
Abstract
Mood disorders are associated with regional brain abnormalities, including reductions in glial cell and neuron number, glutamatergic irregularities, and differential patterns of brain activation. Because astrocytes are modulators of neuronal activity and are important in trafficking the excitatory neurotransmitter glutamate, it is possible that these pathologies are interrelated and contribute to some of the behavioral signs that characterize depression and related disorders. We tested this hypothesis by determining whether depressive-like signs were induced by blocking central astrocytic glutamate uptake with the astrocytic glutamate transporter (GLT-1) inhibitor, dihydrokainic acid (DHK), in behavioral tests that quantify aspects of mood, including reward and euthymia/dysthymia: intracranial self-stimulation (ICSS) and place conditioning. We found that DHK elevated ICSS thresholds, a depressive-like effect that could reflect reduced sensitivity to reward (anhedonia) or increased aversion (dysphoria). However, DHK treatment did not establish conditioned place aversions, suggesting that this treatment does not induce dysphoria. To identify the brain regions mediating the behavioral effects of DHK, we examined c-Fos expression in areas implicated in motivation and emotion. DHK increased c-Fos expression in many of these regions. The dentate gyrus of the hippocampus was robustly activated, which led us to explore whether DHK alters hippocampal learning. DHK impaired spatial memory in the MWM. These findings identify disruption of astrocyte glutamate uptake as one component of the complex circuits that mediate anhedonia and cognitive impairment, both of which are common symptoms of depression. These finding may have implications for the etiology of depression and other disorders that share the features of anhedonia and cognitive impairment.
Similar content being viewed by others


INTRODUCTION
Little progress has been made over the past 20 years in identifying more effective treatments for Major Depressive Disorder (MDD). This may be due, in part, to a lack of knowledge about specific abnormalities present in the depressed brain. Because all currently available antidepressants were discovered serendipitously, the mechanisms of action for effective antidepressant treatment remains largely unknown (Nestler and Carlezon, 2006). This work is designed to help identify the cellular or molecular mediators of behavior that could be novel treatment targets by mimicking behavioral abnormalities reported in depressed humans and evaluating the potential role of these mediators in the etiology of depressive illness.
MDD is associated with distributed regional abnormalities of the brain, including fewer glial cells (Bowley et al, 2002; Cotter et al, 2002; Cotter et al, 2001; Hamidi et al, 2004; Hercher et al, 2009; Müller et al, 2001; Öngür et al, 1998; Rajkowska, 2000; Rajkowska et al, 1999), fewer neurons (Arango et al, 1996; Baumann et al, 2002; Law and Harrison, 2003; Rajkowska et al, 1999), alterations in glutamate function (Paul and Skolnick, 2003; Pittenger et al, 2007; Sanacora et al, 2008; Valentine and Sanacora, 2009), and anomalous patterns of regional brain activation (Milak et al, 2005; Videbech, 2000; Wu et al, 2001). Over- or under-activation in specific brain regions can be associated with specific symptoms of depression, although an overall positive association is observed between severity of depression and activation of a distributed cortico-limbic circuit (Milak et al, 2005). Furthermore, normalization of activity is associated with alleviation of depressive symptoms (Buchsbaum et al, 1997; Kennedy et al, 2001; Mayberg et al, 1997; Mayberg et al, 2000; Mayberg et al, 2005; Pizzagalli et al, 2001; Wu et al, 1999). The role of each of these pathologies in the etiology of mood disorders is unknown. However, because astrocytes are crucial for the uptake of the excitatory neurotransmitter glutamate (Anderson and Swanson, 2000; Danbolt, 2001), it is possible that some of these pathologies are causally interrelated. Recent reports point to a causal link between glutamate and the treatment of depression by showing that ketamine, an antagonist of glutamatergic NMDA receptors, has antidepressant-like effects in humans and laboratory animals (Berman et al, 2000; Maeng et al, 2007; Zarate et al, 2006). Preclinical studies suggest that cortical glial ablation (Banasr and Duman, 2008) and blockade of astrocytic glutamate uptake in the amygdala (Lee et al, 2007) or ventral tegmental area (Herberg and Rose, 1990) can produce some depressive-like effects and that levels of the astrocytic glutamate transporter (GLT-1 aka EAAT2; Anderson and Swanson, 2000; Danbolt, 2001) are decreased in an animal model of depression (Zink et al, 2010). Thus, there is accumulating evidence that glutamate neurotransmission and glial cells could be involved in both the causes and the treatment of depression. Because glial cell reduction has been reported in several limbic regions and increased limbic activation is positively associated with symptom severity, we examined the effects of central blockade of astrocytic glutamate uptake using the GLT-1 inhibitor dihydrokainic acid (DHK; Anderson and Swanson, 2000 Arriza et al, 1994) in rats using tests that can quantify various symptoms of depression.
Features of the depressive syndrome that can be modeled in rats include states of anhedonia (diminished sensitivity to reward) and dysphoria (discomfort, observable as aversion). Intracranial self-stimulation (ICSS) is a paradigm that detects both diminished and enhanced reward states (Carlezon and Chartoff, 2007). Although ICSS is a powerful tool for quantifying motivation, it is difficult to distinguish anhedonia-like behavior from dysphoria-like behavior in this paradigm as both produce the same outcome: treatments that block reward (eg, dopamine antagonists) and induce aversion (eg, acute opiate withdrawal) elevate reward thresholds (Carlezon and Chartoff, 2007). Place conditioning has been widely used to study the rewarding and aversive properties of drugs (for review see Carlezon, 2003; Tzschentke, 2007). We used the effects in this paradigm to interpret the effects in the ICSS paradigm because tests of place conditioning can detect dysphoria (increased aversion) separately from anhedonia (decreased reward). Finally, we used c-Fos immunoreactivity to begin to identify brain areas that may mediate the observed effects. We hypothesized that intracranial DHK infusions would produce a depressive-like phenotype and that DHK would selectively alter the activity of a forebrain circuit related to the processing of emotional stimuli.
MATERIALS AND METHODS
Animals
Seventy-three male Sprague–Dawley rats (Charles River Laboratories Inc.) weighing 275–300 g on arrival were used in these studies. After surgery the rats were housed singly. The rats were maintained on a 12-h light–dark cycle, with food and water available freely. All experiments used separate cohorts of rats, except the activity studies, as detailed below, and were conducted between 1200 and 1800 hours. The procedures were conducted with the approval of the McLean Hospital Institutional Animal Care and Use Committee within the guidelines of The National Research Council's Guide for the Care and Use of Laboratory Animals.
Drugs
DHK (Tocris Bioscience, Ellisville, MO) blocks the uptake of glutamate into astrocytes (Anderson and Swanson, 2000; Arriza et al, 1994), which leads to increased extrasynaptic glutamate. This effect has been shown ex vivo with synaptosomes (Robinson et al, 1991) and in vivo with microdialysis (Fallgren and Paulsen, 1996). DHK has little or no effect at AMPA/kainate receptors where it is 500 times less potent than kainic acid and 250 times less potent than quisqualic acid (Johnston et al, 1979). Doses were derived from preliminary findings and previously published behavioral data (Lee et al, 2007). DHK was dissolved in PBS (pH 7.4) as previously reported (Lee et al, 2007).
Surgery
Rats were implanted with monopolar stainless steel stimulating electrodes (0.25 mm diameter) aimed at the medial forebrain bundle (10 degree angle; from the bregma 3.0 mm anterior±1.6 mm lateral, 7.6 mm ventral from the dura) and/or intracerebroventricular (ICV) cannulae (23 ga.) aimed at the lateral ventricle (from the bregma 0.6 mm posterior, 1.4 mm lateral, 2.4 mm ventral from the dura with additional 1.5-mm injector projection) (Paxinos and Watson, 2004). The rats were anesthetized with Nembutal (65 mg/kg, IP). Small burr holes were made in the skull through which an electrode or guide cannula was lowered to the specified depth under stereotaxic guidance. The electrodes and guide cannulae were fastened to the skull using stainless steel screws and dental acrylic. The rats were allowed 5–7 days of recovery.
Intracranial Self-Stimulation
Intracranial self-stimulation (ICSS) was conducted to assess changes in hedonic state after DHK treatment. The rats were trained on a fixed-ratio-1 (FR1) schedule of reinforcement to obtain brain stimulation as previously described (Tomasiewicz et al, 2008). Sessions began with the delivery of a 5.0-s non-contingent stimulation train of square-wave cathodal pulses (0.1 ms pulse duration, 141 Hz). Each subsequent lever press resulted in the delivery of a 0.5-s pulse train and illumination of the house light for 0.5 s. The stimulation current (100–300 μA) was adjusted for each animal to the lowest value that sustained responding at a rate greater than 1 lever press per second. After stable responding was obtained, the rats were trained to lever press for a series of 15 descending frequencies. At the end of each trial, a 5-s time-out period was imposed. The frequency for each successive trial was decreased by 10% (0.05 log10 units). The completion of all 15 descending frequency trials, termed a ‘pass’, was achieved in 15 min. Four consecutive passes made-up the daily training sessions. The training period typically spanned 6–8 weeks. To characterize the relationship between response strength and reward magnitude, a least-squares best fit line was plotted across the frequencies that maintained responding at 20, 30, 40, 50, and 60% of the maximum response. From this function, the ICSS threshold was defined as the frequency at which the function intersects with the x-axis (theta-0; T0) (Miliaressis et al, 1986), which represents the point at which the stimulation becomes rewarding. The maximum rate of responding (Max Rate) was calculated in parallel to detect impairments in performance. Mean daily ICSS thresholds were determined by averaging the thresholds for the last three passes. Drug test sessions began the day after mean ICSS thresholds varied by <±10% across five training sessions. On the test days rats were allowed to press through three baseline passes, infused ICV with one of four doses (0.0, 12.5, 25.0, 50.0 nmol in 1 μl) of DHK Anderson and Swanson, 2000; Arriza et al, 1994), and then subjected to three additional passes. Each animal was administered each dose in increasing order on separate test days, with treatment days interspersed with baseline recovery days.
Place Conditioning
Place conditioning was assessed to determine whether DHK induced an aversive (dysphoric) state. The unbiased apparatus had three chambers consisting of a neutral middle (12 L × 21 W × 21 H cm) chamber and two conditioning chambers that differed in floor texture, wall coloring, and lighting (24 L × 18 W × 33 H cm; Med Associates, St Albans, VT). The experiment proceeded in three phases: habituation/pre-testing, conditioning, and preference testing. On the first day, rats were given an ICV vehicle infusion before being placed in the conditioning apparatus for 30 min. This day allowed the rats to habituate to the experimental apparatus and the infusion procedures, and provided a reference point for individual rat bias. For conditioning, the rats were infused with vehicle (CS− trials) or DHK (CS+ trials) on alternating days immediately before being confined to one of the two conditioning chambers for 30 min. Treatment and chamber pairing were counterbalanced within groups. Because aversive conditioning is often acquired rapidly, a 30-min preference test was conducted after two conditioning trials (2 CS+, 2 CS−), followed by two additional conditioning trials and a final test. The rats were infused with vehicle before each test and then allowed to explore the apparatus. The time spent in each compartment was monitored using an automated system. The conditioning trial duration (30 min) and the DHK doses (0.0, 25.0, 50.0 nmol in 1 μl) were chosen on the basis of the effects observed in the ICSS study (ie, peak effects within 15 min at 25.0 and 50.0 nmol DHK).
Activity Testing
Because decreases in activity can influence responding in the ICSS paradigm, activity was assessed to determine whether DHK induced motor impairment. Two separate experiments were conducted with lights off or on (15–35 lux depending on position), because it was not clear which would be more comparable to lighting in the ICSS paradigm where the chambers are dark when rats are not responding and illuminated by the house light (45 lux) for 0.5 s after each lever press. The rats were tested 1 week after completion of place conditioning, for lights-off or MWM for lights-on activity testing. The rats were infused with DHK (0.0, 25.0, 50.0 nmol in 1 μl) immediately before being placed in the activity chamber for 30 min. Activity was monitored in automated chambers (43 L × 43 W × 31 H cm; MED Associates, St Albans, VT). Photobeam breaks during 5-min intervals were used to automatically calculate the distance traveled (cm) during each interval.
Immunohistochemistry for c-Fos
Immunohistochemistry for c-Fos was conducted to identify the brain regions involved in the effects of DHK. The methods used were as previously described, with the exception of the primary antibody (Bechtholt et al, 2008). Briefly, rats were perfused (4% paraformaldehyde) and brains were post-fixed overnight and then immersed in sucrose solution until saturated. Non-serial 30 μm coronal sections were collected and incubated in rabbit anti c-Fos antisera (1 : 9000; Calbiochem–EMD Bioscience, Darmstadt, Germany) for 24 h at 4 °C. The sections were then incubated in biotinylated donkey anti-rabbit antiserum (1 : 200; Jackson Labs, West Grove, PA) for 90 min, followed by treatment with the avidin–biotin complex (1 : 600; ABC Elite Kit, Vector Labs, Burlingame, CA) for 90 min at room temperature. The sections were immersed in 0.02% 3,3′-diaminobenzidine-4HCl (DAB; Sigma-Aldrich Co., St Louis, MO) containing 0.01% H2O2 in phosphate buffer for 10–15 min. The sections were mounted and photomicrographs were taken at × 20 magnification (Zeiss, Oberkochen, Germany). The number of labeled cells was counted for each rat and brain area using the Image J software (National Institutes of Health—public domain) in both hemispheres of the three non-serial sections closest to those shown in Figure 1. The average of the six values was recorded. The regions examined were chosen on the basis of their association with depression and/or reward (Bechtholt et al, 2008; Hercher et al, 2009; Nestler and Carlezon, 2006; Phillips et al, 2003). Two anatomically close control regions, dorsal striatum (dStr) and ventral posterolaterlal nucleus of the thalamus (VP), were included that have not been heavily implicated in mood or reward.

Location of the 15 areas counted for c-Fos immunoreactivity indicated on atlas representations of the rat brain (Figures 12, 19, 56, 59, 94, 115 in reference Paxinos and Watson, 2004). Abbreviations are defined in Table 1.
Morris Water Maze
The Morris Water Maze (MWM) was conducted to determine whether DHK induced hippocampus-associated cognitive impairments. The rats were placed in a large circular pool of opaque water (25 °C; 178 cm diameter) and allowed to search for a hidden platform for 90 s. If the platform was not located within 90 s the rat was guided to the platform. The rats remained on the platform for 30 s. Two training trials were conducted per day for seven consecutive days. On the eighth day the platform was removed for a probe test and the rats were allowed to search the pool for 60 s immediately after being infused with vehicle or 50 nmol of DHK. Behavior was recorded and automatically tabulated using an overhead camera and Ethovision software (Noldus, Leesburg, VA).
RESULTS
Effects of DHK on ICSS
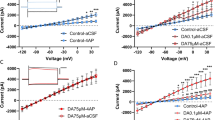
ICV DHK substantially and dose dependently increased the ICSS thresholds (Figure 2). This conclusion was evidenced by a statistically significant dose × time interaction (F(6, 36)=2.9; p<0.05). The main effects of dose and time were not significant. Follow-up comparisons showed that rats treated with DHK required significantly higher minimum stimulation frequencies to maintain responding (T0) in the first 15 min after DHK infusion as compared with the vehicle-treated rats (50.0 nmol, p<0.05; 25.0 nmol, p<0.01).

Effect of ICV DHK (12.5, 25.0, or 50.0 nmol) on ICSS thresholds over 45 min (left panel) and within the first 15 min (right panel) after infusion. Follow-up comparisons showed that the effects of DHK were short in duration and dose-dependent. Significant effects were observed during the first 15 min after DHK infusion (right panel) and increasing doses of DHK yielded greater thresholds. *: Significantly different from the vehicle group. Time × dose interaction: p<0.05. †: Significantly different from the 50.0-nmol group. One symbol: p<0.05; two symbols: p<0.01 (n=7).
Figure 3 shows the effects of DHK on the maximum response rates (Max Rate). Statistical analysis showed a significant main effect of time (F(2, 36)=3.9; p<0.05) and non-significant effects of dose (F(3, 36)=3.0; p=0.059) and the interaction of dose and time (F(6, 36)=1.2; p=0.332). While statistical significance was not reached, a drug-induced suppression of maximal responding is suggested by an apparent flat dose effect of DHK on maximum rate during the first 15 min after DHK infusion (Figure 3).

Effect of ICV DHK (12.5, 25.0, or 50.0 nmol) on ICSS maximum response rate across three passes (left panel) and within the first 15 min (right panel). A significant main effect of time was detected, showing an increase in the maximum response rate over time. However, the time and the DHK dose did not interact. Main effect of time: p<0.05 (n=7).
Figure 4 shows the characteristic rightward shift of the rate–frequency function (increased T0) from a representative rat, which indicates a depressant-like (reward decreasing) effect. In addition, a downward shift is shown (decreased Max Rate), which could be the result of decreased hedonic value of the stimulation (Do Carmo et al, 2009) or reduced performance capability (Carlezon and Chartoff, 2007).

ICSS rate–frequency functions from a representative rat at baseline and after treatment with 50 nmol DHK. Changes in motivation are indicated by lateral shifts of the frequency response curve. Shifts to the right of this frequency response curve indicate that higher frequencies of brain stimulation are required to support lever pressing, suggesting depressed-like mood with an anhedonic or dysphoric state. Shifts to the left indicate that lower frequencies are required to maintain responding, suggesting an elevated mood with a state of enhanced reward. Vertical shifts in the frequency response curve can also be indicative of changes in motivation; however, it is difficult to distinguish these effects from effects on performance capacity. These curves show that DHK caused both a downward and a rightward shift in the rate–frequency functions.
Effects of DHK on Place Conditioning
In contrast to findings in ICSS, DHK did not induce conditioned place aversion (Figure 5). This conclusion was supported by a lack of significant effects of test, dose, or their interaction. That is, no change in aversion score was observed between tests, suggesting that the rats did not avoid the chamber that had been previously paired with DHK infusion.

Effect of ICV DHK (25.0 or 50.0 nmol) on average aversion scores. Neither dose of DHK that increased ICSS thresholds was sufficient to induce conditioned place aversion (n=8–9 per group).
Effects of DHK on Locomotor Activity
As shown in Figure 6, the distance traveled decreased over time during locomotor activity testing with lights off (top panel) or on (bottom panel), and this effect was not affected by DHK infusion. This suggestion was confirmed by statistical analysis showing significant effects of time in both lights off (F(5, 105)=21.8; p=0.001) or lights on (F(5, 87)=19.4; p=0.001) conditions, but no significant effects of dose or dose × time interactions. These null data taken together with ICSS maximum response rate data, suggest that effects observed in ICSS were not the result of sedation.

Effect of ICV DHK (25.0 or 50.0 nmol) on the average distance traveled in cm within 5-min intervals for the 30-min test with the lights off (top panel) or lights on (bottom panel). Inset: Data pooled into two 15-min intervals; timing corresponding to the effects observed in ICSS (n=8–10 per group).
Effects of DHK on c-Fos Expression
Figures 7 and 8 show typical c-Fos photomicrographs of representative brain regions, the dorsal dentate gyrus of the hippocampus (dDG) and the infralimbic prefrontal cortex (IL). Table 1 shows the mean number of c-Fos-positive cells for each dose group, the p-values for the one-way ANOVAs, and abbreviations for all of the examined brain regions. The general effects of DHK were replicated across brain areas of interest; DHK dose responsively increased c-Fos expression in many reward and depression related areas, whereas having no effect in the control regions. Significant one-way ANOVAs for the number of c-Fos-positive profiles were observed for the effects of DHK in the AcbC (F(2, 21)=4.2; p<0.05), AcbSh (F(2, 21)=14.8; p<0.001), BL (F(2, 21)=7.8; p<0.005), Ce (F(2, 21)=23.9; p<0.001), IL (F(2, 18)=23.9; p<0.001), DRV (F(2, 22)=12.1; p<0.001); dDG (F(2, 21)=3.9; p<0.05), and LC (F(2, 20)=18.1; p<0.001). Follow-up comparisons and abbreviations are shown in Table 1. No significant effects were observed in the Pir, dCA1, dCA2, dCA3, dStr, MD, and VP.

Example photomicrographs for the effects of PBS or 50.0 nmol of DHK on c-Fos expression in the dDG. The left side is medial. The bar graphs show the mean number of c-Fos-positive cells in the dDG after treatment with varying doses of DHK. *: Significantly different from the vehicle group; †: significantly different from the 25.0-nmol group.

Example photomicrographs for the effects of PBS or 50.0 nmol of DHK on c-Fos expression in the IL at × 10 (left panels) and × 20 (right panels) magnification. The left side is medial. Outlines in the left panels indicate the area magnified in the right panels. The bar graphs show the mean number of c-Fos-positive cells in the IL after treatment with varying doses of DHK. *: significantly different from the vehicle group; †: significantly different from the 25.0-nmol group.
Effects of DHK on MWM
Groups were well matched before treatment in the MWM. As shown in Figure 9 rats that would later be treated with vehicle or 50 nmol of DHK during the probe test showed similar drug-free acquisition patterns in the MWM task. This was shown by a main effect of training day (F(6, 102)=32.7; p<0.001) and by overlapping the distance traveled to the platform that decreased across training days and reached a plateau over days 6 and 7. During the probe test, rats treated with DHK swam faster (t(17)=3.0; p<0.01), searched farther from the former platform location (t(17)=−8.9; p<0.001), and spent less time in the target quadrant (t(17)=3.3; p<0.005) than the vehicle-treated rats. We noted that the DHK-treated animals spent the majority of their time around the perimeter of the pool, whereas the vehicle-treated animals searched the interior. This finding suggests that DHK induces impairments in spatial navigation.

A line graph showing drug-free acquisition of the MWM task expressed as the average distance traveled (cm) to reach the platform during each of seven training days (top panel). The bottom panels show the effects of DHK during the post-training probe test on velocity (left), mean distance from the platform (middle), and the percent of time spent in the target quadrant (right). DHK significantly increased swim velocity and the mean distance from the platform, but decreased the time spent in the target quadrant. *: Significantly different from the vehicle group (n=9–10 per group).
DISCUSSION
We have shown that blockade of central glial glutamate uptake selectively activates a forebrain circuit involved in the regulation of mood and induces a phenotype with some features of depression. By implication these findings suggest a relationship between a lack of glial cells observed in humans with depression and clinical dysregulation of mood. Here, a lack of astrocytic glial glutamate uptake results in diminished reward value, impaired spatial memory, and activation of brain regions that are associated with motivation. Disruption of glial glutamate uptake is one component of a complex circuit that may mediate anhedonia and cognitive impairment. This finding may provide insight not only into the etiology of depression, but also into the mechanisms underlying the symptoms of other neuropsychiatric illnesses that share these symptoms.
Intracranial DHK increased the ICSS thresholds (T0), the minimum frequency that would sustain responding, suggesting that blocking astrocytic glutamate uptake attenuates brain stimulation reward. This effect is consistent with the induction of an anhedonic-like or dysphoric-like state; two constructs from animal models that represent prominent symptoms of depression (ie, diminished pleasure and depressed or negative mood, respectively). These findings are in agreement with recent work suggesting that glial cells may be involved in the induction or mediation of depressive-like states. For instance, prefrontal cortical glial ablation reportedly decreases sucrose intake, increases response to novelty, and increases immobility in the forced swimming test, effects consistent with a depressant-like effect (Banasr and Duman, 2008). Further, blockade of amygdalar astrocytic glutamate uptake has been reported to decrease social exploration and disrupt circadian rhythms, effects that are also consistent with depression-like symptoms (Lee et al, 2007).
DHK had an effect on the maximal response rate although this was not statistically significant. This effect could be indicative of a performance impairment (Carlezon and Chartoff, 2007), as it has been previously noted that manipulations that interfere with lever pressing, such as muscle relaxation and increasing lever weight, results in lower asymptotic response rates (Miliaressis et al, 1986). It has also been shown that reductions in stimulation intensity or increases in response requirements (eg, changing from an FR1 to an FR10 schedule of reinforcement) can decrease the maximum response rates (Do Carmo et al, 2009), suggesting a relationship between motivation and performance. Examination of frequency–rate curves (eg, Figure 4) shows that both rightward and downward shifts were characteristic of DHK's effect. Together these data indicated that there was a decrease in reward efficacy that could have been accompanied by motor sedation. Consequently, we examined the effects of DHK on locomotor activity, where no signs of sedation were observed. Collectively these data suggest that the threshold-increasing and maximal rate-reducing effects of DHK were not an artifact of performance impairment, but rather the result of reductions in the reward efficacy of the brain stimulation.
Because ICSS provides a relative index of reward (eg, changes in response to rewarding brain stimulation) it is difficult to distinguish between manipulations that induce anhedonia and those that induce dysphoria, as both produce decreases in reward efficacy. In humans, too, although anhedonia and dysphoria may appear together, they represent distinct symptoms that can be observed to varying degrees in different episodes of clinical depression or related illnesses. Place conditioning can be helpful in clarifying the presence of each state because dysphoric effects can be detected as avoidance, without the need for a relative comparison to a reward state. In the present experiments, DHK treatment yielded no significant change in the aversion scores, suggesting that DHK does not provoke discomfort and avoidance of the DHK-paired chamber (ie, no conditioned place aversion). Interpreting these data in combination with the ICSS results make it likely that the effects observed in ICSS were the result of the induction of anhedonia without dysphoria. It should be noted that a lack of an observable effect does not prove that no effect exists, so it is possible that under different conditions DHK would produce dysphoria.
DHK treatment yielded a unique pattern of activation in discrete brain areas that are associated with depression and reward (Hercher et al, 2009; Nestler and Carlezon, 2006; Phillips et al, 2003), without inducing activation in areas that have not been explicitly associated with depression and/or reward. More specifically, c-Fos expression was increased in the AcbC, AcbSh, BL, Ce, IL, DRV, dDG, and LC, but not in the dSTr and VP. In addition to reductions in the volume of some of these mood/reward-related regions (for review see Phillips et al, 2003), specific cellular changes have also been reported in depressed patients (for review see Hercher et al, 2009). A reduction in glial cells or associated markers has been noted in the prefrontal cortex (Cotter et al, 2001, 2002; Öngür et al, 1998; Rajkowska et al, 1999), hippocampus (Müller et al, 2001), and amygdala (Bowley et al, 2002; Hamidi et al, 2004). By contrast, reductions in neurons or their markers have been noted in the the prefrontal cortex (Rajkowska et al, 1999, 2005, 2007), dorsal raphe nucleus (Baumann et al, 2002), and locus coeruleus (Arango et al, 1996). To be clear, the raphe nucleus and the locus coeruleus have not yet been examined for glial abnormalities. These regions are part of a large interconnected circuit with projections from the prefrontal cortex to the ventromedial striatum (Öngür and Price, 2000) and raphe nucleus, and projections from the locus coeruleus to the raphe nucleus (Marcinkiewicz et al, 1989), as well as reciprocal connections between the medial prefrontal cortex, amygdala, and the enorhinal cortex (Öngür and Price, 2000). Because these regions are so highly connected, it is not possible at this time to distinguish between c-Fos expression resulting from the direct effects of blocking glial glutamate uptake in a given locus and expression that is an indirect consequence of activation through projections. Nonetheless, these data begin to characterize how glial deficits can produce depressive-like effects and serve to highlight important candidate regions for further study.
We observed robust activation of the dentate gyrus of the hippocampus after DHK treatment that was accompanied by deficits in hippocampus-dependent spatial navigation. Although little is known about effects in mammals, astrocyte trafficking of glutamate has been heavily implicated in learning and memory consolidation in chicks (for review see Gibbs et al, 2008) and the hippocampus has been extensively studied with respect to depression. In patients with unremitted depression, hippocampal volumes are decreased (Sheline et al, 2003) and numerous cognitive deficits have been documented (Clark et al, 2009). Impaired spatial navigation, which requires the hippocampus, is among these cognitive deficits (Gould et al, 2007). Likewise, animal models of depression show major alterations in hippocampal plasticity and structure (Duman, 2004; Kim et al, 2006) that can be accompanied by impaired spatial navigation (Song et al, 2006; Sun and Alkon, 2004). The present results are in agreement with these many findings and extend them to suggest that similar mechanisms involving astrocyte dysfunction may underlie both cognitive and reward-related symptoms in depression.
One might suspect that DHK-induced cognitive impairment could underlie the decreased responding in the ICSS paradigm and a lack of conditioned place aversion in the place conditioning paradigm. Some evidence suggests otherwise. There is a large published literature showing that many drugs that are well known to impair spatial learning and memory (eg, MK-801; Whishaw and Auer, 1989) actually facilitate, rather than impair, ICSS responding (Corbett, 1989). Furthermore, the unique timing of the effects in this paradigm diminishes the likelihood that learning or memory impairment decreased the responding for rewarding brain stimulation. The frequencies were presented in a descending order for 50 s each. Here, rats responded normally for the highest frequencies, which were presented first, and anhedonia was shown by a lack of responding at lower frequencies. As such, the change in responding for a given frequency occurred during the 5-s timeout and the 0.5-s non-contingent stimulus train that came between trials. This normal initial responding coupled with the rapid transition from responding to not responding suggests that the rats were able to recall the task. For place conditioning, there is evidence in the literature that rats seek novelty (eg, reference Hughes, 1968). If DHK disrupted memory, we would expect the DHK-paired chamber to be novel relative to the vehicle-paired chamber. By extension, we would expect the rats to prefer the DHK-paired environment, for its novelty, relative to the vehicle-paired chamber. The observation that the rats spent equal amounts of time in the DHK- and saline-paired chambers suggests that each was equally familiar, and supports the hypothesis that DHK neither produced dysphoria nor disrupted place conditioning.
Here we suggest an important role for astrocytes in the etiology of anhedonia and cognitive dysfunction in depression. However, these symptoms are not unique to depression as they are shared by a number of neuropsychiatric disorders, including, but not limited to; bipolar disorder, schizophrenia, and Parkinson's disease (Andreasen and Olsen, 1982; Burdick et al, 2007; Cassidy et al, 2000; Gorwood, 2008; Ibarretxe-Bilbao et al, 2009; Isella et al, 2003; Kraus and Keefe, 2007). Of particular interest is bipolar disorder, for which an unspecified reduction in glial cell density and number has also been reported (Öngür et al, 1998; Rajkowska, 2002; Rajkowska et al, 2001). Abnormalities in specific glial cell subtypes have been strongly implicated in schizophrenia and Parkinson's disease, as well, although lack of astrocytes has not been suggested in these disorders (Bernstein et al, 2009; Mena and Garcia de Yebenes, 2008). Nevertheless, there is a suggested relationship between anhedonia, cognitive dysfunction, and glial cells in a number of neuropsychiatric disorders. Thus, our findings may have implications beyond depression. Indeed, a current active discussion in psychiatry is underway as to whether psychiatric disorders are best conceived as separate illnesses with separate pathophysiologies or as illnesses along several dimensions of disorder, such as mood or cognitive disorder, with overlapping underlying pathologies in particular neurotransmitter (eg, glutamate) or cellular (eg, glial) systems shared to varying degrees among the illnesses.
DHK treatment provided a model to mimic some aspects of what might occur clinically as a consequence of a paucity or dysfunction of glial cells. This treatment appeared to induce anhedonia, absent dysphoria, and cognitive impairment suggesting that a lack of astrocytic glutamate uptake could have a causal role in producing specific symptoms of depression. Furthermore, DHK induced the activation of a limited number of brain areas that are thought to be key in the regulation of mood. These findings broaden our understanding of the underlying pathophysiology of depression, highlight possible novel sites of action for the development of new treatments, and may be important for other neuropsychiatric disorders that, like depression, include anhedonia and cognitive impairment as symptoms.
References
Anderson CM, Swanson RA (2000). Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32: 1–14.
Andreasen NC, Olsen S (1982). Negative v positive schizophrenia. Definition and validation. Arch Gen Psychiatry 39: 789–794.
Arango V, Underwood MD, Mann JJ (1996). Fewer pigmented locus coeruleus neurons in suicide victims: preliminary results. Biol Psychiatry 39: 112–120.
Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG (1994). Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci 14: 5559–5569.
Banasr M, Duman RS (2008). Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol Psychiatry 64: 863–870.
Baumann B, Bielau H, Krell D, Agelink MW, Diekmann S, Wurthmann C et al (2002). Circumscribed numerical deficit of dorsal raphe neurons in mood disorders. Psychol Med 32: 93–103.
Bechtholt AJ, Valentino RJ, Lucki I (2008). Overlapping and distinct brain regions associated with the anxiolytic effects of chlordiazepoxide and chronic fluoxetine. Neuropsychopharmacology 33: 2117–2130.
Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS et al (2000). Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 47: 351–354.
Bernstein HG, Steiner J, Bogerts B (2009). Glial cells in schizophrenia: pathophysiological significance and possible consequences for therapy. Expert Rev Neurother 9: 1059–1071.
Bowley MP, Drevets WC, Öngür D, Price JL (2002). Low glial numbers in the amygdala in major depressive disorder. Biol Psychiatry 52: 404–412.
Buchsbaum MS, Wu J, Siegel BV, Hackett E, Trenary M, Abel L et al (1997). Effect of sertraline on regional metabolic rate in patients with affective disorder. Biol Psychiatry 41: 15–22.
Burdick KE, Braga RJ, Goldberg JF, Malhotra AK (2007). Cognitive dysfunction in bipolar disorder: future place of pharmacotherapy. CNS Drugs 21: 971–981.
Carlezon Jr WA (2003). Place conditioning to study drug reward and aversion. Methods Mol Med 84: 243–249.
Carlezon Jr WA, Chartoff EH (2007). Intracranial self-stimulation (ICSS) in rodents to study the neurobiology of motivation. Nat Protoc 2: 2987–2995.
Cassidy F, Ahearn E, Murry E, Forest K, Carroll BJ (2000). Diagnostic depressive symptoms of the mixed bipolar episode. Psychol Med 30: 403–411.
Clark L, Chamberlain SR, Sahakian BJ (2009). Neurocognitive mechanisms in depression: implications for treatment. Annu Rev Neurosci 32: 57–74.
Corbett D (1989). Possible abuse potential of the NMDA antagonist MK-801. Behav Brain Res 34: 239–246.
Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall IP (2002). Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb Cortex 12: 386–394.
Cotter D, Mackay D, Landau S, Kerwin R, Everall I (2001). Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch Gen Psychiatry 58: 545–553.
Danbolt NC (2001). Glutamate uptake. Prog Neurobiol 65: 1–105.
Do Carmo GP, Folk JE, Rice KC, Chartoff E, Carlezon Jr WA, Negus SS (2009). The selective non-peptidic delta opioid agonist SNC80 does not facilitate intracranial self-stimulation in rats. Eur J Pharmacol 604: 58–65.
Duman RS (2004). Depression: a case of neuronal life and death? Biol Psychiatry 56: 140–145.
Fallgren AB, Paulsen RE (1996). A microdialysis study in rat brain of dihydrokainate, a glutamate uptake inhibitor. Neurochem Res 21: 19–25.
Gibbs ME, Hutchinson D, Hertz L (2008). Astrocytic involvement in learning and memory consolidation. Neurosci Biobehav Rev 32: 927–944.
Gorwood P (2008). Neurobiological mechanisms of anhedonia. Dialogues Clin Neurosci 10: 291–299.
Gould NF, Holmes MK, Fantie BD, Luckenbaugh DA, Pine DS, Gould TD et al (2007). Performance on a virtual reality spatial memory navigation task in depressed patients. Am J Psychiatry 164: 516–519.
Hamidi M, Drevets WC, Price JL (2004). Glial reduction in amygdala in major depressive disorder is due to oligodendrocytes. Biol Psychiatry 55: 563–569.
Herberg LJ, Rose IC (1990). Excitatory amino acid pathways in brain-stimulation reward. Behav Brain Res 39: 230–239.
Hercher C, Turecki G, Mechawar N (2009). Through the looking glass: examining neuroanatomical evidence for cellular alterations in major depression. J Psychiatr Res 43: 947–961.
Hughes RN (1968). Behaviour of male and female rats with free choice of two environments differing in novelty. Anim Behav 16: 92–96.
Ibarretxe-Bilbao N, Tolosa E, Junque C, Marti MJ (2009). MRI and cognitive impairment in Parkinson's disease. Mov Disord 24 (Suppl 2): S748–S753.
Isella V, Iurlaro S, Piolti R, Ferrarese C, Frattola L, Appollonio I et al (2003). Physical anhedonia in Parkinson's disease. J Neurol Neurosurg Psychiatry 74: 1308–1311.
Johnston GA, Kennedy SM, Twitchin B (1979). Action of the neurotoxin kainic acid on high affinity uptake of L-glutamic acid in rat brain slices. J Neurochem 32: 121–127.
Kennedy SH, Evans KR, Kruger S, Mayberg HS, Meyer JH, McCann S et al (2001). Changes in regional brain glucose metabolism measured with positron emission tomography after paroxetine treatment of major depression. Am J Psychiatry 158: 899–905.
Kim JJ, Song EY, Kosten TA (2006). Stress effects in the hippocampus: synaptic plasticity and memory. Stress 9: 1–11.
Kraus MS, Keefe RS (2007). Cognition as an outcome measure in schizophrenia. Br J Psychiatry Suppl 50: s46–s51.
Law AJ, Harrison PJ (2003). The distribution and morphology of prefrontal cortex pyramidal neurons identified using anti-neurofilament antibodies SMI32, N200 and FNP7. Normative data and a comparison in subjects with schizophrenia, bipolar disorder or major depression. J Psychiatr Res 37: 487–499.
Lee Y, Gaskins D, Anand A, Shekhar A (2007). Glia mechanisms in mood regulation: a novel model of mood disorders. Psychopharmacology (Berl) 191: 55–65.
Maeng S, Zarate Jr CA, Du J, Schloesser RJ, McCammon J, Chen G et al (2007). Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry 63: 349–352.
Marcinkiewicz M, Morcos R, Chretien M (1989). CNS connections with the median raphe nucleus: retrograde tracing with WGA-apoHRP-Gold complex in the rat. J Comp Neurol 289: 11–35.
Mayberg HS, Brannan SK, Mahurin RK, Jerabek PA, Brickman JS, Tekell JL et al (1997). Cingulate function in depression: a potential predictor of treatment response. Neuroreport 8: 1057–1061.
Mayberg HS, Brannan SK, Tekell JL, Silva JA, Mahurin RK, McGinnis S et al (2000). Regional metabolic effects of fluoxetine in major depression: serial changes and relationship to clinical response. Biol Psychiatry 48: 830–843.
Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C et al (2005). Deep brain stimulation for treatment-resistant depression. Neuron 45: 651–660.
Mena MA, Garcia de Yebenes J (2008). Glial cells as players in Parkinsonism: the ‘good,’ the ‘bad,’ and the ‘mysterious’ glia. Neuroscientist 14: 544–560.
Milak MS, Parsey RV, Keilp J, Oquendo MA, Malone KM, Mann JJ (2005). Neuroanatomic correlates of psychopathologic components of major depressive disorder. Arch Gen Psychiatry 62: 397–408.
Miliaressis E, Rompre PP, Laviolette P, Philippe L, Coulombe D (1986). The curve-shift paradigm in self-stimulation. Physiol Behav 37: 85–91.
Müller MB, Lucassen PJ, Yassouridis A, Hoogendijk WJ, Holsboer F, Swaab DF (2001). Neither major depression nor glucocorticoid treatment affects the cellular integrity of the human hippocampus. Eur J Neurosci 14: 1603–1612.
Nestler EJ, Carlezon Jr WA (2006). The mesolimbic dopamine reward circuit in depression. Biol Psychiatry 59: 1151–1159.
Öngür D, Drevets WC, Price JL (1998). Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci USA 95: 13290–13295.
Öngür D, Price JL (2000). The organization of networks within the orbital and medial prefrontal cortex of rats, monkeys and humans. Cereb Cortex 10: 206–219.
Paul IA, Skolnick P (2003). Glutamate and depression: clinical and preclinical studies. Ann NY Acad Sci 1003: 250–272.
Paxinos G, Watson C (2004). The Rat Brain in Stereotaxic Coordinates—the New Coronal Set 5th edn. Academic Press: New York, NY.
Phillips ML, Drevets WC, Rauch SL, Lane R (2003). Neurobiology of emotion perception II: implications for major psychiatric disorders. Biol Psychiatry 54: 515–528.
Pittenger C, Sanacora G, Krystal JH (2007). The NMDA receptor as a therapeutic target in major depressive disorder. CNS Neurol Disord Drug Targets 6: 101–115.
Pizzagalli D, Pascual-Marqui RD, Nitschke JB, Oakes TR, Larson CL, Abercrombie HC et al (2001). Anterior cingulate activity as a predictor of degree of treatment response in major depression: evidence from brain electrical tomography analysis. Am J Psychiatry 158: 405–415.
Rajkowska G (2000). Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol Psychiatry 48: 766–777.
Rajkowska G (2002). Cell pathology in bipolar disorder. Bipolar Disord 4: 105–116.
Rajkowska G, Halaris A, Selemon LD (2001). Reductions in neuronal and glial density characterize the dorsolateral prefrontal cortex in bipolar disorder. Biol Psychiatry 49: 741–752.
Rajkowska G, Miguel-Hidalgo JJ, Dubey P, Stockmeier CA, Krishnan KR (2005). Prominent reduction in pyramidal neurons density in the orbitofrontal cortex of elderly depressed patients. Biol Psychiatry 58: 297–306.
Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY et al (1999). Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry 45: 1085–1098.
Rajkowska G, O’Dwyer G, Teleki Z, Stockmeier CA, Miguel-Hidalgo JJ (2007). GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology 32: 471–482.
Robinson MB, Hunter-Ensor M, Sinor J (1991). Pharmacologically distinct sodium-dependent L-[3H]glutamate transport processes in rat brain. Brain Res 544: 196–202.
Sanacora G, Zarate CA, Krystal JH, Manji HK (2008). Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov 7: 426–437.
Sheline YI, Gado MH, Kraemer HC (2003). Untreated depression and hippocampal volume loss. Am J Psychiatry 160: 1516–1518.
Song L, Che W, Min-Wei W, Murakami Y, Matsumoto K (2006). Impairment of the spatial learning and memory induced by learned helplessness and chronic mild stress. Pharmacol Biochem Behav 83: 186–193.
Sun MK, Alkon DL (2004). Induced depressive behavior impairs learning and memory in rats. Neuroscience 129: 129–139.
Tomasiewicz HC, Todtenkopf MS, Chartoff EH, Cohen BM, Carlezon Jr WA (2008). The kappa-opioid agonist U69,593 blocks cocaine-induced enhancement of brain stimulation reward. Biol Psychiatry 64: 982–988.
Tzschentke TM (2007). Measuring reward with the conditioned place preference (CPP) paradigm: update of the last decade. Addict Biol 12: 227–462.
Valentine GW, Sanacora G (2009). Targeting glial physiology and glutamate cycling in the treatment of depression. Biochem Pharmacol 78: 431–439.
Videbech P (2000). PET measurements of brain glucose metabolism and blood flow in major depressive disorder: a critical review. Acta Psychiatr Scand 101: 11–20.
Whishaw IQ, Auer RN (1989). Immediate and long-lasting effects of MK-801 on motor activity, spatial navigation in a swimming pool and EEG in the rat. Psychopharmacology (Berl) 98: 500–507.
Wu J, Buchsbaum MS, Gillin JC, Tang C, Cadwell S, Wiegand M et al (1999). Prediction of antidepressant effects of sleep deprivation by metabolic rates in the ventral anterior cingulate and medial prefrontal cortex. Am J Psychiatry 156: 1149–1158.
Wu JC, Buchsbaum M, Bunney Jr WE (2001). Clinical neurochemical implications of sleep deprivation's effects on the anterior cingulate of depressed responders. Neuropsychopharmacology 25 (5 Suppl): S74–S78.
Zarate Jr CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA et al (2006). A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63: 856–864.
Zink M, Vollmayr B, Gebicke-Haerter PJ, Henn FA (2010). Reduced expression of glutamate transporters vGluT1, EAAT2 and EAAT4 in learned helpless rats, an animal model of depression. Neuropharmacology 58: 465–473.
Acknowledgements
This work was supported by grants from and the National Alliance for Research on Schizophrenia and Depression (NARSAD—AJBG), the Jerome Lyle Rappaport Charitable Foundation (AJBG), the Shervert Frazier Research Institute (BMC), the Englehard Foundation (BMC), and the National Institutes of Mental Health (MH087695, AJBG; MH063266, WAC). We thank Elena Chartoff for providing the c-Fos antibody.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors have no conflicts of interest relating to this report and Dr Bechtholt-Gompf, Hali Walther, and Martha Adams have no other potential conflicts to report. Dr Carlezon has a US patent covering the use of kappa antagonists in the treatment of depression (Assignee: McLean Hospital) and is a member of a collaborative group, including Dr Cohen, that has submitted a patent application covering the synthesis and use of salvinorin derivatives (Assignees: McLean Hospital and Temple University). In the last 3 years Dr Carlezon has received compensation for professional services from The American College of Neuropsychopharmacology, Huya Bioscience International, Infinity Pharmaceuticals, Lantheus Medical Imaging, The Society for Neuroscience, and Transcept Pharmaceuticals. Dr Öngür received Riluzole for a study from Sanofi-Aventis during the past 3 years. Dr Cohen has three additional pending patents on pyrimidines to treat bipolar disorders, kappa agonists in bipolar mania, and mitochondrial replacement therapy, and a book on bipolar disorder that will be published by Jossey-Bass/Wiley within the next year.
Rights and permissions
About this article
Cite this article
Bechtholt-Gompf, A., Walther, H., Adams, M. et al. Blockade of Astrocytic Glutamate Uptake in Rats Induces Signs of Anhedonia and Impaired Spatial Memory. Neuropsychopharmacol 35, 2049–2059 (2010). https://doi.org/10.1038/npp.2010.74
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2010.74
