
Key Points
-
New impetus has been given to antiparasitic drug discovery by the sequencing of several parasite genomes, and by the establishment of new public–private partnerships (PPPs) whose focus is specifically on tropical diseases.
-
New drugs for parasitic diseases need to meet product profiles that are defined in conjunction with those responsible for control programmes in the affected countries, and reflect what is required for use in resource-poor settings.
-
Compound screening against selected molecular targets is increasingly being used in the search for new chemical leads. The quality of the compound libraries being assessed is a key factor in determining success.
-
The majority of successful drugs have come from either evaluating known classes of active compounds or from discovering new indications for known drugs. This approach continues to be an important part of any discovery programme for antiparasitic drugs.
-
Lead compounds need to be optimized so that they have the characteristics required to meet product profiles. As in most drug discovery projects, this is often the rate-limiting step. Compounds will need to be tested in several animal models with careful consideration given to formulation.
-
The recent entry of a new synthetic peroxide antimalarial into clinical trials illustrates how public funding for a diverse set of early-stage discovery projects led to a set of good lead compounds that were selected for further development by a PPP. Early-stage discovery research needs further strengthening and will require financial support from public sources.
-
Antiparasitic drug discovery and development is not primarily market-driven, and so partners from organizations with differing cultures and underlying objectives need to work together. The role in such partnerships of researchers, public-health officials and industry leaders from disease-endemic countries needs to be increased.
Abstract
New antiparasitic drugs are urgently needed to treat and control diseases such as malaria, leishmaniasis, sleeping sickness and filariasis, which affect millions of people each year. However, because the majority of those infected live in countries in which the prospects of any financial return on investment are too low to support market-driven drug discovery and development, alternative approaches are needed. In this article, challenges and opportunities for antiparasitic drug discovery are considered, highlighting some of the progress that has been made in recent years, partly through scientific advances, but also by more effective partnership between the public and private sectors.
Similar content being viewed by others
Main
Parasitic diseases continue to take an enormous toll on human health, particularly in tropical regions. The major burden is caused by the PROTOZOA and HELMINTHS listed in Table 1. The drugs used to treat these diseases are far from ideal, and many of them were introduced decades ago. Problems associated with some of the commonly used drugs are noted in the Table 1. As many authors have emphasized, market forces are insufficient to drive the discovery and development of new drugs for these diseases. Of more than 1,300 new drugs introduced for all indications between 1975 and 1999, only 13 were for TROPICAL DISEASES such as those listed in Table 11. In 2000, only about 0.1% of global investment in health research was devoted to drug discovery for selected tropical diseases (malaria, leishmaniasis and trypanosomiasis) and tuberculosis, which together contribute about 5% of the global disease burden2,3.
Several welcome developments during the past few years have given new impetus to antiparasitic drug discovery. These include the publicly-funded sequencing of the genomes of several of the parasites in Tables 1,2, and the establishment of new public–private partnerships (PPPs) whose focus is specifically on tropical diseases4,5, counteracting, at least to some extent, the withdrawal of many large pharmaceutical companies from direct involvement in antiparasitic drug discovery. The injection of new funds into the area of antiparasite research, particularly by the Bill and Melinda Gates Foundation, is also having a significant impact. Some PPPs currently involved in antiparasite drug discovery are the Medicines for Malaria Venture (MMV), the Drugs for Neglected Diseases initiative (DNDi) and the Institute for One World Health (IOWH)4. These PPPs combine a pharmaceutical industry-derived approach to drug discovery and development with the disease-specific knowledge and experience of public healthcare organizations.
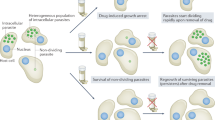
In light of these opportunities, we discuss some challenges to the discovery of new antiparasitic drugs, defined as the work leading up to the definition of a drug development candidate (Fig. 1). Drug discovery is an iterative process which, depending on the strategy used, typically comprises several discrete stages: target identification and validation; assay development; screening (whole cell or molecular target-based) to identify hits (Box 1); procurement/synthesis and assessment of analogues to develop structure–activity relationships (SAR) and identify leads; iterative medicinal chemistry to optimize leads; and preclinical development prior to clinical evaluation. We describe different approaches to antiparasitic drug discovery, discuss the promise of high-throughput screening (HTS) on new molecular targets and emphasize the importance of lead optimization. Finally, we mention the contributions that different types of partnership can make to the discovery process.

Drug discovery is an iterative process involving discrete stages. This often begins with basic exploratory biology and biochemistry to identify molecular targets. In other cases compounds are tested, without knowledge of the target, for activity against the whole parasite. Compounds (actual or potential inhibitors) are assayed for activity against the target, if known, and for activity against the whole parasite. (Inhibitors of the target are often used to validate the target.) Compounds active against the whole parasite are defined as hits (see Box 1) that can be considered for further testing in animal models of the disease. Other tests that monitor the compounds' pharmacokinetic properties are also initiated at this stage. Compounds that are active in the animal models and considered to be 'druggable' are defined as leads (see Box 1). Lead compounds generally require optimization for efficacy and good pharmaceutical properties. Note the importance of medicinal chemistry in both identifying an appropriate lead molecule and in the more time-consuming iterative process (thick arrows) of lead optimization. Early pharmacokinetic studies are also emphasized in this diagram. The process of optimization for pharmaceutical properties (adsorption, distribution, metabolism and excretion (ADME)) and lack of overt drug toxicity, as well as for efficacy against the target organism, is crucial. Once a compound reaches the stage at which it can be considered for testing in human patients, it is defined as a drug candidate. From there it enters the preclinical and then clinical studies of a typical drug development pathway. Adapted from Ref. 4.
Challenges in antiparasitic drug discovery
Drug discovery for parasitic diseases is not intrinsically more costly or technically demanding than for other indications. Generally, for infectious (including parasitic) diseases, preclinical models tend to be more predictive, and clinical trials less complex and costly, than for non-infectious, chronic disorders. It has been estimated that the cost of bringing a new antimalarial to market is about US$300 million, compared with the cost for a new drug averaged over all indications of at least US$500 million. The risk of failure in Phase II clinical trials is estimated to be 50% for a new antimalarial, which is lower than the corresponding risk for a non-infectious disease4,6.
Antiparasitic drug discovery is not primarily driven by the commercial need to introduce novel compounds. Historically, many antiparasite drugs were first developed for other indications. This opportunistic approach of capitalizing on knowledge gained from work on non-parasitic indications has been very successful, as described in the next section, and has clear advantages in terms of cost reduction. However, the approach does not favour the introduction of chemically novel agents and might be reaching a point of diminishing returns, particularly as a result of widespread resistance to certain drug classes. It does not fully exploit new knowledge of parasite genome sequences, leading to the view that “the next big challenge in tropical diseases is determining the best way to translate the insights obtained from genomics into new, robust chemical leads that can form the basis of innovative drug discovery”4.
A second major challenge in this arena is that multiple organizations with vastly differing cultures and underlying objectives need to work together. As a result of the abandonment of in-house discovery research for antiparasitics by many large pharmaceutical companies, a key factor in drug development for neglected diseases has been the formation of effective partnerships for 'virtual drug discovery'4,5,7. Much progress in recent years has been made by effectively re-engaging the private biopharmaceutical industry in the effort. Public support for early drug discovery feeds into the drug development projects undertaken by industry in partnership with public-health organizations. Increasing the role in such partnerships of researchers, public-health and industry leaders in the disease-endemic countries remains a challenge. Some examples of partnerships of different types are discussed below.
Finally, for the 'neglected' diseases, drug discovery is principally field-driven — that is, designed to meet the needs of disease-control programmes in the field. This generally means an emphasis on low cost of goods, short treatment regimes and the ability to use the drug safely in the absence of close medical supervision. It is therefore vitally important to define, in conjunction with those responsible for control programmes in the affected countries, desired product profiles based on what is required for use in resource-poor settings (Table 3). Optimizing lead compounds so that they have the characteristics required to meet product profiles is the rate-limiting factor in preclinical drug development.
Approaches to drug discovery
Different basic approaches to drug discovery for tropical diseases, as reviewed recently for malaria8 or tuberculosis9, can be classed as short-to-medium term (based on exploiting existing compounds or compound classes) or long-term (requiring discovery of new chemical classes). The approaches we describe are illustrated by examples, with emphasis on the diseases in Table 1 other than malaria. They aim at the discovery of pure chemical entities: the possible advantages of, and problems associated with, encouraging the wider use of traditional medicines, particularly for malaria, have been discussed elsewhere10,11.
Combinations of existing drugs. Combinations of existing drugs (Table 1), such as eflornithine and melarsoprol for African trypanosomiasis12, or praziquantel and oxamniquine for schistosomiasis13, offer possibilities of synergy, reduced toxicity, shorter treatment regimens and slowing the development of resistance. In particular, extensive use of drug combinations is being made in malaria therapy8, and the rationale for this has been recently reviewed14. Wherever possible, fixed-dose combinations are being developed to increase patient compliance, particularly using artemisinin-like compounds as one of the components. Combinations of an antimalarial drug with a RESISTANCE REVERSER are also being considered8.
New indications for existing drugs. An attractive short-term strategy offering major savings in development time and expense involves extending the indications of drugs that were first developed for other indications. Historically, this 'piggy-back' approach has been very successful; many antiparasite drugs first entered development for other indications (see case histories in Table 4). For example, DB289, which was initially used to treat Pneumocystis pneumonia, is now in clinical trials as a potential oral treatment for malaria and early-stage African trypanosomiasis15,16. A downside of this strategy can be the reluctance of pharmaceutical companies to allow their products to be tested in a non-commercial patient class and so risk uncovering toxicities that might blight the economic potential of their drugs. For example, several companies have been reluctant to permit clinical trials of antifungal triazoles against Chagas' disease, in spite of their demonstrated activity against Trypanosoma cruzi in animal models17.
Improvements to known drugs and compound classes. In the medium-term, analogues of existing antiparasitics (Table 5) might prove effective. For malaria, novel analogues of pyrimethamine are being specifically designed to overcome drug resistance resulting from mutations in dihydrofolate reductase18, whereas analogues of amodiaquine with potentially reduced toxicity are being investigated19. Ferroquine contains a quinoline nucleus similar to chloroquine but with a novel ferrocenic group in its side chain. It has excellent activity against malaria parasites, including those resistant to chloroquine20. In the anthelmintic area, moxidectin, an analogue of ivermectin, is being pursued for the treatment of lymphatic filariasis and onchocerciasis21. This compound, already licensed as a veterinary product, has very different pharmacokinetic properties from ivermectin, and this difference is expected to result in improved efficacy against onchocerciasis and other FILARIAL infections.
Focused sample collections. An alternative, and arguably a more productive, approach to screening large libraries of compounds against whole parasites (see below) is to screen focused sample collections. Here the emphasis is on identifying compounds with either defined biological effects against related parasites, or biochemical activity against isoenzymes or receptors related to known molecular targets of other organisms. This strategy is particularly important in the search for new antifilarials and schistosomicides, for which screening capacity is limited by the supply of relevant test helminths. For example, in laboratories funded by the United Nations Children's Fund/United Nations Development Programme/World Bank/World Health Organization Special Programme for Research and Training in Tropical Diseases (TDR), it has only been possible to evaluate some 1,000 samples per year against Onchocerca and SCHISTOSOMES. To improve the quality of the test compounds, efforts are being made to source samples with existing anthelmintic properties — for example, from crop protection and animal-health companies.
Work on parasite genome sequences coupled with biochemical investigations has pinpointed enzymes such as protein farnesyl transferases, cysteine proteases, histone deacetylases and fatty-acyl synthases (Table 6) as potential drug targets for malaria and TRYPANOSOMATID diseases. Investigators working in both academia and the pharmaceutical industry have established compound collections focused on inhibitors of such enzyme classes. Opportunities to access these compounds and evaluate them for their antiparasitic activity need to be exploited, as the compounds can be a valuable source of new leads. As an example, MMV has in its portfolio a lead-optimization project based on inhibitors of protein farnesyl transferase that originated with compounds from Yale University and from a cancer chemotherapy programme at Bristol-Myers Squibb22.
De novo discovery: whole parasite assays
Longer-term strategies aim to discover novel active substances unrelated to known drugs. In the biopharmaceutical industry, molecular target identification and HTS dominate much of the early drug discovery process. However, for the parasitic diseases there has been, and still is, a valuable alternative approach based on screening and analysing compounds for their activity against whole parasites. Screening diverse compound collections on whole parasites in vitro has been steadily declining during the past two decades but is now undergoing a renaissance, due mainly to assay improvements. This is particularly true for test systems using Plasmodium falciparum, T. brucei, T. cruzi and Leishmania species. Screening now often relies on the use of parasites transfected with reporter genes, such as those encoding green fluorescent protein, β-lactamase, or β-galactosidase, to enable easy, rapid detection of antiparasitic activity. Some progress has also been made in adapting protozoal screens to work in 384-well plates, especially with P. falciparum.
One recent example in which the capacity to test large numbers of compounds against whole parasites has been developed is at the Harvard University Institute of Chemistry and Cell Biology, Initiative for Chemical Genetics (ICCB-ICG), where whole parasites are used in an HTS format to assess tens of thousands of samples (see Further information). The Belgian company Tibotec, working with support from TDR, also developed assays based on whole parasites in 384-well plates in order to analyse relatively large numbers of compounds. For example, in one project, Tibotec analysed 10,000 compounds (purchased from a commercial compound supplier) for activity against four parasites (P. falciparum, T. brucei, L. donovani and T. cruzi) and a mammalian cell line (to assess cytotoxicity). Numerous active compounds were detected and further investigated by the TDR network of drug discovery laboratories. The hits were subjected to more detailed analysis, involving accurate IC50 determinations against whole parasites, measurement of general cytotoxicity and, for the most promising compounds, in vivo assessment in animal models of the relevant parasitic infection. This led to the identification of one compound as a novel lead active against malaria. Further optimization via analogue synthesis is now required to try to identify a credible development candidate. These data illustrate the high rate of attrition in lead identification and the need to screen large compound collections against whole cells in order to have a reasonable chance of success.
The quality of the compound libraries being assessed is a key factor in determining the success rate of screening (see also below). Screening libraries of natural products has special advantages for parasitic diseases, as well as other infectious diseases and cancer. Natural products are attractive because their structural diversity is remarkable and, for the parasitic diseases in particular, medicinal plants can be potential sources of novel pharmacophores23. For example, the antimalarial artemisinins were first isolated from a traditional Chinese medicine24. Other natural products whose chemistry is currently being explored in focused drug discovery programmes include manzamines25, chalcones26 and borrelidins27, all of which have antimalarial activity in animal models. Large-scale screening of natural products against other parasites has been less exploited. The possibilities (and problems) of this approach are illustrated by PX-6518, a glycosylated saponin whose antileishmanial activity was detected in a screen of some 10,000 plant extracts, but whose development was stopped due to toxicity concerns28. This case illustrates the difficulties in pursuing natural products, which are often chemically complex and provide few opportunities for rapidly investigating SAR by synthesising or procuring analogues. Converting the original natural product to a metabolically robust, orally bioavailable drug can be extremely challenging, with very long time lines. Because many natural products are produced as biological defence mechanisms, cytotoxicity is also a common problem. Many plant products are produced both at specific times in the growing cycle and in different parts of the organism, and this can lead to difficulties in sourcing sufficient material for study. Nevertheless, because of the amazing diversity of plant species and the heavy reliance on herbal remedies in tropical/subtropical disease-endemic countries, pursuing natural products as antiparasitics remains an attractive proposition.
De novo discovery: molecular targets and HTS
Although HTS against molecular targets has become the preferred mode of early drug discovery for much of the biopharmaceutical industry, it is has only recently been used to any wide extent in the search for new drugs for the neglected parasitic diseases. This strategy is expected to increase in importance as the genome sequences of the relevant parasites become available, and as HTS facilities and compound libraries become more accessible to research groups in academia.
Target identification and validation. Access to parasite genome sequences offers exciting opportunities for drug discovery based on the identification and validation of new molecular drug targets. The numbers (hundreds of thousands) of compounds that can be screened in a typical HTS campaign based on a molecular target far exceed the throughput possible using assays based on whole-cell parasite viability. This is particularly true for screening against helminths, for which throughput in whole-parasite assays is at least ten times less than for protozoa, which therefore makes the identification and validation of molecular targets for helminths an especially important and valuable approach for de novo drug discovery.
Nevertheless, one should not overestimate the number of suitable drug targets that parasite genomes could encode. The Plasmodium genome contains about 5,000 genes, of which Yeh et al.29 estimated about 200 (4%) might encode suitable drug targets, using a computational algorithm that identified enzymes that catalyse 'chokepoint' reactions (those that uniquely either consume a specific substrate or produce a specific product). Among these were about 30 that were not significantly similar to any human enzyme. This compares favourably with Hopkins and Groom's estimate30 that less than 1,500 (5%) of the ∼30,000 genes in the human genome are suitable drug targets, using quite different criteria.
Experience with the genome-based discovery of new antibacterials also suggests that enthusiasm for this approach should be based on a realistic view of its limitations. None of the 18 new antibacterials now in clinical trials were discovered through a genomics programme31,32. Numerous potential targets have been identified and explored, but the limiting factor in developing new antibacterials is clearly not the characterization of compounds that are active against new targets. Rather, the limiting factor is the conversion of such compounds into drug candidates that are optimized not only for activity but also for other desirable pharmaceutical and physicochemical properties33,34,35.
Ideally, targets selected for a screening campaign should be genetically and/or chemically validated, biochemically and structurally characterized, open to selective inhibition without a tendency for the parasite to develop resistance, and technically amenable to screening large numbers of compounds (Box 2). Some parasite-specific points should be noted. First, parasite species that differ from the human pathogen are commonly used in validating hits and searching for leads (Table 7). For example, P. berghei is widely used in animal models of malaria. This requires consideration of homologous targets from different parasite species, as discussed in the next section. Second, development of resistance (for example, to dihydrofolate reductase) is well documented and the resistance potential of parasites to new chemical leads should therefore be investigated early in development. Third, kinetic factors can strongly affect target suitability: for example, inhibition of trypanosomal ornithine decarboxylase might only be successful because the turnover rates of the human and parasite enzymes differ such that the parasite is unable to regenerate enzyme fast enough to survive its irreversible blockade36. This example highlights the importance of a detailed knowledge of parasite biochemistry in target selection.
Potential parasite drug targets are being validated using chemical (Table 6) or genetic methods (for example, gene-expression profiling following drug treatment, RNA interference (RNAi) or genetic knockout techniques) for most of the protozoa in Table 1. About 20 potential targets with known inhibitors have been identified for P. falciparum29, fewer for other parasites (Table 6). In vitro culture and manipulation of the helminths in Table 1 is technically more demanding than for protozoa. The use of the readily available, free-living NEMATODE Caenorhabditis elegans as a model organism37, coupled with RNAi methodology, has been recommended for systematic identification of new targets in Onchocerca and Brugia species38.
HTS and compound libraries. Examples of the relatively few HTS campaigns that have been conducted using parasite enzymes include lactate dehydrogenase, peptide deformylase, glyceraldehyde-3-phosphate dehydrogenase, enoyl-ACP reductase (Fab I) and trypanothione reductase. Not all of these campaigns have yielded hits worth pursuing and several of these targets have since been downgraded — for example, lactate dehydrogenase (Table 6). As more parasite molecular targets are identified from genomics programmes, the use of HTS campaigns is expected to increase. The choice of which compound collections to screen is crucial. If the target is related to one already being pursued by pharmaceutical companies for other indications — for example, protein kinases relevant to oncology — then it would be feasible and desirable to conduct the campaign using a small, focused compound collection (perhaps 500–1,000 compounds) based around chemical scaffolds known to provide inhibitors of such protein classes. However, in situations in which the target has not been well studied, then recourse must be made to screening large, diverse compound or natural product libraries, often numbering in excess of 100,000 samples. The advent of commercial suppliers of large compound collections now enables academic institutions to organize screening campaigns using either focused or diverse collections. In all cases chemical libraries should undergo rigorous triaging to ensure 'drug-likeness' and to eliminate compounds likely to be generally toxic, mutagenic, highly reactive, unstable or intractable to chemical modification. A surprising number of non-drug-like molecules still permeate many libraries, due to the difficulty of writing general formulae that will ensure their detection and removal without eliminating many other useful compounds. For example, MICHAEL ACCEPTORS are often present and in general these can be expected to be susceptible to nucleophilic attack, thereby rendering them nonspecific in their biological actions. However, this class contains some notable exceptions such as the vinylsulphones. Here at least one member of this potent series of parasite cysteine protease inhibitors has good target specificity and is being developed under the auspices of IOWH for treatment of Chagas' disease39 (Table 6). In addition, because of their common use as synthetic intermediates, most libraries also contain aromatic or heterocyclic nitro compounds. These compound types are undesirable for screening against parasites, as they often show good activity, particularly against protozoa, due to bio-reduction and formation of reactive free radicals. However, such properties are also commonly associated with mutagenicity, and on balance it seems reasonable to exclude these compounds from screening libraries.
The creation of chemical libraries has been greatly furthered by advances in new technologies relating to combinatorial and parallel synthesis40. Such libraries can be based on proprietary or non-proprietary compounds. Companies might have patents, or be seeking patent protection, on proprietary compounds in their libraries. This issue is important because several pharmaceutical companies are now allowing academia-driven HTS campaigns to be conducted against parasite proteins using their sample collections. Difficulties can arise when the chemical structures of the hits need to be released for further study if such compounds belong to commercially sensitive chemical series. Such problems generally do not arise when the compounds in the library have been purchased from commercial suppliers and are considered non-proprietary. It will still be advisable, however, to run patent searches on compounds that are being considered as the basis of a lead-optimization programme, to gain information on the chemical class as well as to note any claims that could interfere with future commercial development.
Moving from hits to leads to drug candidates
The progression from 'hit' to 'lead' to 'drug candidate' (Fig. 1; Box 1) follows the same general pattern for the discovery of antiparasitics as for other drugs. Compounds are selected for improved efficacy and pharmaceutical properties by studies of analogues and iterative medicinal chemistry. The structure of the target molecule, if known, can be very helpful in directing medicinal chemistry efforts18. An advantage for the discovery of new antiparasitic drugs is the existence of good, highly predictive in vitro and in vivo assays for activity, which often use the same parasitic organism that infects the human patient. However, protocols are not standardized and it is often not straightforward to compare results from different laboratories. Techniques are always evolving and are also being adapted to achieve higher throughput (for example, by the use of reporter genes to allow fluorescence-based assays to be used instead of microscopy). A recent paper has reviewed the models used for antimalarial drug discovery and has recommended a streamlined process for evaluating new compounds41. Table 7 lists some of the commonly used in vitro and in vivo models in antiparasitic disease discovery.
Although the parasitic strains used in laboratory tests are often the same or very similar to those infecting the human patient there are certain cases where important differences exist. The standard animal models for malaria infection use P. berghei, P. chabaudi, P. yoelii or (less often) P. vinckei rather than the Plasmodium species that infect humans. The T. brucei brucei parasites used in the initial African trypanosomiasis tests differ from the T. b. rhodesiense and gambiense subspecies that cause human disease. The Onchocerca gutturosa worms used as in vitro models for onchocercal infection are parasites of cattle rather than humans. These differences can be especially crucial when a molecular target-based drug discovery strategy is followed; for example, the cysteine proteases of P. vinckei (vinckepains) differ from those of P. falciparum (falcipains), so that both types of protease had to be expressed and studied in a falcipain-based programme42. Genetically modified parasites in which the pathogen target replaces the model target gene would be one solution to this problem.
An important consideration in choosing the appropriate animal model is the desired product profile (Table 3). Most of the diseases require testing in several types of animal model. The primary models generally reflect the acute form of a disease, whereas the secondary, more complex assays represent the chronic or drug-resistant disease for which treatment is being sought. For example, compounds would not be considered useful leads for late-stage African trypanosomiasis or chronic Chagas' disease unless they showed activity in the corresponding secondary infection model (Table 7). The primary models also serve to filter out compounds before entering the time-consuming and expensive chronic tests, in which infected animals are typically followed for 6 weeks or longer.
The helminth models offer particular challenges. In vitro assays of antischistosome activity are not standardized, and it is not clear how well these will predict activity in animal models. For most in vitro helminth screens, readout is based on modulation of parasite motility, although dye reduction can be used as a secondary criterion to assess worm viability. There is reasonable, but not complete, correlation between the motility and dye reduction readouts. Genetically modified C. elegans might increase throughput in early discovery. In addition, for Onchocerca, the primary animal model is based on microfilariae and not on the adult worms, yet the main need is a drug that would act against the adult worms (macrofilariae). Time-consuming and relatively expensive secondary assays are required to select good lead compounds for onchocerciasis or lymphatic filariasis.
For all the studies in animals, the impact of formulation on the activity of compounds needs to be considered. In order to give compounds the maximum chance of success (seeing at least some activity), it is usual to initially test compounds in water-based formulations containing 10% dimethylsulphoxide (DMSO). This can have a significant impact on oral bioavailability, particularly with water-insoluble compounds. DMSO-containing formulations are not normally acceptable for the assessment of toxicity and it is important to retest active compounds in non-DMSO-containing vehicles, such as 'standard suspending vehicle' (SSV)43.
Importance of lead optimization. As outlined in Fig. 1, lead optimization is an iterative process in which medicinal chemistry is used to design and synthesize new compounds, and these are evaluated for improved properties. Increasingly, as the cycle depicted in Fig. 1 is traversed, costs escalate and time frames expand, with the lead-optimization stage being the point most crucial in constraining the drug discovery process. This stage is essentially an exercise in problem-solving in which bioavailability, metabolic and toxicity considerations come into play in the selection of a robust drug candidate. It is at this stage that involvement of the pharmaceutical industry becomes highly desirable. Lead optimization has been the most disregarded and under-funded section of the antiparasite drug discovery process during the past two decades. The advent of PPPs has dramatically improved the situation in recent years by re-engaging the pharmaceutical industry and by providing funding.
An example highlighting the role of lead optimization in producing a drug candidate is that of the antimalarial synthetic peroxide OZ277 (RBx-11160), which has now entered clinical trials44. Under the TDR 'malperox' programme45, meetings were organized for TDR-funded biologists and chemists to discuss the key issues in the optimization of artemisinin-like compounds. More than 1,000 semi-synthetic and synthetic artemisinins from at least seven different laboratories were evaluated. Biological data were reported back to the chemists to assist them in the optimization process. One of the most promising projects concerned a series of synthetic peroxides. Since 2000 this project has been funded by a newly formed PPP, MMV. Under MMV, a strong 'virtual' discovery team was established that links chemists in the United States with parasitologists in Switzerland and pharmacokineticists in Australia. An optimized drug candidate was selected in 2003 and taken to an Indian pharmaceutical company (Ranbaxy) for scaled-up production to provide the material now being assessed in clinical trials44.
This scenario illustrates how public funding for a diverse set of early-stage discovery projects led to a set of good lead compounds that were selected for further development by a PPP. The PPP managed the subsequent successful development of the lead compound into a drug candidate by using its funding power and its ability to bring together the necessary expertise, particularly from the pharmaceutical sector.
Partnerships for drug discovery
The role of PPPs in drug development for neglected diseases has been extensively discussed4,5,46,47. They have had encouraging successes in moving drug candidates into clinical trials, and in stimulating discussion of how industry can contribute to this process48. However, drug discovery is more risky than development, and the PPPs in Table 6 have adopted portfolio approaches that often emphasize development. Basic research and early discovery research are supported primarily by public funds. Early discovery research can involve collaborations between different public institutions as well as public–private agreements. We give some examples below of collaborative early discovery research supported by public funds, and in some cases also by in-kind contributions from industry.
The sequencing of parasite genomes involved the establishment of international genome networks (Table 2). To help translate the resulting knowledge into new drugs, TDR, through its Pathogenesis and Applied Genomics group, is supporting the establishment of regional networks in South America, Asia and Africa for training in bioinformatics and its applications to parasite genome studies. This includes the identification of potential drug targets. Through its Genomics and Discovery Research group TDR can follow up potential targets with validation studies and assay development. With validated targets and suitable assay formats, the principal investigators can seek partners with expertise in HTS and access to compound collections. These partners can be located in academia — for example, at ICCB-ICG Harvard mentioned above, or at the Walter & Eliza Hall Institute of Medical Research in Melbourne, Australia. Alternatively, an industrial partner can be involved, as in the case of the screening of plasmodial lactate dehydrogenase (Table 6), or the ongoing collaboration between TDR and Serono, which is enabling scientists from two disease-endemic countries to screen enzyme targets from Plasmodium and other parasites. Several other collaborative networks aim to link suppliers of natural products with academic laboratories that can evaluate their antiparasite activity (see, for example, the Multilateral Initiative on Malaria (MIM) and the University of Mississippi websites in Further information). The Kitasato Institute in Japan has been screening thousands of natural products from its own stocks27 and some 30,000 synthetic compounds from Japanese pharmaceutical companies for antimalarial activity under a broad collaboration between WHO/TDR, the Japanese Ministry of Health & Welfare and 14 Japanese pharmaceutical companies.
These collaborations bring together, for a specific purpose, groups that have particular expertise in different areas of drug discovery. In such collaborations, a formal agreement should define the project's objectives and each party's rights and obligations; such agreements typically require preferential pricing of any resulting products for developing countries, and define how any intellectual property rights are to be protected and made available4. Projects from these early drug discovery collaborations, if successful, can become candidates for support by PPPs, whose experience and concept of management through focused project development teams are tailored to advancing discovery projects further.
Outlook
Although, as stated in the introduction, very few new drugs were approved for the tropical diseases between 1975 and 1999, there has been a burst of activity since 2000, with more than 20 new agents developed or in development for parasitic diseases49 (Table 6). This burst of activity is largely attributed to a new spirit of partnership between the public and private sectors, with industry and public-health interests more closely aligned. Public–private agreements, and in particular the PPP model of project management, together with increased philanthropic funding, have increased the numbers of antiparasite drug candidates reaching the clinic during the past 5 years. Even if many of these candidates are combinations or drugs already used for other indications, the pipeline of discovery projects is much richer than a decade ago. The pharmaceutical industry's historically diminished involvement is being compensated, at least partially, by their involvement in PPPs, by individual agreements concerning specific projects and, more recently, by the foundation of industry-backed research institutes with specific drug discovery mandates for indications such as malaria, tuberculosis and dengue4.
In order to continue to be successful, the PPPs will need to identify new compounds for development. There are concerns that most of the 'low-hanging fruit' have been picked, and that early-stage discovery research needs further strengthening in both the public and private sectors, with financial support from public sources. The success of the PPP model should not obscure the fact that they need a thriving background of discovery-oriented research, itself largely dependent on public funding. Examples of relevant publicly funded early-stage research include parasite genetics and biochemistry, molecular target identification and validation, HTS against these targets, screening compounds against whole parasites, and chemistry to progress hits to lead compounds. Once good lead compounds are identified, it will be easier to attract new sources of funding — for example, from PPPs. They can provide the financial resources and expertise in drug development necessary to turn leads into drug candidates (as exemplified by the OZ compound discussed earlier44).
The disease-endemic countries are playing an increasingly important role in the discovery of new drugs. Some countries, such as Brazil, China, India or South Korea, already have a drug-manufacturing industry and institutions involved in drug discovery research. Other countries have research institutes with expertise in, for example, natural product chemistry, but as yet lack a pharmaceutical industry capable of moving from compounds in discovery all the way through the drug development pathway as illustrated in Fig. 1. There is a growing awareness (see, for example, Ref. 50) that the countries most affected by these diseases need to be actively involved in the solutions, including research to develop new and better treatments: a country's capacity to respond to the threat of disease is closely linked to its research capacity51. Both private and public support are increasing: for example, the Gates Foundation recently awarded a $20-million grant to science academies in Nigeria, South Africa and Uganda, and Britain's Department for International Development is planning to increase its spending on research and development in Africa. TDR has for 30 years supported research capacity strengthening in the developing countries, with this being a key component of its mission. Many of the leaders in tropical disease research now come from the disease-endemic countries and were supported by TDR in the early stages of their careers. This core of experts and expertise that exists in the disease-endemic countries needs to be encouraged and more actively engaged, not only in early drug discovery projects, but also in more advanced drug discovery and development projects like those being supported by the PPPs.
References
Trouiller, P. et al. Drug development for neglected diseases: a deficient market and a public-health policy failure. Lancet 359, 2188–2194 (2002). Review of the history of the discovery and development of drugs for neglected diseases, making it clear that non-market-driven mechanisms are required to stimulate new drug development
Global Forum for Health Research. The 10/90 Report on Health Research, 2003–2004 (Global Forum for Health Research, Geneva, 2004).
Médecins Sans Frontières Access to Essential Medicines Campaign. Fatal Imbalance: The crisis in Research and Development for Drugs for Neglected Diseases (Médecins Sans Frontières, Geneva, 2001).
Nwaka, S. & Ridley, R. G. Virtual drug discovery and development for neglected diseases through public-private partnerships. Nature Rev. Drug Discov. 2, 919–928 (2003). A clear discussion of the role and importance of public-private partnerships.
Kettler, H. E. & Marjanovic, S. Engaging biotechnology companies in the development of innovative solutions for diseases of poverty. Nature Rev. Drug Discov. 3, 171–176 (2004).
Medicines for Malaria Venture. Annual Report 2002 [online], <http://www.mmv.org/filesupld/53.pdf> (2003).
Ridley, R. G. Research on infectious diseases requires better coordination. Nature Med. 10 (Suppl.), S137–S140 (2004).
Rosenthal, P. J. Antimalarial drug discovery: old and new approaches. J. Exp. Biol. 206, 3735–3744 (2003).
Barry, C. E. III & Duncan K. Tuberculosis — strategies towards anti-infectives for a chronic disease. Drug Discov. Today: Ther. Strat. (in the press).
Willcox, M. L. & Bodeker, G. Traditional herbal medicines for malaria. BMJ 329, 1156–1159 (2004).
Basu, P. Trading on traditional medicines. Nature Biotechnol. 22, 263–265 (2004).
Mpia, B. & Pepin, J. Combination of eflornithine and melarsoprol for melarsoprol-resistant Gambian trypanosomiasis. Trop. Med. Int. Health 7, 775–779 (2002).
TDR. TDR Sixteenth Programme Report: Progress 2001–2002 [online], < http://www.who.int/tdr/publications/publications/pr16.htm> (2003).
Kremsner, P. G. & Krishna, S. Antimalarial combinations. Lancet 364, 285–294 (2004). Thoughtful analysis of the current combination therapies used in the treatment/prophylaxis of malaria.
Medicines for Malaria Venture. Annual Report 2003 [online], <http://www.mmv.org/FilesUpld/184.pdf> (2004). Review of the MMV portfolio of projects. A more up-to-date Annual Report for 2004 is now available. Much information on anti-parasite drug development projects is also available from the MMV, DNDi and IOWH web sites cited in Table 5.
Yeates, C. DB-289 Immtech International. IDrugs 6, 1086–1093 (2003).
Urbina, J. A. & Docampo, R. Specific chemotherapy of Chagas disease: controversies and advances. Trends Parasitol. 19, 495–501 (2003).
Yuvaniyama, J. et al. Insights into antifolate resistance from malarial DHFR-TS structures. Nature Struct. Biol. 10, 357–365 (2003).
O'Neill, P. M. et al. Isoquine and related amodiaquine analogues: a new generation of improved 4-aminoquinoline antimalarials. J. Med. Chem. 46, 4933–4945 (2003).
Biot, C. et al. Insights into the mechanism of action of ferroquine. Relationship between physicochemical properties and antiplasmodial activity. Mol. Pharm. 2, 185–193 (2005). An example of how successful veterinary drugs have been identified that have the potential to be important drugs for the treatment of human diseases.
Cotreau, M. M. et al. The antiparasitic moxidectin: safety, tolerability, and pharmacokinetics in humans. J. Clin. Pharmacol. 43, 1108–1115 (2003).
Nwaka, S., Riopel, L., Ubben, D. & Craft, J. C. Medicines for Malaria Venture new developments in antimalarials. Travel Med. and Inf. Disease 2, 161–170 (2004).
Tagboto, S. & Townson, S. Antiparasitic properties of medicinal plants and other naturally occurring products. Adv. Parasitol. 50, 199–295 (2001).
Klayman, D. L. Qinghaosu (artemisinin): an antimalarial drug from China. Science 228, 1049–1055 (1985).
Ang, K. K., Holmes, M. J., Higa, T., Hamann, M. T. & Kara, U. A. In vivo antimalarial activity of the β-carboline alkaloid manzamine A. Antimicrob. Agents Chemother. 44, 1645–1649 (2000).
Lui, M., Wilairat, P., Croft, S. L., Tan, L. C. A. & Go, M. L. Structure–activity relationships of antileishmanial and antimalarial chalcones. Bioorg. Med. Chem. 11, 2729–2738 (2003).
Otoguro, K. et al. In vitro and in vivo antimalarial activities of a non-glycosidic 18-membered macrolide antibiotic, borrelidin, against drug-resistant strains of Plasmodia. J. Antibiot. (Tokyo) 56, 727–729 (2003). An example of one type of computational analysis that may be helpful in deciding which parasite targets are worth pursuing.
Maes, L et al. In vitro and in vivo activities of a triterpenoid saponin extract (PX-6518) from the plant Maesa balansae against visceral leishmania species. Antimicrob. Agents Chemother. 48, 130–136 (2004).
Yeh, I., Hanekamp, T., Tsoka, S., Karp, P. D. & Altman, R. B. Computational analysis of Plasmodium falciparum metabolism: organizing genomic information to facilitate drug discovery. Genome Research 14, 917–924 (2004).
Hopkins, A. L. & Groom, C. R. The druggable genome. Nature Rev. Drug Discov. 1, 727–730 (2002).
Bush, K., Macielag, M. & Weidner-Wells, M. Taking inventory: antibacterial agents currently at or beyond phase 1. Curr. Opin. Microbiol. 7, 466–476 (2004).
Overbye, K. M. & Barrett, J. F. Antibiotics: where did we go wrong? Drug Discov. Today 10, 45–52 (2005).
Thomson, C. J., Power, E., Ruebsamen-Waigmann, H. & Labischinski, H. Antibacterial research and development in the 21(st) century — an industry perspective of the challenges. Curr. Opin. Microbiol. 7, 445–450 (2004).
Gribbon, P. & Sewing, A. High-throughput drug discovery: what can we expect from HTS? Drug Discov. Today 10, 17–22 (2005).
Bleicher, K. H., Bohm, H. J., Muller, K. & Alanine, A. I. Hit and lead generation: beyond high-throughput screening. Nature Rev. Drug Discov. 2, 369–378 (2003).
Phillips, M. A., Coffino, P. & Wang, C. C. Cloning and sequencing of the ornithine decarboxylase gene from Trypanosoma brucei: implications for enzyme turnover and selective difluoromethylornithine inhibition. J. Biol. Chem. 262, 8721–8727 (1987). Useful modus operandi for how genomics data can be used in the selection of molecular targets for helminth drug discovery.
Jones, A. K., Buckingham, S. D. & Sattelle, D. B. Chemistry-to-gene screens in Caenorhabditis elegans. Nature Rev. Drug Discov. 4, 321–330 (2005). A recommended approach and set of procedures in the discovery path to new anti-malarial drugs, much of which is applicable to the discovery of other anti-parasitic drugs.
Behm, C. A., Bendig, M. M., McCarter, J. P. & Sluder, A. E. RNAi-based discovery and validation of new drug targets in filarial nematodes. Trends Parasitol. 21, 97–100 (2005).
Sajid, M. & McKerrow, J. H. Cysteine proteases of parasitic organisms. Mol. Biochem. Parasitol. 120, 1–21 (2002).
Musonda, C. C. & Chibale, K. Application of combinatorial and parallel synthesis methodologies to antiparasite drug discovery. Curr. Med. Chem. 11, 2519–2533 (2004). Excellent example of a successful lead optimization program made all the more noteworthy by the fact that the lead compounds are 1,2,4 - trioxolanes (ozonides), a chemical class which would not normally be considered drug-like.
Fidock, D. A., Rosenthal, P. J., Croft, S. L., Brun, R. & Nwaka, S. Antimalarial drug discovery: efficacy models for compound screening. Nature Rev. Drug Discov. 3, 509–520 (2004).
Lee, B. J. et al. Antimalarial activities of novel synthetic cysteine protease inhibitors. Antimicrob. Agents Chemother. 47, 3810–3814 (2003).
Peters, W., Fleck, S. L, Robinson, B. L., Stewart, L. B & Jefford, C. W. The chemotherapy of rodent malaria. LX. The importance of formulation in evaluating the blood schizontocidal activity of some endoperoxide antimalarials. Ann. Trop. Med. Parasitol. 96, 559–573 (2002).
Vennerstrom, J. L. et al. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature 430, 900–904 (2004).
Bendig, M. Graduate of the TDR 'malperox' programme reaches the clinics. TDRnews 73, Oct (2004).
Widdus, R. Public–private partnerships for health: their main targets, their diversity, and their future directions. Bull. World Health Organ. 79, 713–720 (2001).
Nishtar, S. Public–private 'partnerships' in health — a global call to action. Health Res. Policy Syst. 2, 5 (2004). A wide-ranging overview with emphasis on the need for effective coordination of research and control activities.
Webber, D. & Kremer, M. Perspectives on stimulating industrial research and development for neglected infectious diseases. Bull. World Health Organ. 79, 735–741 (2001).
Horton, J. Drug development for tropical diseases — present situation, future perspectives. Trends Parasitol. 19, P06 (2003).
Editorial. Africa 2005. Nature 433, 669 (2005).
Breman, J. G., Alilio, M. S. & Mills, A. Conquering the intolerable burden of malaria: what's new, what's needed: a summary. Am. J. Trop. Med. Hyg. 71 (2 Suppl.), 1–15 (2004).
World Health Organization. The World Health Report 2004 [online], <http://www.who.int/whr/en/> (2004).
Burri, C. & Brun, R. Eflornithine for the treatment of human African trypanosomiasis. Parasitol. Res. 90 (Supp. 1, S49–S52 (2003).
Lell, B. et al. Fosmidomycin, a novel chemotherapeutic agent for malaria. Antimicrob. Agents Chemother. 47, 735–738 (2003).
Jha, T. et al. Miltefosine, an oral agent, for the treatment of Indian visceral leishmaniasis. N. Engl. J. Med. 341, 1795–1800 (1999).
Medicines for Malaria Venture. Annual Report 2003 [online], <http://www.mmv.org/FilesUpld/184.pdf> (2003).
Triglia, T., Menting, J. G., Wilson, C. & Cowman, A. F. Mutations in dihydropteroate synthase are responsible for sulfone and sulfonamide resistance in Plasmodium falciparum. Proc. Natl Acad. Sci. USA 94, 13944–13949 (1997).
Hudson, A. T. et al. 566C80: a potent broad spectrum anti-infective agent with activity against malaria and opportunistic infections in AIDS patients. Drugs Exp. Clin. Res. 17, 427–435 (1991).
Looareesuwan, S., Chulay, J. D., Canfield, C. J. & Hutchinson, D. B. Malarone (atovaquone and proguanil hydrochloride): a review of its clinical development for treatment of malaria.
Malarone Clinical Trials Study Group. Am. J. Trop. Med. Hyg. 60, 533–541 (1999).
Fry, M. & Pudney, M. Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4'-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80). Biochem. Pharmacol. 43, 1545–1553 (1992).
Jomaa, H. et al. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science 285, 1573–1576 (1999).
Borrmann, S. et al. Fosmidomycin–clindamycin in Plasmodium falciparum infections in African children. J. Infect. Dis. 189, 901–908 (2004).
Kuzoe, F. A. Current situation of African trypanosomiasis. Acta Trop. 54, 153–162 (1993).
Urbina J. A., New chemotherapeutic approaches for the treatment of Chagas disease (American trypanosomiasis). Expert Opin. Ther. Patents 13, 661–669 (2003).
Buckner, F. et al. A class of sterol 14-demethylase inhibitors as anti-Trypanosoma cruzi agents. Proc. Natl Acad. Sci. USA 100, 15149–15153 (2003).
Martin, M. B. et al. Bisphosphonates inhibit the growth of Trypanosoma brucei, Trypanosoma cruzi, Leishmania donovani, Toxoplasma gondii and Plasmodium falciparum: a potential route to chemotherapy. J. Med. Chem. 44, 909–916 (2001).
Martin, R. J. Modes of action of anthelmintic drugs. Vet. J. 154, 11–34 (1997).
Lacey, E. Mode of action of benzimidazoles. Parasitol. Today, 6, 112–115 (1990).
Omura, S. & Crump, A. The life and times of ivermectin — a success story. Nature Rev. Microbiol. 2, 984–989 (2004).
Cully, D. F., Wilkinson, H., Vassilatis, D. K., Etter, A. & Arena, J. P. Molecular biology and electrophysiology of glutamate-gated chloride channels of invertebrates. Parasitology 113 (Suppl.) S191–S200 (1996).
Baldwin, J. et al. High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J. Biol. Chem. 280, 21847–21853 (2005).
Kemp, L. E., Bond, C. S. & Hunter, W. N. Structure of 2C-methyl-D-erythritol 2,4-cyclodiphosphate synthase: An essential enzyme for isoprenoid biosynthesis and target for antimicrobial drug development. Proc. Natl Acad. Sci. USA 99, 6591–6596 (2002).
Rosenthal, P. J. Cysteine proteases of malaria parasites. Int. J. Parasitol. 34, 1489–1499 (2004).
Gelb, M. H. et al. Protein farnesyl and N-myristoyl transferases: piggy-back medicinal chemistry targets for the development of antitrypanosomatid and antimalarial therapeutics. Mol. Biochem. Parasitol. 126, 155–163 (2003).
Ohkanda, J. et al. Design and synthesis of peptidomimetic protein farnesyltransferase inhibitors as anti-Trypanosoma brucei agents. J. Med. Chem. 47, 432–445 (2004).
Waller, R. F. et al. Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proc. Natl Acad. Sci. USA 95, 12352–12357 (1998).
Morita, Y. S., Paul, K. S. & Englund, P. T. Specialised fatty acid synthesis in African trypanosomes: myristate for GPI anchors. Science 288, 140–143 (2000).
Surolia, N. & Surolia, A. Triclosan offers protection against blood stages of malaria by inhibiting enoyl-ACP reductase of Plasmodium falciparum. Nature Med. 7, 167–173 (2001).
Meinnel, T. Peptide deformylase of eukaryotic protists: a target for new antiparasitic agents? Parasitol. Today 16, 165–168 (2000).
Joet, T. & Krishna, S. The hexose transporter of Plasmodium falciparum is a worthy drug target. Acta Trop. 89, 371–374 (2004).
Wyllie, S., Cunningham, M. L. & Fairlamb, A. H. Dual action of antimonial drugs on thiol redox metabolism in the human pathogen Leishmania donovani. J. Biol. Chem. 279, 39925–39932 (2004).
Saravanamuthu, A. et al. Two interacting binding sites for quinacrine derivatives in the active site of trypanothione reductase: a template for drug design. J. Biol. Chem. 279, 29493–29500 (2004).
Daubenberger, C. A. et al. Identification and recombinant expression of glyceraldehyde-3-phosphate dehydrogenase of Plasmodium falciparum. Gene 246, 255–264 (2000).
Kron, M., Petridis, M., Milev, Y., Leykam, J. & Hartlein, M. Expression, localization and alternative function of cytoplasmic asparaginyl-tRNA synthetase in Brugia malayi. Mol. Biochem. Parasitol. 129, 33–39 (2003).
Cameron, A. et al. Identification and activity of a series of azole-based compounds with lactate dehydrogenase-directed anti-malarial activity. J. Biol. Chem. 279, 31429–31439 (2004).
Tripathi, A. K. et al. An α-proteobacterial type malate dehydrogenase may complement LDH function in Plasmodium falciparum. Cloning and biochemical characterization of the enzyme. Eur. J. Biochem. 271, 3488–3502 (2004).
Blackman, M. J. Proteases involved in erythrocyte invasion by the malaria parasite: function and potential as chemotherapeutic targets. Curr. Drug Targets 1, 59–83 (2000).
Doerig, C., Endicott, J. & Chakrabarti, D. Cyclin-dependent kinase homologues of Plasmodium falciparum. Int. J. Parasitol. 32, 1575–1585 (2002).
Keenan, S. M., Geyer, J. A., Welsh, W. J., Prigge, S. T. & Waters, N. C. Rational inhibitor design and iterative screening in the identification of selective plasmodial cyclin-dependent kinase inhibitors. Comb. Chem. Highthroughput Screen. 8, 27–38 (2005).
Kasekarn, W., Sirawaraporn, R., Chahomchuen, T., Cowman, A. F. & Sirawaraporn, W. Molecular characterization of bifunctional hydroxymethyldihydropterin pyrophosphokinase-dihydropteroate synthase from Plasmodium falciparum. Mol. Biochem. Parasitol. 137, 43–53 (2004).
Fries, D. S. & Fairlamb, A. H. in Burger's Medicinal Chemistry and Drug Discovery Sixth Edn Vol. 5: Chemotherapeutic Agents (ed. Abraham, D. J.) Ch. 15 (J. Wiley & Sons, Chichester, 2003).
Fumarola, L., Spinelli, R. & Brandonisio, O. In vitro assays for evaluation of drug activity against Leishmania spp. Res. Microbiol. 155, 224–230 (2004).
Lorente, S. O. et al. Novel azasterols as potential agents for treatment of leishmaniasis and trypanosomiasis. Antimicrob. Agents Chemother. 48, 2937–2950 (2004).
Raz, B., Iten, M., Grether-Buhler, Y., Kaminsky, R. & Brun, R. The Alamar Blue assay to determine drug sensitivity of African trypanosomes (T.b. rhodesiense and T.b. gambiense) in vitro. Acta Trop. 68, 139–147 (1997).
Ansede, J. H. et al. O-alkoxyamidine prodrugs of furamidine: in vitro transport and microsomal metabolism as indicators of in vivo efficacy in a mouse model of Trypanosoma brucei rhodesiense infection. J. Med. Chem. 47, 4335–4338 (2004).
Yardley, V. & Croft, S. L. In vitro and in vivo activity of amphotericin B-lipid formulations against experimental Trypanosoma cruzi infections. Am. J. Trop. Med. Hyg. 61, 193–197 (1999).
Yan, W. & Moreno, S. N. J. J. Immunol. Methods 220, 123–128 (1998).
Urbina, J. A. et al. Parasitological cure of acute and chronic experimental Chagas disease using the long-acting experimental triazole TAK-187. Activity against drug-resistant Trypanosoma cruzi strains. Int. J. Antimicrob. Agents 21, 39–48 (2003).
Cheever, A. W., Lenzi, J. A., Lenzi, H. L. & Andrade, Z. A. Experimental models of Schistosoma mansoni infection. Mem. Inst. Oswaldo Cruz 97, 917–940 (2002).
Liang, Y. S., Coles, G. C., Doenhoff, M. J. & Southgate V. R. In vitro responses of praziquantel-resistant and -susceptible Schistosoma mansoni to praziquantel. Int. J. Parasitol. 31, 1227–1235 (2001).
Cioli, D. et al. Determination of ED50 values for praziquantel in praziquantel-resistant and -susceptible Schistosoma mansoni isolates. Int. J. Parasitol. 34, 979–987 (2004).
Utzinger, J., Chollet, J., Tu, Z., Xiao, S. & Tanner, M. Comparative study of the effects of artemether and artesunate on juvenile and adult Schistosoma mansoni in experimentally infected mice. Trans. R. Soc. Trop. Med. Hyg. 96, 318–323 (2002).
Farah, I. O., Kariuki, T. M., King, C. L. & Hau, J. An overview of animal models in experimental schistosomiasis and refinements in the use of non-human primates. Lab. Anim. 35, 205–212 (2001).
Townson, S. The development of a laboratory model for onchocerciasis using Onchocerca gutturosa: in vitro culture, collagenase effects, drug studies and cryopreservation. Trop. Med. Parasitol. 39, 475–479 (1988).
Suswillo, R. R. and Denham, D. A. A new system for testing for filaricidal activity using transplanted adult Brugia in the jird. J. Parasitol. 63, 591–592 (1977).
Court, J. P., Stables, J. N., Lees, G. M., Martin-Short, M. R. & Rankin, R. Dipetalonema viteae and Brugia pahangi transplant infections in gerbils for use in antifilarial screening. J. Helminthol. 62, 1–9 (1988).
McCall, J. W., Dzimianski, M. T., Supakorndej, P. & Jun, J. J. The antifilarial activity of moxidectin against patent infections of Brugia pahangi in dogs. Am. J. Trop. Med. Hygiene 61 (Suppl.), 444 (1999).
Townson, S., Dobinson, A., Connelly, C. & Muller, R. Chemotherapy of Onchocerca lienalis microfilariae in mice: a model for the evaluation of novel compounds for the treatment of onchocerciasis. J. Helminthol. 62, 181–194 (1988).
Strote, G., Weiland, S., Darge, K. & Comley, J. C. W. In vitro assessment of the activity of anthelmintic compounds on adults of Onchocerca volvulus. Acta Liedensia 59, 285–296 (1990).
Acknowledgements
We thank A. Fairlamb for helpful comments, and all members of TDR-funded laboratories (particularly R. Brun, S. Croft, L. Maes, K. Otoguro and S. Townson) that contributed to TDR-supported work reported here.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Glossary
- PROTOZOA
-
Single-celled eukaryotic organisms with nuclei that show some characteristics usually associated with animals, most notably mobility and heterotrophy. A few are important parasites.
- HELMINTH
-
A multicellular organism, generally longer than it is wide or deep, commonly called a worm. There are three major groups causing parasitic diseases in humans: nematodes, flukes, and tapeworms.
- TROPICAL DISEASES
-
Information on the parasitic diseases discussed here (see Table 1), their pathology, treatment, and incidence can be found on the WHO/TDR web site (http://www.who.int/tdr/) and by linking to the section titled Disease Watch (www.who.int/tdr/dw/default.html). The Disease Watch pages include links to articles originally published in Nature Reviews Microbiology.
- RESISTANCE REVERSER
-
A compound that will alter a cell's properties to make it sensitive to a certain drug to which it has developed resistance.
- FILARIAL WORMS
-
Long, hair-like nematodes of which the adults (macrofilariae) live in the blood or tissues of vertebrates. In some species, the larvae (microfilariae) may be found in the blood. Examples of diseases caused by filarial worms include lymphatic filariasis and onchocerciasis.
- SCHISTOSOME
-
A group of flukes of the genus Schistosoma, many of which are parasitic in the blood of humans and other mammals.
- TRYPANOSOMATID PARASITES
-
A group of flagellated protozoal parasites of the order Trypanosomatidae, transmitted to the vertebrate bloodstream, lymph, and spinal fluid by certain insects and often causing diseases such as African trypanosomiasis, Chagas' disease, and leishmaniasis.
- NEMATODE
-
A group of organisms also known as roundworms. They reproduce by laying eggs, or larvae which hatch from their eggs inside the body of the female worm. They are among the most common multicellular parasites of humans and include the filarial worms.
- KINETOPLASTIDS
-
A group of flagellated protozoa, including the trypanosomatids (see below), that are distinguished by the presence of a kinetoplast, a DNA-containing granule located within the single mitochondrion and associated with the flagellar bases.
- MICHAEL ACCEPTOR
-
A compound containing an activated carbon-carbon double bond susceptible to nucleophilic attack.
Rights and permissions
About this article
Cite this article
Pink, R., Hudson, A., Mouriès, MA. et al. Opportunities and Challenges in Antiparasitic Drug Discovery. Nat Rev Drug Discov 4, 727–740 (2005). https://doi.org/10.1038/nrd1824
Issue Date:
DOI: https://doi.org/10.1038/nrd1824
