through its E3 ubiquitin ligase activity127. Thus, the number of different effector mechanisms used by members of the TRIM family continues to grow.
The myxoma resistance proteins (MXs) are also antiviral effector molecules involved at an early stage in type I IFN- and IFNλ-induced host defence against orthomyxoviruses (such as influenza and Thogoto viruses), bunyaviruses, togaviruses and rhabdoviruses128. Human and mouse MX1, as well as mouse MX2, exhibit antiviral activity128,129. Mouse MX1 localizes to promyelocytic leukaemia (PML) nuclear bodies and restricts nuclear viruses, whereas both human MX1 and mouse MX2 are cytosolic proteins that target cytoplasmic viruses128. Human MX1 exhibits the broadest range of antiviral activity, targeting all the infectious genera of the Bunyaviridae family (that is, orthobunyaviruses, hantaviruses, phleboviruses and nairoviruses) as well as coxsackievirus and hepatitis B virus128. This fits with its expression in human endothelial cells, hepatocytes, plasmacytoid dendritic cells, peripheral blood mononuclear cells and other myeloid cells.
Current mechanistic models propose that GTPase-driven MX protein oligomers form ring-like structures to trap viral nucleocapsids and associated polymerases128,130. Such interactions may occur when MX proteins recognize incoming viral ribonuclear particle complexes that are destined for nuclear import or non-nuclear sites of replication130. Results from recent crystallography experiments suggest that disordered loops within an elongated MX1 helical 'stalk' may dock with negatively charged nucleocapsids to mediate entrapment130.
Structural analogies with the MX proteins could also underpin the antiviral activity reported for dynamin-like GBPs against vesicular stomatitis virus (VSV), encephalomyocarditis virus, hepatitis C virus and influenza A virus18,131. Human GBP1, GBP3 and a novel splice isoform termed GBP3ΔC (which lacks part of the C-terminal helical domain) (Table 1) appear to be dependent on GTP binding but not hydrolysis for their effects, suggesting that oligomerization is important for the antiviral activity of GBPs. This evolutionary adaptation may allow GBPs to avoid viral antagonists such as the NS5B protein of hepatitis C virus, which can interfere with their catalytic activity132.
Inhibiting viral replication. Once viruses uncoat, they establish cytoplasmic or nuclear sites of replication (which for Retroviridae includes chromosomal integration). The landmark discoveries of IFN-induced, RNA-activated protein kinase (PKR) and 2′-5′ oligoadenylate synthase 1 (OAS1), OAS2 and OAS3 (and OASL in humans) provided early insights regarding how viral RNA substrates are targeted (reviewed in Ref. 8). PKR possesses RNA-binding motifs at its N-terminus that engage both double-stranded RNA (dsRNA) and single-stranded RNA (ssRNA) (Table 3); the viral uncapped RNAs that are recognized by PKR often have limited duplexed regions and 5′ triphosphate moieties, which enable the enzyme to distinguish them from host, capped RNA species8. Once activated, PKR phosphorylates eukaryotic translation initiation factor 2α (EIF2α) to block viral and host protein translation, a process that is thought to be under intense positive selection to avoid the emergence of viral mimics of the substrate EIF2α133. Likewise, the recognition of dsRNA by OAS enzymes results in the production of 2′-5′ oligoadenylates, which when polymerized activate the latent endoribonuclease RNase L to degrade viral RNA transcripts. Lastly, the exonuclease ISG20 (IFN-stimulated gene 20 kDa protein) degrades RNA transcripts belonging to VSV, influenza virus and encephalomyocarditis virus8.
Some IFN-dependent enzymes edit viral RNAs instead of degrading them. APOBEC3 (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide 3) and ADAR1 (adenosine deaminase, RNA-specific 1) are site-specific cytidine and adenosine deaminases, respectively. APOBEC3 converts cytidine to uridine in dsRNA, whereas ADAR1 catalyses the deamination of adenosine to inosine8,134. These incorporations lead to RNA destabilization and hypermutation after reverse transcription to cause lethal genome mutations in retroviruses such as HIV-1 (Ref. 135). Different APOBEC isoforms (APOBEC3F and APOBEC3G) exhibit distinct mechanisms involving the processing of long terminal repeats, and they may also interact with RNA and/or the Gag protein from HIV-1 to prevent the packaging of these molecules into viral particles8,135 (Fig. 4).
Another IFN-inducible retroviral restriction factor termed SAM-domain- and HD-domain-containing protein 1 (SAMHD1) was found more recently in macrophages and dendritic cells136,137,138, providing some explanation as to why HIV-1 inefficiently transduces mononuclear phagocytes. SAMHD1 contains a nucleotide-phosphohydrolase domain that hydrolyses deoxynucleotides from the cellular pool (Table 3), and depleting this nucleotide supply is currently posited to limit HIV-1 reverse transcriptase activity138 (Fig. 4).
Non-nucleotide targets are also subject to IFN-mediated inhibition. The ubiquitin-like modifier ISG15 restricts influenza viruses, herpesviruses, Sindbis virus, HIV-1, human papillomavirus (HPV) and Ebola virus in cells activated by type I IFNs139,140,141, and many of these viruses cause lethal infection in Isg15−/− mice139. ISG15 acts by conjugating target viral (and cellular) proteins in a process termed ISGylation140. ISGylation substrates include many newly synthesized viral proteins, such as the influenza A virus protein NS1 and the HPV capsid proteins L1 and L2, which are needed for replication and host evasion, and the HIV-1 protein Gag and the Ebola virus protein VP40, which are involved in viral budding140,141. ISGylation can interfere with modification of these viral proteins by ubiquitin, which would otherwise help to activate their functions141.
Nitrosylation is another post-translational modification that inhibits viruses. NO released by IFN-induced NOS2 blocks DNA viruses — including poxviruses (such as ectromelia virus and vaccinia virus), herpesviruses (such as HSV-1 and Epstein–Barr virus) and rhabdoviruses (such as VSV) — as well some RNA viruses (such as coxsackie B3 virus)7,27,142,143,144. Where examined, the loss of antiviral effector function in Nos2−/− mice coincided with heightened susceptibility to viral infection (Table 2). The processes targeted by NO include early and late viral protein synthesis, as well as S-nitrosylation of structural proteins (in the case of VSV) or cysteine proteases (in the case of coxsackie B3 virus). They also extend to DNA replication (in the case of vaccinia virus) and to RNA or DNA synthesis via the inhibition of an immediate-early gene encoding the transactivator Zta (in the case of Epstein–Barr virus)27,142,143,144. Thus, replicative viral DNA and RNA, as well as viral proteins, serve as direct targets for IFN-mediated modification and inactivation.
Preventing viral assembly, budding and release. Following replication, viral DNA, RNA and structural proteins are packaged into nascent virions for budding and release. At least two recently described IFN-induced proteins — tetherin and viperin — affect late-stage export.
Tetherin (also known as CD317 and BST2) is a viral restriction factor that prevents the release of HIV-1 particles from infected macrophages, where it also serves as a target for the HIV-1 protein Vpu145,146. In addition, it prevents the release of filovirus, arenavirus and herpesvirus particles in response to type I IFN or IFNγ stimulation147 in macrophages and plasmacytoid dendritic cells.
The mature tetherin protein is a type II transmembrane disulphide-linked dimer. Its C-terminal ectodomain is modified by a glycophosphatidylinositol (GPI) linkage, and its N-terminal cytoplasmic domain contains YxY motifs for binding the clathrin adaptor proteins AP1 and AP2 during the endocytic internalization of tethered virus for lysosomal delivery147 (Table 3). This topology may enable the association of tetherin with lipid rafts and virion lipids so that it can be incorporated into HIV-1 particles. The secondary rather than primary structure of tetherin is thought to dictate its antiviral activity148, with the N-terminal and coiled-coil regions within the tetherin ectodomain minimally required for viral retention149 (Fig. 4; Table 3).
Viperin (also known as RSAD2) was originally shown to be induced by type I and II IFN signalling in human cytomegalovirus-infected skin cells and in mice infected with lymphocytic choriomeningitis virus150. Viperin contains an S-adenosyl methionine (SAM) domain and an N-terminal amphipathic helix that contributes to its antiviral activity by helping viperin to associate with ER membranes or lipid droplets151,152 (Fig. 4; Table 3), where it interferes with the assembly and egress of influenza virus and hepatitis C virus particles. This may occur through the disruption of ER-derived lipid rafts that transport viral envelope proteins to the plasma membrane, possibly via the inhibition of farnesyl pyrophosphate synthase, which is involved in cholesterol and isoprenoid synthesis151,152. Recent work also demonstrates that viperin inhibits dengue virus, HIV-1 and West Nile virus, although whether it uses similar mechanisms remains untested150,153.
Numerous IFN-inducible restriction factors therefore target each stage of the viral life cycle in a variety of cell types, ensuring broad protective coverage to combat this diverse group of pathogens.
Conclusions and future directions
An avalanche of information has emerged over the last 15 years on the sensory apparatus and signalling cascades that mobilize innate immunity in response to infection6. By contrast, little is known about the cell-autonomous effector mechanisms that confer sterilizing immunity. How do we actually kill intracellular pathogens, or at least restrict their growth? Remarkably, such mechanisms seem to operate across most vertebrate cells, an inheritance foretold by the defence repertoires of plants and lower organisms1,2, but with the added features of expansive diversification and induction by IFNs in larger, long-lived chordates3,8.
Recent applications of systems biology have begun to unearth new IFN-induced antiviral factors (such as IFITMs)13, and genome-wide in silico identification coupled with traditional loss-of-function approaches has revealed proteins with novel antibacterial activities (such as GBPs)16. This list will continue to grow as we probe the interface between vertebrate hosts and microbial pathogens using large-scale unbiased methods154,155, in some cases with the assistance of government centres dedicated to the systematic study of infection (see Ref. 156).
As next-generation informatics takes hold, we are likely to find new IFN-inducible proteins with unique and perhaps unusual functions in host defence. For example, such proteins could protect the nucleus from retroviral insertion or bacterial factors157; defend gap junctions from bacterial cell-to-cell spread158,159; alter microbial or host cell metabolism160; participate in pathogen-selective forms of autophagy161; or use different forms of nucleotide-directed defence (such as microRNAs or interference with small non-coding microbial RNAs) instead of protein activity162. Such candidates would expand the reach of IFNs beyond toxic gases, lytic peptides, ion transporters, DNases and RNases as the main cell-intrinsic means by which to bring infection under control. They may also reinforce the idea that synergy between IFN-induced genes is more than the sum of their individual parts, one of the founding doctrines of systems biology154,155,156.
Other outstanding questions include the identity of the membrane signals, signatures and structures that allow the recruitment of effectors to intact or damaged pathogen compartments for their eventual removal, a topic in which the IFN-inducible IRGs and GBPs will play a leading part. In fact, it was previously proposed that these and related proteins could provide a physical bridge between the detection and disposal of this particular class of organisms18,163. Now is the time to test such predictions by modern methods. To do so should help to build a more complete picture of intracellular defence at the single-cell level. It will also better define what constitutes the IFN-induced resistome.
References
Jones, J. D. & Dangl, J. L. The plant immune system. Nature 444, 323–329 (2006).
Marathe, R., Guan, Z., Anandalakshmi, R., Zhao, H. & Dinesh-Kumar, S. P. Study of Arabidopsis thaliana resistome in response to cucumber mosaic virus infection using whole genome microarray. Plant Mol. Biol. 55, 501–520 (2004).
Beutler, B. et al. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu. Rev. Immunol. 24, 353–389 (2006).
Mascie-Taylor, C. G. & Karim, E. The burden of chronic disease. Science 302, 1921–1922 (2003).
Bezbradica, J. J. & Medzhitov, R. Integration of cytokine and heterologous receptor signaling pathways. Nature Immunol. 10, 333–339 (2009).
Kawai, T. & Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650 (2011).
Nathan, C. & Shiloh, M. U. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl Acad. Sci. USA 97, 8841–8848 (2000).
Sadler, A. & Williams, B. R. G. Interferon-inducible antiviral effectors. Nature Rev. Immunol. 8, 559–568 (2008).
Flannagan, R. S., Cosío, G. & Grinstein, S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nature Rev. Microbiol. 7, 355–366 (2009).
Nathan, C. F., Murray, H. W., Weibe, M. E. & Rubin, B. Y. Identification of IFN-γ as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J. Exp. Med. 158, 670–689 (1983). This paper was the first to describe IFNγ as the long sought after macrophage-activating factor required for defence against intracellular pathogens (see also the historical note in reference 11).
MacMicking, J. D. Macrophage activation and host defense. Cell Host Microbe 5, 1–3 (2009).
Hertzog, P., Forster, S. & Samarajiwa, S. Systems biology of interferon responses. J. Interferon Cytokine Res. 31, 5–11 (2011).
Brass, A. L. et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139, 1243–1254 (2009).
Kumar, D. et al. Genome-wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis. Cell 140, 731–743 (2010). References 13 and 14 were among the first to use genome-wide small interfering RNA screening against viruses and bacteria to identify new type I and II IFN effectors.
Schoggins, J. W. et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 (2011). In this study, a comprehensive gain-of-function screen discovered new type I IFN antiviral factors against several major human pathogens, including hepatitis C virus.
Kim, B. H. et al. A family of IFN-γ-inducible 65-kD GTPases protects against bacterial infection. Science 332, 717–721 (2011). In this study, family-wide loss-of-function screens and gene-targeted mice were used to identify the 65 kDa GBPs as new IFNγ-inducible proteins that combat intracellular bacterial infection.
Krombach, F. et al. Cell size of alveolar macrophages: an interspecies comparison. Environ. Health Perspect. 105 (Suppl. 5), 1261–1263 (1997).
MacMicking, J. D. IFN-inducible GTPases and immunity to intracellular pathogens. Trends Immunol. 25, 601–609 (2004).
Martens, S. & Howard, J. The IFN-inducible GTPases. Annu. Rev. Immunol. 22, 558–598 (2006).
Kumar, Y. & Valdivia, R. H. Leading a sheltered life: intracellular pathogens and maintenance of vacuolar compartments. Cell Host Microbe 5, 593–601 (2009).
Tiwari, S., Choi, H. P., Matsuzawa, T., Pypaert, M. & MacMicking, J. D. Targeting of the GTPase Irgm1 to the phagosomal membrane via PtdIns(3,4)P2 and PtdIns(3,4, 5)P3 promotes immunity to mycobacteria. Nature Immunol. 10, 907–917 (2009).
MacMicking, J. D., Taylor, G. A. & McKinney, J. D. Immune control of tuberculosis by IFN-γ-inducible LRG-47. Science 302, 654–659 (2003). The first report of an IFN-inducible GTPase regulating membrane traffic to compartmentalized pathogens for cell-autonomous defence.
Singh, S. B., Davis, A. S., Taylor, G. A. & Deretic, V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313, 1438–1441 (2006).
Thurston, T. L., Ryzhakov, G., Bloor, S., von Muhlinen, N. & Randow, F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nature Immunol. 10, 1215–1221 (2009).
Thurston, T. L., Wandel, M. P., von Muhlinen, N., Foeglein, A. & Randow, F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418 (2012). References 24 and 25 identify IFN-induced ubiquitin- and glycan-recognition receptors, respectively, that help to bridge the detection of cytosolic bacteria with sequestration by the autophagy pathway.
Nathan, C. & Ding, A. SnapShot: reactive oxygen intermediates (ROI). Cell 140, 952–952. e2 (2010).
Karupiah, G. et al. Inhibition of viral replication by interferon-γ-induced nitric oxide synthase. Science 261, 1445–1448 (1993).
Lambeth, J. D. NOX enzymes and the biology of reactive oxygen. Nature Rev. Immunol. 4, 181–189 (2004).
Fields, K. A. & Hackstadt, T. The chlamydial inclusion: escape from the endocytic pathway. Annu. Rev. Cell Dev. Biol. 18, 221–245 (2002).
Oliveira Gde, A., Lieberman, J. & Barillas-Mury, C. Epithelial nitration by a peroxidase/NOX5 system mediates mosquito antiplasmodial immunity. Science 335, 856–859 (2012).
Sonoda, J. et al. Nuclear receptor ERRα and coactivator PGC-1β are effectors of IFN-γ-induced host defense. Genes Dev. 21, 1909–1920 (2007).
West, A. P. et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472, 476–480 (2011).
Bustamente, J. et al. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nature Immunol. 12, 213–221 (2011).
Gardet, A. et al. LRRK2 is involved in the IFN-γ response and host response to pathogens. J. Immunol. 185, 5577–5585 (2010).
Moskwa, P. et al. A novel host defense system of airways is defective in cystic fibrosis. Am. J. Respir. Crit. Care Med. 175, 174–183 (2007).
Botteauxa, A., Hosteb, C., Dumontb, J. E., Van Sandeb, J. & Allaouia, A. Potential role of Noxes in the protection of mucosae: H2O2 as a bacterial chemorepellant. Microbes Infect. 11, 537–544 (2009).
Gattas, M. V. et al. Oxidative epithelial host defense is regulated by infectious and inflammatory stimuli. Free Radic. Biol. Med. 47, 1450–1458 (2009).
Flores, M. V. et al. Dual oxidase in the intestinal epithelium of zebrafish larvae has antibacterial properties. Biochem. Biophys. Res. Commun. 400, 164–168 (2010).
MacMicking, J. D. et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell 81, 641–650 (1995).
Pollock, J. D. et al. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nature Genet. 9, 202–209 (1995). References 39 and 40 describe the first gene-targeted mice deficient for IFN-inducible effector proteins.
Shiloh, M. U. et al. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity 10, 29–38 (1999).
Ng, V. H., Cox, J. S., Sousa, A. O., MacMicking, J. D. & McKinney, J. D. Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol. Microbiol. 52, 1291–1302 (2004).
Singh, R. et al. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 322, 1392–1395 (2008).
Myers, J. T., Tsang, A. W. & Swanson, J. A. Localized reactive oxygen and nitrogen intermediates inhibit escape of Listeria monocytogenes from vacuoles in activated macrophages. J. Immunol. 171, 5447–5453 (2003).
Bagshaw, R. D., Mahuran, D. J. & Callahan, J. W. A proteomic analysis of lysosomal integral membrane proteins reveals the diverse composition of this organelle. Mol. Cell. Proteomics 4, 133–143 (2005).
Trost, M. et al. The phagosomal proteome in interferon-γ-activated macrophages. Immunity 30, 143–154 (2009). A comprehensive proteomic study that identified IFN-inducible proteins that are recruited to the phagosome.
Wild, P. et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333, 228–233 (2011).
Mostowy, S. et al. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J. Biol. Chem. 286, 26987–26995 (2011).
Cemma, M., Kim, P. K. & Brumell, J. H. The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy 7, 341–345 (2011).
Yoshikawa, Y. et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nature Cell Biol. 11, 1233–1240 (2009).
Shenoy, A. R. et al. Emerging themes in IFN-γ-induced macrophage immunity by the p47 and p65 GTPase families. Immunobiology 212, 771–784 (2008).
Coers, J. et al. Chlamydia muridarum evades growth restriction by the IFN-γ-inducible host resistance factor Irgb10. J. Immunol. 180, 6237–6245 (2008).
Al-Zeer, M. A., Al-Younes, H. M., Braun, P. R., Zerrahn, J. & Meyer, T. F. IFN-γ-inducible Irga6 mediates host resistance against Chlamydia trachomatis via autophagy. PLoS ONE 4, e4588 (2009).
Lapaquette, P., Glasser, A. L., Huett, A., Xavier, R. J. & Darfeuille-Michaud, A. Crohn's disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell. Microbiol. 12, 99–113 (2010).
Nelson, D. E. et al. Chlamydial IFN-γ immune evasion is linked to host infection tropism. Proc. Natl Acad. Sci. USA 101, 10658–10663 (2005).
Bernstein-Hanley, I. et al. The p47 GTPases Igtp and Irgb10 map to the Chlamydia trachomatis susceptibility locus Ctrq-3 and mediate cellular resistance in mice. Proc. Natl Acad. Sci. USA 103, 14092–14097 (2006).
Henry, S. C. et al. Impaired macrophage function underscores susceptibility to Salmonella in mice lacking Irgm1 (LRG-47). J. Immunol. 179, 6963–6972 (2007).
Miyairi, I. et al. The p47 GTPases Iigp2 and Irgb10 regulate innate immunity and inflammation to murine Chlamydia psittaci infection. J. Immunol. 179, 1814–1824 (2007).
Singh, S. B. et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nature Cell Biol. 12, 1154–1165 (2010).
Brest, P. et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn's disease. Nature Genet. 43, 242–245 (2011).
Lippmann, J. et al. Dissection of a type I interferon pathway in controlling bacterial intracellular infection in mice. Cell. Microbiol. 13, 1668–1682 (2011).
Hunn, J. P. et al. Regulatory interactions between IRG resistance GTPases in the cellular response to Toxoplasma gondii. EMBO J. 27, 2495–2509 (2008). This paper introduced the concept of regulatory and effector members of the IRG family in the targeting of the pathogen vacuole.
Khaminets, A. et al. Coordinated loading of IRG resistance GTPases on to the Toxoplasma gondii parasitophorous vacuole. Cell. Microbiol. 12, 939–961 (2010).
Cai, Q. & Sheng, Z.-H. Uncovering the role of Snapin in regulating autophagy-lysosomal function. Autophagy 7, 445–447 (2011).
Grégoire, I. P. et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 7, e1002422 (2011).
McCarroll, S. A. et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nature Genet. 40, 1107–1112 (2008).
Intemann, C. D. et al. Autophagy gene variant IRGM -261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog. 5, e1000577 (2009).
King, K. Y. et al. Polymorphic allele of human IRGM1 is associated with susceptibility to tuberculosis in African Americans. PLoS ONE 6, e16317 (2011).
Khor, B., Gardet, A. & Xavier, R. J. Genetics and pathogenesis of inflammatory bowel disease. Nature 474, 307–317 (2011).
Rupper, A. C. & Cardelli, J. A. Induction of guanylate binding protein 5 by gamma interferon increases susceptibility to Salmonella enterica serovar Typhimurium-induced pyroptosis in RAW 264.7 cells. Infect. Immun. 76, 2304–2315 (2008).
Tietzel, I., El-Haibi, C. & Carabeo, R. A. Human guanylate binding proteins potentiate the anti-Chlamydia effects of interferon-γ. PLoS ONE 4, e6499 (2009).
Modiano, N., Lu, Y. E. & Cresswell, P. Golgi targeting of human guanylate-binding protein-1 requires nucleotide binding, isoprenylation, and an IFN-γ-inducible cofactor. Proc. Natl Acad. Sci. USA 102, 8680–8685 (2005).
Britzen-Laurent, N. et al. Intracellular trafficking of guanylate-binding proteins is regulated by heterodimerization in a hierarchical manner. PLoS ONE 5, e14246 (2010).
Nantais, D. E., Schwemmle, M., Stickney, J. T., Vestal, D. J. & Buss, J. E. Prenylation of an interferon-γ-induced GTP-binding protein: the human guanylate binding protein, huGBP1. J. Leukoc. Biol. 60, 423–431 (1996).
Traver, M. K. et al. Immunity-related GTPase M (Irgm) proteins influence the localization of guanylate-binding protein 2 (Gbp2) by modulating macroautophagy. J. Biol. Chem. 286, 30471–30480 (2011).
Virreira Winter, S. et al. Determinants of GBP recruitment to Toxoplasma gondii vacuoles and the parasitic factors that control it. PLoS ONE 6, e24434 (2011). An elegant study demonstrating heterotypic GBP interactions that dictate the targeting of T. gondii by members of this IFN-inducible GTPase family.
Alonso, S., Pethe, K., Russell, D. G. & Purdy, G. E. Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc. Natl Acad. Sci. USA 104, 6031–6036 (2007).
Ponpuak, M. et al. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity 32, 1–13 (2010).
Kim, J. Y. & Ozato, K. The sequestosome 1/p62 attenuates cytokine gene expression in activated macrophages by inhibiting IFN regulatory factor 8 and TNF receptor-associated factor 6/NF-κB activity. J. Immunol. 182, 2131–2140 (2009).
Korioth, F., Gieffers, C., Maul, G. G. & Frey, J. Molecular characterization of NDP52, a novel protein of the nuclear domain 10, which is redistributed upon virus infection and interferon treatment. J. Cell Biol. 130, 1–13 (1995).
Lu, F. T. et al. Expression and function of galectin-3, a β-galactoside-binding lectin, in human monocytes and macrophages. Am. J. Pathol. 147, 1016–1028 (1995).
Shahnazari, S. et al. A diacylglycerol-dependent signaling pathway contributes to regulation of antibacterial autophagy. Cell Host Microbe 8, 137–146 (2010).
Jabado, N. et al. Natural resistance to intracellular infections: natural resistance-associated macrophage protein 1 (Nramp1) functions as a pH-dependent manganese transporter at the phagosomal membrane. J. Exp. Med. 192, 1237–1248 (2000). This study defined the elusive cation transporter function of NRAMP1 at the phagosomal membrane of macrophages.
Nairz, M. et al. Interferon-γ limits the availability of iron for intramacrophage Salmonella typhimurium. Eur. J. Immunol. 38, 1923–1936 (2008).
White, C., Lee, J., Kambe, T., Fritsche, K. & Petris, M. J. A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J. Biol. Chem. 284, 33949–33956 (2009).
Wagner, D. et al. Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis-, and Mycobacterium smegmatis-containing phagosomes indicates pathogen-induced microenvironments within the host cell's endosomal system. J. Immunol. 174, 1491–1500 (2005).
Govoni, G., Gauthier, S., Billia, F., Iscove, N. N. & Gros, P. Cell-specific and inducible Nramp1 gene expression in mouse macrophages in vitro and in vivo. J. Leukoc. Biol. 62, 277–286 (1997).
Zaharik, M. L. et al. The Salmonella enterica serovar typhimurium divalent cation transport systems MntH and SitABCD are essential for virulence in an Nramp1G169 murine typhoid model. Infect. Immun. 72, 5522–5525 (2004).
Wagner, D. et al. Changes of the phagosomal elemental concentrations by Mycobacterium tuberculosis Mramp. Microbiology 151, 323–332 (2005).
Sibley, L. D. Invasion and intracellular survival of protozoan parasites. Immunol. Rev. 240, 72–91 (2011).
Diefenbach, A. et al. Type 1 interferon (IFNα/β) and type 2 nitric oxide synthase regulate the innate immune response to a protozoan parasite. Immunity 8, 77–87 (1998).
Holscher, C. et al. Defective nitric oxide effector functions lead to extreme susceptibility of Trypanosoma cruzi-infected mice deficient in γ-interferon receptor or inducible nitric oxide synthase. Infect. Immun. 66, 1208–1215 (1998).
Mellouk, S. et al. Nitric oxide-mediated antiplasmodial activity in human and murine hepatocytes induced by gamma interferon and the parasite itself: enhancement by exogenous tetrahydrobiopterin. Infect. Immun. 62, 4043–4046 (1994).
Scharton-Kersten, T. M., Yap, G., Magram, J. & Sher, A. Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii. J. Exp. Med. 185, 1261–1273 (1997).
Collazo, C. M. et al. Inactivation of LRG-47 and IRG-47 reveals a family of interferon γ-inducible genes with essential, pathogen-specific roles in resistance to infection. J. Exp. Med. 194, 181–188 (2001).
Zhao, Y. et al. Virulent Toxoplasma gondii evade immunity-related GTPase-mediated parasite vacuole disruption within primed macrophages. J. Immunol. 182, 3775–3781 (2009).
Halonen, S. K., Taylor, G. A. & Weiss, L. M. Gamma interferon-induced inhibition of Toxoplasma gondii in astrocytes is mediated by IGTP. Infect. Immun. 69, 5573–5576 (2001).
Martens, S. et al. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog. 1, e24 (2005).
Ling, Y. M. et al. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J. Exp. Med. 203, 2063–2071 (2006).
Zhao, Y., Wilson, D., Matthews, S. & Yap, G. S. Rapid elimination of Toxoplasma gondii by gamma interferon-primed mouse macrophages is independent of CD40 signaling. Infect. Immun. 75, 4799–4803 (2007).
Liesenfeld, O. et al. The IFN-γ-inducible GTPase, Irga6, protects mice against Toxoplasma gondii but not against Plasmodium berghei and some other intracellular pathogens. PLoS ONE 6, e20568 (2011).
Santiago, H. C. et al. Mice deficient in LRG-47 display enhanced susceptibility to Trypanosoma cruzi infection associated with defective hemopoiesis and intracellular control of parasite growth. J. Immunol. 175, 8165–8172 (2005).
Zhao, Z. et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe 4, 458–469 (2008).
Degrandi, D. et al. Extensive characterization of IFN-induced GTPases mGBP1 to mGBP10 involved in host defense. J. Immunol. 179, 7729–7740 (2007). Together with reference 16, this study provides a comprehensive genomic, molecular and cellular description of the expanded GBP family.
Fentress, S. J. et al. Phosphorylation of immunity-related GTPases by a Toxoplasma gondii secreted kinase promotes macrophage survival and virulence. Cell Host Microbe 8, 484–495 (2010).
Pfefferkorn, E. R. Interferon γ blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophan. Proc. Natl Acad. Sci. USA 81, 908–912 (1984). This study first showed the importance of tryptophan degradation in restricting the growth of T. gondii.
Carlin, J. M., Borden, E. C. & Byrne, G. I. Interferon-induced indoleamine 2,3-dioxygenase activity inhibits Chlamydia psittaci replication in human macrophages. J. Interferon Res. 9, 329–337 (1989).
Roshick, C., Wood, H., Caldwell, H. D. & McClarty, G. Comparison of gamma interferon-mediated antichlamydial defense mechanisms in human and mouse cells. Infect. Immun. 74, 225–238 (2006).
Daubener, W. et al. Restriction of Toxoplasma gondii growth in human brain microvascular endothelial cells by activation of indoleamine 2,3-dioxygenase. Infect. Immun. 69, 6527–6531 (2001).
Mao, R. et al. Indoleamine 2,3-dioxygenase mediates the antiviral effect of gamma interferon against hepatitis B virus in human hepatocyte-derived cells. J. Virol. 85, 1048–1057 (2011).
Meisel, R. et al. Human but not murine multipotent mesenchymal stromal cells exhibit broad-spectrum antimicrobial effector function mediated by indoleamine 2,3-dioxygenase. Leukemia 25, 648–654 (2011).
Knubel, C. P. et al. Indoleamine 2,3-dioxigenase (IDO) is critical for host resistance against Trypanosoma cruzi. FASEB J. 24, 2689–2701 (2010).
Divanovic, S. et al. Opposing biological functions of tryptophan catabolizing enzymes during intracellular infection. J. Infect. Dis. 205, 152–161 (2012).
Siegrist, F., Eberling, M. & Certa, U. The small interferon-induced transmembrane genes and proteins. J. Interferon Cytokine Res. 31, 183–197 (2011).
Lu, J. et al. The IFITM proteins inhibit HIV-1 infection. J. Virol. 85, 2126–2137 (2011).
Huang, I. C. et al. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 7, e1001258 (2011).
Weidner, J. M. et al. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J. Virol. 84, 12646–12657 (2010).
Yount, J. S. et al. Palmitoylome profiling reveals S-palmitoylation-dependent antiviral activity of IFITM3. Nature Chem. Biol. 6, 610–614 (2010).
Feeley, E. M. et al. IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog. 7, e1002337 (2011).
McNab, F. W., Rasjbaum, R., Stoyle, J. P. & O'Garra, A. Tripartite-motif proteins and innate immune regulation. Curr. Opin. Immunol. 23, 46–56 (2011).
Uchil, P. D., Quinlin, B. D., Chan, W.-T., Luna, J. M. & Mothes, W. TRIM E3 ligases interfere with the early and late stages of the retroviral life cycle. PLoS Pathog. 4, e16 (2008).
Stemlau, M. et al. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature 427, 848–853 (2004). This was the first report of TRIM-mediated activity against HIV-1, which led to the idea that members of the TRIM family can act as intrinsic antiviral factors.
Pertel, T. et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472, 361–365 (2011). This study adds a second major function for TRIM5 in innate immune signalling cascades following retroviral infection.
Gao, B., Duan, Z., Xu, W. & Xiong, S. Tripartite motif-containing 22 inhibits the activity of hepatitis B virus core promoter, which is dependent on nuclear-located RING domain. Hepatology 50, 424–433 (2009).
Eldin, P. et al. TRIM22 E3 ubiquitin ligase activity is required to mediate antiviral activity against encephalomyocarditis virus. J. Gen. Virol. 90, 536–545 (2009).
Taylor, R. T. et al. TRIM79α, an interferon-stimulated gene product, restricts tick-borne encephalitis virus replication by degrading the viral RNA polymerase. Cell Host Microbe 10, 185–196 (2011).
Mallery, D. L. et al. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc. Natl Acad. Sci. USA 107, 19985–19990 (2010).
Haller, O. & Kochs, G. Human MxA protein: an interferon-induced dynamin-like GTPase with broad antiviral activity. J. Interferon Cytokine Res. 31, 79–87 (2011).
Mordstein, M. et al. Interferon-λ contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 4, e1000151 (2008).
Gao, S. et al. Structural basis of oligomerization in the stalk region of dynamin-like MxA. Nature 465, 502–506 (2010).
Nordmann, A., Wixler, L., Boergeling, Y., Wixler, V. & Ludwig, S. A new splice variant of the human guanylate-binding protein 3 mediates anti-influenza activity through inhibition of viral transcription and replication. FASEB J. 26, 1290–1300 (2012).
Itsui, Y. et al. Antiviral effects of the interferon-induced protein guanylate binding protein 1 and its interaction with the hepatitis C virus NS5B protein. Hepatology 50, 1527–1537 (2009).
Elde, N. C., Child, S. J., Geballe, A. P. & Malik, H. S. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 457, 485–489 (2009). An insightful study revealing the evolutionary adaptations undergone by PKR to avoid the emergence of viral mimics of its substrate EIF2α.
Harris, R. S. et al. DNA deamination mediates innate immunity to retroviral infection. Cell 113, 803–809 (2003). The first demonstration of the DNA-deaminating activity of an APOBEC protein against retroviral infection.
Mbisa, J. L., Bu, W. & Pathak, V. K. APOBEC3F and APOBEC3G inhibit HIV-1 DNA integration by different mechanisms. J. Virol. 84, 5250–5259 (2010).
Laguette, N. et al. SAMHD1 is the dendritic-and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474, 654–657 (2011).
Hrecka, K. et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474, 658–661 (2011).
Goldstone, D. C. et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 480, 379–382 (2011).
Lenschow, D. J. et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl Acad. Sci. USA 104, 1371–1376 (2007). This study comprehensively demonstrates the importance of ISG15 as an inhibitory mechanism against phylogenetically diverse viruses.
Durfee, L. A., Lyon, N., Seo, K. & Huibregtse, J. M. The ISG15 conjugation system broadly targets newly synthesized proteins: implications for the antiviral function of ISG15. Mol. Cell 38, 722–732 (2010).
Skaug, B. & Chen Z. J. Emerging role of ISG15 in antiviral immunity. Cell 143, 187–190 (2010).
Bi, Z. & Reiss, C. S. Inhibition of vesicular stomatitis virus infection by nitric oxide. J. Virol. 69, 2208–2213 (1995).
Mannick, J. B., Asano, K., Izumi, K., Kieff, E. & Stamler, J. S. Nitric oxide produced by human B lymphocytes inhibits apoptosis and Epstein-Barr virus replication. Cell 79, 1137–1146 (1994).
Zaragoza, C. et al. The role of inducible nitric oxide synthase in the host response to Coxsackievirus myocarditis. Proc. Natl Acad. Sci. USA 95, 2469–2474 (1998).
Neil, S. J. et al. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451, 425–431 (2008). The first report on the role of tetherin in preventing HIV-1 release.
Van Damme, N. et al. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3, 245–252 (2008).
Evans, D. T., Serrano-Moreno, R., Singh, R. K. & Guatelli, J. C. BST-2/tetherin: a new component of the innate immune response to enveloped viruses. Trends Microbiol. 18, 388–396 (2010).
Perez-Caballero, D. et al. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 139, 499–511 (2009). A well-designed study that demonstrates that the domain structure of tetherin dictates its antiviral tethering activity.
Hinz, A. et al. Structural basis of HIV-1 tethering to membranes by the BST-2/Tetherin ectodomain. Cell Host Microbe 7, 314–323 (2010).
Seo, J. Y., Yaneva, R. & Cresswell, P. Viperin: a multifunctional, interferon-inducible protein that regulates virus replication. Cell Host Microbe 10, 534–539 (2011).
Hinson, E. R. & Cresswell, P. The antiviral protein, viperin, localizes to lipid droplets via its N-terminal amphipathic helix. Proc. Natl Acad. Sci. USA 106, 20452–20457 (2009).
Wang, X., Hinson, E. R. & Cresswell, P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe 2, 96–105 (2007).
Stzretter, K. J. et al. The interferon-inducible gene viperin restricts West Nile virus pathogenesis. J. Virol. 85, 11557–11566 (2011).
Shapira, S. D. & Hacohen, N. Systems biology approaches to dissect mammalian innate immunity. Curr. Opin. Immunol. 23, 71–77 (2011).
Amit, I., Regev, A. & Hacohen, N. Strategies to discover regulatory circuits of the mammalian immune system. Nature Rev. Immunol. 11, 873–880 (2011).
Aderem, A. et al. A systems biology approach to infectious disease research: innovating the pathogen–host research paradigm. mBio 2, 1–4 (2011).
Bierne, H. & Cossart, P. When bacteria target the nucleus: the emerging family of nucleomodulins. Cell. Microbiol. 23 Feb 2012 (doi:10.1111/j.1462-5822.2012.01758.x).
Schnoor, M., Betanzos, A., Weber, D. A. & Parkos, C. A. Guanylate-binding protein-1 is expressed at tight junctions of intestinal epithelial cells in response to interferon-γ and regulates barrier function through effects on apoptosis. Mucosal Immunol. 2, 33–42 (2009).
Bonazzi, M. & Cossart, P. Impenetrable barriers or entry portals? The role of cell–cell adhesion during infection. J. Cell Biol. 195, 349–358 (2011).
Blanc, M. et al. Host defense against viral infection involves interferon mediated down-regulation of sterol biosynthesis. PLoS Biol. 9, e1000598 (2011).
Orvedahl, A. et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 480, 113–117 (2011).
Pedersen, I. M. et al. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 449, 919–923 (2007).
MacMicking, J. D. Immune control of phagosomal bacteria by p47 GTPases. Curr. Opin. Microbiol. 8, 74–82 (2005).
Gomez, L. M. et al. A polymorphism in the inducible nitric oxide synthase gene is associated with tuberculosis. Tuberculosis 87, 288–294 (2008).
Li, X. L. et al. An essential role for the antiviral endoribonuclease, RNase-L, in antibacterial immunity. Proc. Natl Acad. Sci. USA 105, 20816–20821 (2008).
Acknowledgements
The author apologizes to colleagues whose work has not been cited owing to space constraints. J.D.M. is supported by the US National Institutes of Health (R01AI068041-06), a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease Award (1007845), a Crohn's and Colitis Foundation of America Senior Research Award (R09928) and a W.W. Winchester Award.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The author declares no competing financial interests.
Supplementary information
Supplementary Figure 1
IFN-inducible enzymes in oxidative and nitrosative defense. (PDF 277 kb)
Related links
Glossary
- Autophagy
-
A specialized process involving the degradative delivery of a portion of the cytoplasm or of damaged organelles to the lysosome. Internalized pathogens can also be eliminated by this pathway.
- Reactive oxygen species
-
(ROS). Aerobic organisms derive their energy from the reduction of oxygen. The metabolism of oxygen, and in particular its reduction through the mitochondrial electron-transport chain, generates by-products such as superoxide (O2−) and downstream intermediates such as hydrogen peroxide (H2O2) and hydroxyl radicals (·OH). These three species are referred to as ROS. ROS can damage important intracellular targets, such as DNA, lipids or proteins.
- Reactive nitrogen species
-
(RNS). Nitric oxide (NO) chemistry is complex because of the extreme reactivity of NO, which can result in the formation of different reactive nitrogen intermediates (RNI) depending on the amount of NO that is produced by cells. At low concentrations, NO reacts directly with metals and other radicals. At higher concentrations, indirect effects prevail, and these include several oxidation or nitrosylation reactions with oxygen that result in the production of various congeners. NO and related RNI are effective antimicrobial agents and signal-transducing molecules.
- Phagolysosomes
-
Intracellular vesicles that result from the fusion of phagosomes, which enclose extracellular material that has been ingested, with lysosomes, which contain lytic enzymes and antimicrobial peptides.
- NADPH oxidases
-
Enzyme systems that consist of multiple cytosolic and membrane-bound subunits. The complex is assembled in activated phagocytic cells on the plasma and phagosomal membranes. NADPH oxidase uses electrons from NADPH to reduce molecular oxygen to form superoxide anions. Superoxide anions are enzymatically converted to hydrogen peroxide, which in neutrophils can undergo further conversion by myeloperoxidase to hypochloric acid, a highly toxic and microbicidal agent.
- Respiratory burst
-
The process by which molecular oxygen is reduced by the NADPH oxidase system to produce reactive oxygen species.
- Chronic granulomatous disease
-
An inherited disorder caused by defective oxidase activity in the respiratory burst of phagocytes. It results from mutations in any of five genes that are necessary to generate the superoxide radicals required for normal phagocyte function. Affected patients suffer from increased susceptibility to recurrent infections.
- Galectins
-
Lectins that bind a wide variety of glycoproteins and glycolipids containing β-galactoside. They have extracellular and intracellular functions, including the regulation of apoptosis, RAS signalling, cell adhesion and angiogenesis.
- SNARE proteins
-
(Soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins). A class of proteins that is required for membrane fusion events that occur in the course of vesicle trafficking and secretion.
- ISGylation
-
The attachment of the ubiquitin-like modifier ISG15 to either pathogen or host protein targets to regulate their function rather than stimulate degradation.
- MicroRNAs
-
Single-stranded RNA molecules of approximately 21–23 nucleotides in length that are thought to regulate the expression of other genes.
Rights and permissions
About this article
Cite this article
MacMicking, J. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol 12, 367–382 (2012). https://doi.org/10.1038/nri3210
Published:
Issue Date:
DOI: https://doi.org/10.1038/nri3210
This article is cited by
-
TRIM32 reduced the recruitment of innate immune cells and the killing capacity of Listeria monocytogenes by inhibiting secretion of chemokines
Gut Pathogens (2023)
-
Changes in ADAR RNA editing patterns in CMV and ZIKV congenital infections
BMC Genomics (2023)
-
Transcriptome analysis of a newly established mouse model of Toxoplasma gondii pneumonia
Parasites & Vectors (2023)
-
Intracellular lifestyle of Chlamydia trachomatis and host–pathogen interactions
Nature Reviews Microbiology (2023)
-
Vγ9+Vδ2+ T cell control of Listeria monocytogenes growth in infected epithelial cells requires butyrophilin 3A genes
Scientific Reports (2023)
