
Key Points
-
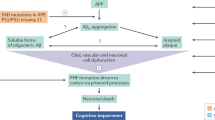
A relatively large and diverse set of human proteins are known to form fibrils that accumulate in tissue in association with several different diseases. It is widely accepted that these fibrils — called amyloid — are the causative agent of the related diseases. Another common attribute of amyloid diseases is that beyond the age of forty, the risk of developing an amyloid disease greatly increases. This is true for all known amyloid diseases with the exception of the familial type.
-
On a molecular level, amyloid fibrils resemble an aircraft cable, with 3–6 filaments wrapped around one another to form the fibril. All amyloid fibrils studied to date are composed of individual filaments that are made of a lamellar cross-?-sheet, which contains thousands of non-covalently associated protein subunits.
-
The most common strategy that is being pursued in the development of small-molecule amyloid inhibitors involves finding molecules that prevent the initiation of the conformational changes or endoproteolytic processing that lead to amyloid formation. A more difficult approach might be to discover compounds that prevent amyloid formation by intercepting the misfolded protein.
-
Intermediates in amyloid assembly might be responsible for some, if not all, of the pathology. Therefore, 'amyloid-fibril inhibitors' could, in theory, lead to more pronounced pathology owing to the accumulation of soluble disease-associated quaternary structures.
-
Small-molecule inhibitors that stabilize the native conformation of transthyretin (TTR) have been discovered using functional screens and structure-based methods. Several of these small-molecule inhibitors efficiently prevent TTR amyloid fibril formation in vitro, and are being tested for their in vivo activity.
Abstract
Amyloid diseases are a large group of a much larger family of misfolding diseases. This group includes pathologies as diverse as Alzheimer's disease, immunoglobulin-light-chain disease, reactive amyloid disease and the familial amyloid polyneuropathies. These diseases are generally incurable at present, although some drugs are known to transiently slow the progression of Alzheimer's disease. As we increase our understanding of the causative mechanisms of these disorders, the likelihood of success for a given therapeutic strategy will become clearer. This review will look at small-molecule and macromolecular approaches for intervention in amyloid diseases other than Alzheimer's disease, although select examples from Alzheimer's disease will be discussed.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

References
Kisilevsky, R. Biology of disease amyloidosis: a familiar problem in light of current pathogenic developments. Lab. Inv. 49, 381–390 (1983).
Pepys, M. B. in Immunological Diseases (ed. Samter, M.) 631–674 (Little, Brown and Co., Boston/Toronto, 1988).
Stevens, F. J. & Kisilevsky, R. Immunoglobulin light chains, glycosaminoglycans, and amyloid. Cell. Mol. Life Sci. 57, 441–449 (2000).
Sipe, J. D. Amyloidosis. Annu. Rev. Biochem. 61, 947–975 (1992).
Jacobson, D. R. & Buxbaum, J. N. Genetic aspects of amyloidosis. Adv. Human Genetics 20, 69–123 (1991).
Sipe, J. D. Amyloidosis. Crit. Rev. Clin. Lab. Sci. 31, 325–354 (1994).
Benson, M. D. & Wallace, M. R. in The Metabolic Basis of Inherited Disease (eds. Scriver, C. R., Beaudet, A. L., Sly, W. S. & Valle, D.) 2439 (McGraw Hill, New York, 1989).
Benson, M. D. Familial amyloidotic polyneuropathy. Trends Biochem. Sci. 12, 88–92 (1989).
Kelly, J. W. Alternative conformations of amyloidogenic proteins govern their behavior. Curr. Opin. Struct. Biol. 6, 11–17 (1996).
Fink, A. L. Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold. Des. 3, R9–R23 (1998).
Sipe, J. D. Serum amyloid A. From fibril to function. Current status. Amyloid 7, 10–12 (2000).
Pepys, M. B. & Hawkins, P. N. Human lysozyme gene mutation causes hereditary systemic amyloidosis. Nature 362, 553–557 (1993).
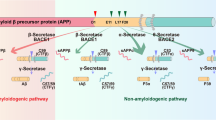
Golde, T. E., Estus, S., Younkin, L. H., Selkoe, D. J. & Younkin, S. G. Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science 255, 728–730 (1992).
Shoji, M. et al. Production of the Alzheimer amyloid β-protein by normal proteolytic processing. Science 258, 126–129 (1992).
Vassar, R. et al. β-Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741 (1999).
Kimberly, W. T., Xia, W., Rahmati, T., Wolfe, M. S. & Selkoe, D. J. The transmembrane aspartates in presenilin 1 and 2 are obligatory for γ-secretase activity and amyloid β-protein generation. J. Biol. Chem. 275, 3173–3178 (2000).
Chen, C. D. et al. Furin initiates gelsolin familial amyloidosis in the Golgi through a defect in Ca2+ stabilization. EMBO J. 20, 6277–6287 (2001).
Kelly, J. W. & Lansbury, P. T. J. A chemical approach to elucidate the mechanism of transthyretin and β-protein amyloid fibril formation. Amyloid: Int. J. Exp. Clin. Invest. 1, 186–205 (1994).
Moyer, B. D. & Balch, W. E. A new frontier in pharmacology: the endoplasmic reticulum as a regulated export pathway in health and disease. Emerging Therap. Targets 5, 165–176 (2001).
Kelly, J. W. Amyloid fibril formation and protein misassembly: a structural quest for insights into amyloid and prion diseases. Structure 5, 595–600 (1997).
Kelly, J. W. The environmental dependency of protein folding best explains prion and amyloid diseases. Proc. Natl Acad. Sci. USA 95, 930–932 (1998).
Westermark, P., Sletten, K., Johansson, B. & Cornwell, G. G. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc. Natl Acad. Sci. USA 87, 2843–2845 (1990).
Saravia, M. J. M., Birken, S., Costa, P. & Goodman, D. S. Amyloid fibril protein in familial polyneuropathy, Portugese type: definition of molecular abnormality in transthyretin (prealbumin). J. Clin. Inv. 74, 104–119 (1984).
Selkoe, D. J. Alzheimer's disease: genotypes, phenotype and treatments. Science 275, 630–631 (1997).
Kiuru, S. Gelsolin-related familial amyloidosis, Finnish type (FAF), and its variants found worldwide. Amyloid 5, 55–66 (1998).
Hurle, M. R., Helms, L. R., Li, L., Chan, W. & Wetzel, R. A role for destabilizing amino acid replacements in light chain amyloidosis. Proc. Natl Acad. Sci. USA 91, 5446–5450 (1994).
Khurana, R. et al. Partially folded intermediates as critical precursors of light chain amyloid fibrils and amorphous aggregates. Biochemistry 40, 3525–3535 (2001).
Wanker, E. E. Protein aggregation in Huntington's and Parkinson's disease: implications for therapy. Mol. Med. Today 6, 387–391 (2000).
Wanker, E. E. Protein aggregation and pathogenesis of Huntington's disease: mechanisms and correlations. Biol. Chem. 381, 937–942 (2000).
Scherzinger, E. et al. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington's disease pathology. Proc. Natl Acad. Sci. USA 96, 4604–4609 (1999).
Conway, K. A., Harper, J. D. & Lansbury, P.T. Jr. Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson's disease are typical amyloid. Biochemistry 39, 2552–2563 (2000).
Goldberg, M. S. & Lansbury, P. T. Jr. Is there a cause-and-effect relationship between α-synuclein fibrillization and Parkinson's disease? Nature Cell Biol. 2, E115–E119 (2000).
Bruijn, L. I. et al. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281, 1851–1854 (1998).
Caughey, B. Transmissible spongiform encephalopathies, amyloidoses and yeast prions: common threads? Nature Med. 6, 751–754 (2000).
Prusiner, S. B. & DeArmond, S. J. Prion protein amyloid and neurodegeneration. Amyloid 2, 39–65 (1995).
Hammarstrom, P., Schneider, F. & Kelly, J. W. Trans-suppression of misfolding in an amyloid disease. Science 293, 2459–2461 (2001).This manuscript shows the molecular basis of interallelic trans-suppression of misfolding. Incorporation of T119M subunits into a tetramer also composed of disease-associated subunits stabilizes the tetramer and slows rate-limiting dissociation.
Selkoe, D. J. Amyloid β-protein and the genetics of Alzheimer's disease. J. Biol. Chem. 271, 18295–18298 (1996).
Schehr, R. S. Amyloid hypothesis of Alzheimer's rides high – for now. Nature Biotechnol. 15, 19–20 (1997).
White, J. T. & Kelly, J. W. Support for the multigenic hypothesis of amyloidosis: the binding stoichiometry of retinol-binding protein, vitamin A, and thyroid hormone influences transthyretin amyloidogenicity in vitro. Proc. Natl Acad. Sci. USA 98, 13019–13024 (2001).This publication shows that ligands that bind to amyloidogenic proteins can have a dramatic influence on amyloidogenicity. More than seventy gene products control the two ligands that bind to TTR.
Meyer, M. R. et al. APOE genotype predicts when – not whether – one is predisposed to develop Alzheimer disease. Nature Genet. 19, 321–322 (1998).
Roses, A. D. & Saunders, A. M. APOE is a major susceptibility gene for Alzheimer's disease. Curr. Opin. Biotechnol. 5, 663–667 (1994).
Hammarstrom, P., Jiang, X., Deechongkit, S. & Kelly, J. W. Anion shielding of electrostatic repulsions in transthyretin modulates stability and amyloidosis: insight into the chaotrope unfolding dichotomy. Biochemistry 40, 11453–11459 (2001).
Jimenez, J. L. et al. Cryo-electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. EMBO J. 18, 815–821 (1999).
Blake, C. & Serpell, L. Synchrotron X-ray studies suggest that the core of the transthyretin amyloid fibril is a continuous β-sheet helix. Structure 4, 989–998 (1996).
Serpell, L. C. et al. Examination of the structure of the transthyretin amyloid fibril by image reconstruction from electron micrographs. J. Mol. Biol. 254, 113–118 (1995).
Serpell, L. C. et al. The protofilament substructure of amyloid fibrils. J. Mol. Biol. 300, 1033–1039 (2000).
Sunde, M. et al. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 273, 729–739 (1997).
Lovat, L. B., Hohenester, E., Westermark, P., Wood, S. P. & Pepys, M. B. in Amyloid Amyloidosis 1998, Proc. 8th Int. Symp. Amyloidosis (eds Kyle, R. A. & Gertz, M. A.) 29–31 (Parthenon Publishing, New York, 1999).
Rydh, A. et al. Serum amyloid P component scintigraphy in familial amyloid polyneuropathy: regression of visceral amyloid following liver transplantation. Eur. J. Nucl. Med. 25, 709–713 (1998).
Klein, W. L., Krafft, G. A. & Finch, C. E. Targeting small Aβ oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci. 24, 219–224 (2001).
Volles, M. J. et al. Vesicle permeabilization by protofibrillar α-synuclein: implications for the pathogenesis and treatment of Parkinson's disease. Biochemistry 40, 7812–7819 (2001).
Colon, W. & Kelly, J. W. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry 31, 8654–8660 (1992).
Lai, Z., Colon, W. & Kelly, J. W. The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate which can self-assemble into amyloid. Biochemistry 35, 6470–6482 (1996).
McCutchen, S. L., Lai, Z., Miroy, G., Kelly, J. W. & Colon, W. Comparison of lethal and non-lethal transthyretin variants and their relationship to amyloid disease. Biochemistry 34, 13527–13536 (1995).
Jiang, X. et al. An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry 40, 11442–11452 (2001).
Lashuel, H. A., Lai, Z. & Kelly, J. W. Characterization of the transthyretin acid denaturation pathways by analytical ultracentrifugation: implications for wild-type, V30M, and L55P amyloid fibril formation. Biochemistry 37, 17851–17864 (1998).
Liu, K., Cho, H. S., Lashuel, H. A., Kelly, J. W. & Wemmer, D. E. A glimpse of a possible amyloidogenic intermediate of transthyretin. Nature Struct. Biol. 7, 754–757 (2000).
Booth, D. R. et al. Instability, unfolding and aggregation of human lysosozyme variants underlying amyloid fibrillogenesis. Nature 385, 787–793 (1997).
Wetzel, R. Mutations and off-pathway aggregation of proteins. Trends Biotechnol. 12, 193–198 (1994).
Dobson, C. M. Protein misfolding, evolution and disease. Trends Biochem. Sci. 24, 329–332 (1999).
Miroy, G. J. et al. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc. Natl Acad. Sci. USA 93, 15051–15056 (1996).
Peterson, S. A. et al. Inhibiting transthyretin conformational changes that lead to amyloid fibril formation. Proc. Natl Acad. Sci. USA 95, 12956–12960 (1998).
Klabunde, T. et al. Rational design of potent human transthyretin amyloid disease inhibitors. Nature Struct. Biol. 7, 312–321 (2000).Structure-based ligand design was used to create novel inhibitors, which were than characterized by X-ray crystallography.
Petrassi, H. M., Klabunde, T., Sacchettini, J. & Kelly, J. W. Structure-based design of N-phenyl phenoxazine transthyretin amyloid fibril inhibitors. J. Am. Chem. Soc. 122, 2178–2192 (2000).Structure-based ligand design was used to conceive of N-phenylphenoxazines as inhibitors of TTR amyloid-fibril formation.
Oza, V. B., Petrassi, H. M., Purkey, H. E. & Kelly, J. W. Synthesis and evaluation of anthranilic acid-based transthyretin amyloid fibril inhibitors. Bioorg. Med. Chem. Lett. 9, 1–6 (1999).
Baures, P. W., Peterson, S. A. & Kelly, J. W. Discovering transthyretin amyloid fibril inhibitors by limited screening. Bioorg. Med. Chem. 6, 1389–1401 (1998).A simple turbidity assay is described to identify TTR amyloid-fibril inhibitors by screening.
Baures, P. W., Oza, V. B., Peterson, S. A. & Kelly, J. W. Synthesis and evaluation of inhibitors of transthyretin amyloid formation based on the nonsteroidal anti-inflammatory drug flufenamic acid. Bioorg. Med. Chem. 7, 1339–1347 (1999).
Kelly, J. W. et al. Transthyretin quaternary and tertiary structural changes facilitate misassembly into amyloid. Adv. Protein Chem. 50, 161–181 (1997).
Krebs, M. R. H. et al. Formation and seeding of amyloid fibrils from wild-type hen lysozyme and a peptide fragment from the β-domain. J. Mol. Biol. 300, 541–549 (2000).
Chiti, F. et al. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl Acad. Sci. USA 96, 3590–3594 (1999).
Kirkitadze, M. D., Condron, M. M. & Teplow, D. B. Identification and characterization of key kinetic intermediates in amyloid-β-protein fibrillogenesis. J. Mol. Biol. 312, 1103–1119 (2001).
Allsop, D. et al. Modulation of β-amyloid production and fibrillization. Biochem. Soc. Symp. 67, 1–14 (2001).
Howlett, D. R. Aβ Oligomerization: a therapeutic target for Alzheimer's disease. Curr. Med. Chem. Immunol. Endocr. Metab. Agents 1, 25–38 (2001).
Bohrmann, B. et al. Self-assembly of β-amyloid 42 is retarded by small molecular ligands at the stage of structural intermediates. J. Struct. Biol. 130, 232–246 (2000).
Findeis, M. A. & Molineaux, S. M. Design and testing of inhibitors of fibril formation. Methods Enzymol. 309, 476–488 (1999).Evaluation methods are described to understand the mechanistic basis by which small molecules prevent the maturation of protein aggregates into amyloid fibrils.
LeVine, H. & Scholten, J. D. Screening for pharmacologic inhibitors of amyloid fibril formation. Methods Enzymol. 309, 467–476 (1999).
Moore, C. L. & Wolfe, M. S. Inhibition of β-amyloid formation as a therapeutic strategy. Expert Opin. Ther. Pat. 9, 135–146 (1999).
Bandiera, T., Lansen, J., Post, C. & Varasi, M. Inhibitors of Aβ peptide aggregation as potential anti-Alzheimer agents. Curr. Med. Chem. 4, 159–170 (1997).
Glenner, G. G. et al. Creation of amyloid fibrils from Bence Jones proteins in vitro. Science 174, 712–714 (1971).
Shirahama, T. & Cohen, A. S. Intralysosomal formation of amyloid fibrils. Am. J. Pathol. 81, 101–116 (1975).
Shirahama, T. et al. Amyloid enhancing factor-loaded macrophages in amyloid fibril formation. Lab. Inv. 62, 61–68 (1990).
Yamazaki, T., Koo, E. H. & Selkoe, D. J. Trafficking of cell-surface amyloid β-protein precursor II. Endocytosis, recycling, and lysosomal targeting detected by immunolocalization. J. Cell Sci. 109, 999–1008 (1996).
Yang, A. J., Chandswangbhuvana, D., Shu, T., Henschen, A. & Glabe, C. G. Intracellular accumulation of insoluble, newly synthesized Aβ n–42 in amyloid precursor protein-transfected cells that have been treated with Aβ 1–42. J. Biol. Chem. 274, 20650–20656 (1999).
Jiang, X., Buxbaum, J. N. & Kelly, J. W. The V122I cardiac variant of transthyretin increases the velocity of rate-limiting tetramer dissociation resulting in accelerated amyloidosis. Proc. Natl Acad. Sci. USA 98, 14943–14948 (2001).
Liu, K. et al. Deuterium–proton exchange on the native wild-type transthyretin tetramer identifies the stable core of the individual subunits and indicates mobility at the subunit interface. J. Mol. Biol. 303, 555–565 (2000).
Purkey, H. E., Dorrell, M. I. & Kelly, J. W. Evaluating the binding selectivity of transthyretin amyloid fibril inhibitors in blood plasma. Proc. Natl Acad. Sci. USA 98, 5566–5571 (2001).
Oza, V. B. et al. Synthesis, structure and activity of diclofenac analogs as transthyretin amyloid fibril inhibitors. J. Med. Chem. 45, 321–332 (2002).
Suhr, O. B. Transplantation for amyloidosis. Rinsho Byori 48, 329–335 (2000).
Anderson, O. & Wallin, G. in Second International Symposium On Familial Amyloidotic Polyneuropathy and Other Transthyretin Related Disorders 49 (Skelleftea, Sweden, 1992).
Wei, Y. et al. Disruption of protein–protein interactions: design of a synthetic receptor that blocks the binding of cytochrome c to cytochrome cperoxidase. J. Chem. Soc. Chem. Commun. 1580–1581 (2001).
Schenk, D. et al. Immunization with amyloid-β attenuates Alzheimer disease-like pathology in the PDAPP mouse. Nature 400, 173–177 (1999).Describes the immunization approach to the amelioration of Alzheimer's-associated amyloid disease.
Lee, V. M. Y. Aβ immunization: moving Aβ peptide from brain to blood. Proc. Natl Acad. Sci. USA 98, 8931–8932 (2001).
Coelho, T. et al. A strikingly benign evolution of FAP in an individual found to be a compound heterozygote for two TTR mutations: TTR Met30 and TTR Met119. J. Rheumatol. 20, 179–179 (1993).
Coelho, T. et al. Compound heterozygotes of transthyretin Met30 and transthyretin Met119 are protected from the devastating effects of familial amyloid polyneuropathy. Neuromusc. Disord. 6, 27 [AU: 1 pge OK?] (1996).
Koepf, E. K. et al. Characterization of the structure and function of W to F WW domain variants: identification of a natively unnfolded protein that folds upon ligand binding. Biochemistry 38, 14338–14351 (1999).
Weggen, S. et al. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 414, 212–216 (2001).
Kim, S.-H. et al. Furin mediates enhanced production of fibrillogenic ABri peptides in familial British dementia. Nature Neurosci. 2, 984–988 (1999).
Fan, J.-Q. Potential drug therapies for lysosomal storage disorders. Front. Biotechnol. Pharmacol. 2, 275–291 (2001).
Fan, J.-Q., Ishii, S., Asano, N. & Suzuki, Y. Accelerated transport and maturation of lysosomal α-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nature Med. 5, 112–115 (1999).Misfolding and mistransport can be corrected by small molecules that bind to, and mediate proper folding of, proteins. This approach was used to correct Fabry's disease – a lysosomal storage disorder.
Perrier, V. et al. Mimicking dominant negative inhibition of prion replication through structure-based drug design. Proc. Natl Acad. Sci. USA 97, 6073–6078 (2000).
Kisislevsky, R. et al. Arresting amyloidosis in vivo using small molecule anionic sulfates or sulfonates: implications for Alzheimer's disease. Nature Med. 1, 143–148 (1995).
Ancsin, J. B. & Kisilevsky, R. The heparin/heparan sulfate-binding site on APO-serum amyloid A. Implications for the therapeutic intervention of amyloidosis. J. Biol. Chem. 274, 7172–7181 (1999).
Ancsin, J. B. & Kisilevsky, R. in Amyloid Amyloidosis 1998, Proc. 8th Int. Symp. Amyloidosis (eds Kyle, R. A. & Gertz, M. A.) 77–79 (Parthenon Publishing, New York 1999).
Morgan, D. et al. Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature 408, 982–985 (2000).
Peretz, D. et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 412, 739–743 (2001).
Acknowledgements
J.W.K. thanks the members of his laboratory, who made the discoveries outlined in this review possible, and appreciates the financial support from the National Institutes of Health, The Skaggs Institute of Chemical Biology and the Lita Annenberg Hazen Foundation. J.C.S. thanks his laboratory and the Robert A. Welch Foundation for financial support.
Author information
Authors and Affiliations
Related links
Related links
DATABASES
LocusLink
OMIM
familial amyloidosis of Finnish
familial amyotrophic lateral sclerosis
haemodialysis-related amyloidosis
hereditary cerebral amyloid angiopathy
transthyretin-based amyloid diseases
FURTHER INFORMATION
Encyclopedia of Life Sciences
LINKS
Glossary
- EXTRACELLULAR MATRIX
-
A network of polysaccharides and proteoglycans that templates an assembly of cells to assist in function and impart strength.
- ENDOPROTEOLYTIC PROCESSING
-
Refers to proteolytic cleavage of internal amide bonds in a protein — that is, all amides except those that connect the carboxy- and amino-terminal residues.
- AMYLOIDOGENIC
-
Refers to a property of a protein that allows it to form amyloid fibrils in a human.
- TRANSTHYRETIN
-
(TTR.) An amyloidogenic plasma protein that normally transports the retinol-binding protein–vitamin A complex and thyroxine.
- MULTIGENIC DISEASE
-
A disease with a course that is influenced by the expression of more than one gene.
- DENATURATION STRESS
-
A change in the aqueous environment of a protein, such as the pH or the dielectric constant, that causes it to denature.
- QUATERNARY STRUCTURE
-
The association of proteins in a geometrically specific manner through non-covalent interactions.
- FREE-ENERGY MINIMUM
-
Refers to the most stable conformation of a macromolecule.
- ENDOCYTIC PATHWAY
-
The vesicular pathway by which molecules are taken into a cell.
- ANALYTICAL ULTRACENTRIFUGATION
-
A method for determining quaternary structure that uses a centrifuge fitted with a UV detector to quantify concentration gradients.
- STOICHIOMETRY
-
Refers to molar proportions in a reaction or assembly.
- MULTIDENTATE
-
A binding interaction in which one partner can bind to two or more sites on another partner.
- HYDROPHOBIC INTERACTION
-
A favourable interaction between nonpolar substructures or surfaces in aqueous solution that is characterized by a large change in heat capacity.
- ELECTROSTATIC INTERACTION
-
A non-covalent dipole–dipole or induced dipole–dipole interaction that can be stabilizing or destabilizing.
- SEEDED POLYMERIZATION
-
A mechanism of polymerization in which the quaternary structure or seed is formed in a rate-determining fashion. This is followed by rapid addition of monomer to afford a high-molecular-weight assembly such as an amyloid fibril.
- MIMETIC
-
A molecule that mimics another.
- SULPHONATED AROMATICS
-
An aromatic ring that is functionalized with a sulphonate functional group.
Rights and permissions
About this article
Cite this article
Sacchettini, J., Kelly, J. Therapeutic strategies for human amyloid diseases. Nat Rev Drug Discov 1, 267–275 (2002). https://doi.org/10.1038/nrd769
Issue Date:
DOI: https://doi.org/10.1038/nrd769
This article is cited by
-
Accessing Methyl Groups in Proteins via 1H-detected MAS Solid-state NMR Spectroscopy Employing Random Protonation
Scientific Reports (2019)
-
Designed peptides that assemble into cross-α amyloid-like structures
Nature Chemical Biology (2018)
-
EGCG-mediated Protection of the Membrane Disruption and Cytotoxicity Caused by the ‘Active Oligomer’ of α-Synuclein
Scientific Reports (2017)
-
Detergent-induced aggregation of an amyloidogenic intrinsically disordered protein
Journal of Chemical Sciences (2017)
-
A cytotoxic amyloid oligomer self-triggered and NIR-enhanced amyloidosis therapeutic system
Nano Research (2015)
