
Abstract
Given the mucosal transmission of HIV-1, we compared whether a mucosal vaccine could induce mucosal cytotoxic T lymphocytes (CTLs) and protect rhesus macaques against mucosal infection with simian/human immunodeficiency virus (SHIV) more effectively than the same vaccine given subcutaneously. Here we show that mucosal CTLs specific for simian immunodeficiency virus can be induced by intrarectal immunization of macaques with a synthetic-peptide vaccine incorporating the LT(R192G) adjuvant. This response correlated with the level of T-helper response. After intrarectal challenge with pathogenic SHIV-Ku2, viral titers were eliminated more completely (to undetectable levels) both in blood and intestine, a major reservoir for virus replication, in intrarectally immunized animals than in subcutaneously immunized or control macaques. Moreover, CD4+ T cells were better preserved. Thus, induction of CTLs in the intestinal mucosa, a key site of virus replication, with a mucosal AIDS vaccine ameliorates infection by SHIV in non-human primates.
Similar content being viewed by others

Main
The mucosal surface is a major natural route of HIV-1 entry and the gut is a major site of HIV and simian immunodeficiency virus (SIV) replication1. Thus, a successful HIV/SIV vaccine will likely need to induce mucosal immunity2,3,4,5. Recent studies6,7,8,9 of viral mucosal transmission indicate that cytotoxic T lymphocytes (CTLs) are an important component of the mucosal immune barrier. Therefore, we investigated whether CTLs generated in the gut mucosa by mucosal immunization may be more effective than CTLs generated elsewhere by systemic immunization.
In previous murine studies, we designed and tested prototype HIV synthetic-peptide vaccines consisting of a multideterminant peptide containing a cluster of overlapping helper epitopes (a cluster peptide)10 colinearly synthesized with an epitope for neutralizing antibodies and CTLs (refs. 3,11). This peptide vaccine elicited neutralizing antibodies, CTLs and helper T cells12,13 as did a similar vaccine studied by others in mice and monkeys14,15,16. This vaccine, with cholera toxin (CT) adjuvant, was more effective given intrarectally than intranasally or intragastrically in eliciting both mucosal and systemic CTLs (ref. 2). Intrarectal immunization generated strong CTL responses in both mucosa and spleen, whereas subcutaneous (s.c.) immunization elicited only systemic CTLs (ref. 2). Such compartmentalization was also seen in mice by others17, but has not been demonstrated in primates. Intrarectally immunized mice were resistant to mucosal transmission of a recombinant vaccinia expressing HIV-1 gp160 (vPE16)2,6. Protection was CD8-dependent and required CTL in the mucosa6, suggesting that to prevent mucosal transmission, an AIDS vaccine would need to induce CTL in the mucosa itself.
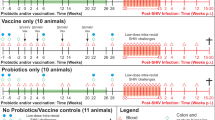
Here we investigated whether these results would translate to retroviral infection in non-human primates. Mucosal and systemic immunization with the same AIDS vaccine have not been compared previously side-by-side in primates, and the role of gut mucosal CTL in clearing virus from this major SIV reservoir has not been examined. We again used a peptide vaccine to avoid viral vectors that might spread when comparing routes to determine the impact of CTL on a limited number of epitopes, and to avoid inducing enhancing antibodies. We made analogous peptide vaccines, that had the same helper cluster peptide segments, but with several CTL epitopes from SIV Gag and Pol presented by the rhesus major histocompatibility (MHC) class I molecule, Mamu-A*01 (refs. 18–20) (Fig. 1). The cluster helper peptides were broadly recognized by helper T cells from humans, mice and macaques3,10,12 (and data not shown). All the animals except the controls received a mixture of all 4 peptides. As a less toxic adjuvant, we substituted for CT a mutant E. coli labile toxin LT(R192G)21,22, that we had found more effective in inducing a Th1 cytokine pattern and stronger CTL response7. For comparison with animals immunized intrarectally with peptides and LT(R192G) (group C), other animals were immunized s.c. with the peptides in Montanide ISA 51 (ref. 13) (group B), or intrarectally with LT(R192G) alone (group A) (Fig. 1). For intrarectal challenge, we chose simian/human immunodeficiency virus (SHIV)-Ku2 (refs. 23,24), a pathogenic strain expressing HIV-1 IIIB gp160, including the helper epitopes, and SIV Gag and Pol, including the CTL epitopes.

a, Peptide vaccine constructs. The helper portion is underlined. A mixture of all 4 peptides was used in all the immunized animals. b, Scheme of immunizations and challenge, and groupings of Mamu-A*01-positive macaques.
Induction of mucosal CTLs and helper T cells in primates
We first compared the CTLs and helper responses to the peptide vaccine delivered mucosally or s.c. after each immunization cycle (Fig. 1). After the first cycle, three of five intrarectally immunized macaques (group C) (694L, 698L, and 699L) had CTLs specific for the immunodominant Gag181 CL10 SIV (CTPYDINQML) epitope in mesenteric lymph nodes (MLN), and in two animals we obtained a large enough colonic biopsy to measure modest CTL responses in the colon (data not shown). All four s.c.-immunized macaques (group B) had substantial CL10-specific CTLs in the axillary lymph nodes (ALN), and two had these CTLs in peripheral blood mononuclear cells (PBMCs). Thus, one cycle of four intrarectal doses did not induce any higher a CTL response than four s.c. doses in the respective target sites of each immunization route.
However, after two cycles of four intrarectal vaccine doses, three of the five macaques (694L, 695L and 699L) made substantial Gag CL10 SIV-specific CTL responses in the MLN and colonic lamina propria as well as in systemic lymphoid tissues, that is, the ALN and PBMCs (Fig. 2). These animals also had CTLs to the Pol and other Gag epitope in the MLN (Fig. 2) and similarly in ALN (data not shown). Although macaques 697L and 698L failed to manifest a response against Gag 181 CL10 in any tissue, macaque 698L did mount a strong CTL response in the MLN (Fig. 2) and ALN (data not shown) against the SIV Gag 372 epitope, which was also part of the vaccine. Overall, SIV-specific Mamu-A*01-restricted CTLs were found both in mucosa and in systemic lymphoid tissues of four of five macaques given two cycles of intrarectal vaccine.

▪, lysis of targets with specific peptide; □, lysis of control targets without peptide. E:T, effector to target ratio.
All four macaques (701L-704L) given two cycles of s.c. vaccine displayed substantial CL10 SIV Gag-specific CTLs in the ALN, and in the two macaques with the highest of these responses (701L and 702L), in PBMCs and the MLN as well (Fig. 3). These latter two macaques also responded to the Gag 372 and the Pol 143 epitopes in the ALN (Fig. 3). However, none of the control macaques (group A) exhibited CTL responses in any tissue (data not shown).

▪, lysis of targets with specific peptide; □, lysis of control targets without peptide.
To further evaluate the CTL responses in immunized macaques, we quantified the number of CL10 Gag-specific CD8+ T cells by stimulating in vitro with CL10 Gag-specific peptide and analyzing the CD8+ CD3-gated T cells by flow cytometry using Mamu-A*01/C9M tetramers. Although the number of tetramer-positive cells in freshly isolated tissues from immunized animals was not significantly greater than in control animals (data not shown), macaque 694L of group C with the highest level of colonic CTLs showed a substantial number of tetramer-positive cells in stimulated cells from the colon (9%), MLN (2%) and ALN (9%). The rank of tetramer-positive cells generally correlated with that of CTLs. Similarly, in macaques 701L and 702L, with the highest CTL levels among s.c.-immunized macaques, tetramer-positive cell levels in the ALN were higher than in other group B macaques (17 and 25% versus 6 and 5%). Thus, tetramer staining and CTL lytic activity gave the same rank order of response for macaques in both the intrarectal- and s.c.-immunized groups.
To assess helper responses, we measured in vitro T-cell proliferation induced by the helper peptides in MLN or ALN cells after two vaccine cycles (Fig. 4a). Although the macaques were all selected for Mamu-A*01, they were outbred for MHC class II and therefore heterogeneous in their helper T-cell response. In group C, the highest helper T-cell proliferative responses were noted in macaques 694L, 695L and 698L, which displayed the highest CTL responses, whereas macaque 697L, which exhibited no CTLs, showed only background proliferative responses (stimulation index ∼1). Only in macaque 699L was a disparity between proliferation and cytolytic activity noted. A similar pattern was seen in group B, in that macaques 701L and 702L with the highest CTL responses also displayed the highest proliferative responses. This correlation between levels of vaccine-induced proliferative response and CTLs (Fig. 4b) (P < 0.002), which has not been previously examined in a primate, suggests that T-helper responses have an important role in the induction of optimal CTL responses in both mucosal and systemic tissues, and underscores the importance of this vaccine component.

a, In vitro proliferative response against T-helper peptides PCLUS3 (▪) and PCLUS6.1 (□) after 8 intrarectal (694L-699L) or s.c. (701L-704L) immunizations. The stimulation index was calculated as the ratio of thymidine incorporation into cellular DNA in wells with antigen over that in control wells with medium alone. b, Correlation of proliferative response to PCLUS3 with CTL response. ○, group C MLN; ●, group B ALN; r = 0.87; P = 0.002.
Notably, a Mamu-A*01-restricted CTL epitope has been identified in PCLUS3 (YAPPISGQI)25. We think it unlikely that CD8+ T cells represent a major part of the proliferative response, because unless exogenous IL-2 is added, most of this response is from CD4+ T cells. To confirm this interpretation, using antibodies to CD4 or CD8 and magnetic beads, we depleted the PBMCs of two representative animals that had remaining cells. The proliferative response was enriched in the CD8-depleted population and diminished in the CD4-depleted population, confirming that approximately 65–93% of the response was due to CD4+ T cells (data not shown). In addition, after the second cycle, we detected no antibodies to PCLUS6.1 in any of the animals, nor to PCLUS3 in any of group C, but we detected low levels against PCLUS3 in three group B macaques (701L, 703L and 704L) (data not shown).
Vaccine effect on intrarectal challenge with SHIV-Ku2
Eight weeks after the two cycles of peptide vaccine, all of the macaques were given an additional dose of vaccine plus adjuvant or adjuvant alone and then, two weeks later (week 25), were exposed intrarectally to approximately ten 50% animal infectious doses (AID50) of pathogenic SHIV-Ku2 (ref. 23, 24) (one ill macaque was removed; see Methods). One macaque in each of groups A and C did not become viremic (as detected by the NASBA assay for viral RNA) whereas the remaining 9 macaques exhibited a peak plasma viral load between 1 × 105–107 mRNA copies per ml within 3 weeks following challenge exposure (Fig. 5). However, by the more sensitive PCR assay for proviral DNA in cells, we were able to detect infection in spleen cells in these two animals later at necropsy. As neither of these animals had a detectable immune response, it is unlikely that lack of viremia was due to an immune response, but probably represents a technical difficulty in obtaining reproducible infection in macaques by mucosal challenge. Therefore in addition to analyzing the data on the whole group, we have also examined the subset of animals that did become viremic, as only in the latter group could the effect of immunization on plasma viral load kinetics be followed.

a–c,Viral load is expressed as viral RNA copies per ml plasma (by NASBA assay) versus time after challenge for macaque groups A–C. a, Group A, immunized intrarectally with LT(R192G) only. ●, 745L; +, 746L; ▵, 747L. b, Group B, immunized s.c. with HIV/SIV peptides in Montanide ISA51. ▵, 701L; ♦, 702L; ●, 703L; +, 704L. c, Group C, intrarectally immunized with the HIV/SIV peptides plus LT(R192G). ♦, 694L; ▵, 695L; ⋄, 698L; ●, 697L. By a non-parametric repeated measures ANOVA, intrarectal group C viral titers were significantly lower than those of s.c. group B (P = 0.013), the critical comparison, and showed a trend toward difference from those of group A (P = 0.14), whereas s.c. group B was not significantly different from control group A (P = 0.92). Examining only macaques with detectable viremia, as the kinetics of viral load could not be followed in the two others, the significance levels improved for group C versus A (P < 0.01), and the differences remained significant for group C versus B (P = 0.031) and not significant for group B versus A (P = 0.65). The overall comparison of A versus B versus C was significant (P = 0.017). d, Typical time-course of mean viral load in the viremic subset macaques of groups A (circles), B (triangles), and C (squares), curve-fit as described in the Methods. Fit parameters: group A, b = 0.076, c = 0.0005; group B, b = 0.071, c = 0.0003; group C, b = 0.0693. Log(V) is log10 of the viral load. e, Time-course of mean CD3+CD8+ T lymphocyte counts per μl in the subsets of macaques in groups A (○), B (▵), and C (▪) that were actually viremic, parallel to d. f and g, At the several time- points during the steady-state phase of the infection process (35–176 d), PBMCs were analyzed by FACScan for the absolute number of CD3+CD8+ or CD4+ cells per ml, and plotted as a scatter diagrams comparing group C (▪) with group A (○) in f or with group B (▵) in g. In both cases, in the vertical dimension, group C exhibited larger numbers of CD3+CD4+ cells than groups A or B (P = 0.005 and P = 0.004, respectively, by the Mann–Whitney test). The absolute numbers of CD3+CD8+ cells were higher in group C than group A (P = 0.01), but not significantly different between group C and group B.
The outcome of SHIV infection in intrarectally immunized macaques (group C) differed substantially from the s.c. immunized (group B) or control macaques (group A). In the peak viral load, the differences between groups were not statistically significant (Fig. 5a–c). However, in all the group C macaques that became viremic, the viral load decreased substantially between days 20 and 70 after challenge, and the viral load reached a 'set point' below the level of detection (2000 mRNA copies/ml) before day 80. By contrast, in group B, one animal had a set point greater than 1 × 105 copies per ml; two fluctuated between 2 × 103 and 3 × 104 copies per ml; and one fluctuated up to 1 × 104 on day 63, but fell below the detection limit after day 70 (Fig. 5a–c).
Statistical analysis showed that the concentration of SIV mRNA in the circulation dropped with similar half-lives (t1/2 of 10 days, 9.8 days and 9.2 days in groups C, B and A, respectively); however, in macaques immunized intrarectally, viral mRNA fell to an undetectable level by day 63, and remained undetectable subsequently (Fig. 5d). At day 63, early in the set- point, groups B and C were statistically different according to the Wilcoxon rank-sum test (P < 0.05). Moreover, the viral loads at all time points measured during the steady-state or set-point phase (days 63–200) showed a significant difference between the intrarectal group C and the s.c. group B by a non-parametric repeated measures ANOVA26,27 (see Fig. 5 legend). This comparison remained statistically significant even when the number of time-points in the analysis was reduced. Thus, despite small sample sizes, these results suggest that mucosal immunization was more effective than s.c. immunization.
In addition to plasma viral load, we also assessed the number of both CD4+ and CD8+ T cells and clinical course. The former was assessed by plotting scatter plots of the absolute number of CD4+ and CD8+ T cells in all peripheral blood specimens taken at various times starting on day 35 after viral challenge in all viremic macaques. By inspection, CD4 counts for group C are mostly higher than those for group A (Fig. 5f; P = 0.005) or group B (Fig. 5g; P = 0.004). Although CD8+ T-cell counts differed less (horizontal axis), some expansion of CD8+ T cells was noted in a kinetic plot (Fig. 5e). Clinically, in each of groups A and B the animal with the highest viral load developed evidence of opportunistic infection (746L, gingivitis and pneumonia; 704L, severe gingivitis, blepharitis, facial dermatitis and pathologic evidence of pulmonary pneumocystis). None of the animals in group C showed evidence of AIDS-like illness.
Two hundred days after SHIV-Ku2 virus challenge, we killed the macaques and determined CTL responses in colon and ALN, and viral loads in gut tissue. We could measure CTL activity in freshly obtained cells without stimulation in vitro, as has been seen in HIV infection28. Notably, compared with control target background, intrarectally immunized group C macaques that exhibited viral set-points below the level of detection (694L, 695L and 698L) consistently exhibited substantially higher CL10 SIV Gag-specific CTL activity in the colon than s.c. immunized group B macaques (Fig. 6a), and control group A had almost none. In contrast, while CTL levels in ALN were also increased after viral challenge, in this case the increase in both groups was equivalent. In both cases, macaques that made a strong CTL response after vaccination were the ones that showed activity ex vivo 200 days after viral infection. These results suggest that the vaccine-primed CTL memory cells were reactivated by the SHIV infection, as recently reported for a DNA vaccine29.

Intestinal tissue and ALN were obtained at the time of necropsy, 200 d after the challenge with pathogenic SHIV-ku2. a and b, Freshly isolated lymphocytes from colonic lamina propria (a) and ALN (b) were used as effector cells, without restimulation in vitro, at an effector-to-target ratio of 100:1. Results show specific lysis of peptide-pulsed targets less that of control unpulsed targets. c and d, Viral RNA load in the colon (c) and jejunum (d) were determined on tissues obtained at the same time-point. The lower limit of detection was 2000 copies per 6 mg tissue, corresponding to a log10 value of 3.3 on the ordinate. In the colon (c), although the numbers are small, group C differs significantly by the χ2 median test from group A (P = 0.014) and from group B (P = 0.005). In the jejunum (d), group C is different from A (P = 0.008) and from group B (P = 0.05). In all cases, groups A and B are not significantly different from each other.
These data suggest an explanation for the lower plasma set- points in the intrarectally immunized macaques as the gut mucosa is a major site of viral replication1; we therefore hypothesized that higher levels of CTLs at this site might lead to better control of infection. Such control of virus in the gut mucosa has not been previously examined. To test this new hypothesis, we measured virus levels in the colon and jejunum at the time of necropsy, 200 days after challenge (Fig. 6c and d). Notably, none of the group C animals had detectable virus (< 2000 copies) in colon or jejunum, whereas the group A and B animals all had levels 10–100-fold over the detection limit (except for animal 703L, that also had the lowest plasma viral load in group B). Thus, the difference in viral load in the major gut reservoir is even more pronounced than the difference in viremia. Group C was significantly different from groups B and A (see figure legend). In the colon, virus levels were largely inversely related to CTL levels (Fig. 6a and c). These results indicate that the better control of plasma viremia in the intrarectally immunized macaques was mediated by a higher level of CTLs in the gut. Virus was more effectively cleared from this major site of replication, which seeds the blood stream. This key role of gut mucosal CTLs in control of HIV/SIV infection has not been previously recognized.
Discussion
In these studies we demonstrated by several independent criteria that intrarectally administered synthetic multiepitope HIV/SIV peptide vaccine with mutant LT (R192G) as an adjuvant induces a mucosal CTL response. This novel vaccine provided better protection against intrarectal SHIV infection than a s.c.-administered vaccine comprising the same peptides that induce as high or higher systemic CTL responses. Reduction of viremia correlated with preservation of CD4+ T-cell counts, with post-infection CTL activity in the gut and with clearance of SHIV from the gut mucosal tissues. These data represent the first evidence from non-human primates for the concept that HIV/SIV-specific CTL responses in the mucosa are at least as important as systemic CTLs in protection against HIV/SIV infection. This is not only because of the importance of mucosal transmission, but also because the gut is a major reservoir of virus. This fundamental concept will be important for the optimal design of effective AIDS vaccines.
Neither of the immunization regimens led to CTL responses sufficient to reduce initial viral expansion and dissemination. Nevertheless, intrarectal immunization with the peptide vaccine did have a positive impact on infection during the plateau phase, in that the set point actually dropped below the level of detection. The difference in viremia between intrarectal and s.c. groups was significant whether or not one included the two animals that never became viremic, although without these two, the smaller number of animals reduced the power of the test and made the difference, although still significant, statistically less robust. Because these animals had no detectable immune response, their lack of viremia is likely due to inadequate intrarectal challenge, and in any case, prevented study of the kinetics of their viremia.
Intrarectal immunization also led to greater preservation of CD4+ T cells than s.c. immunization. This is another measure of infection outcome, independent of viral load, indicating that intrarectal immunization is superior to s.c. immunization in protection against intrarectal infection.
We hypothesize that rectal immunization with peptide vaccine was able to greatly reduce viral replication in the main site of viral burden, the mucosal tissues, because mucosal immunization induced mucosal memory CTLs. Indeed, protection correlated best with CTLs present in the colon after challenge, and the difference between intrarectal and s.c. immunization in reducing viral burden in the gut was even more pronounced than that in plasma (Fig. 6). A recent study of a systemically administered DNA vaccine showed a similar reduction in plasma viral set-point, but only when given with a chimeric interleukin-2/immunogloblulin construct29. Mucosal immunization may achieve the result without cytokine amplification because it focuses the CTLs in the major site of viral replication. Other studies have shown that animals previously infected with SIV, which is known to infect the gut and could induce mucosal immunization, were protected against mucosal transmission of SIV (refs. 8,30). Murphey-Corb et al.8 found that during SIV infection, protection correlated in some animals with CTLs in the jejunum. Targeted iliac lymph node immunization has also protected against mucosal SIV infection (ref. 4) A recent study of DNA/modified vaccinia Ankara (MVA) prime-boost strategy also showed reduction of viral load31. Here we establish the importance of mucosal immunization by a direct comparison of mucosal and systemic immunization against SIV/HIV in a primate, and by examining the effect on viral levels in the gut reservoir.
Such focusing of CTLs in the gut tissues where HIV and SIV replicate depends not only on immunization through a mucosal route, as we have done, but also on the existence of a compartmentalized mucosal CTL response. We2,32 and others17 have seen this compartmentalization in mice, but not in primates. This asymmetry in trafficking suggests that mucosal immunization, which can induce CTLs in both compartments (in contrast to systemic immunization), may be effective at eliminating both systemic as well as mucosal virus. Our results also suggest that this compartmentalization applies to non-human primates, although definitive proof is lacking because we could not obtain colonic biopsies from the s.c.-immunized macaques before challenge. Furthermore, post-challenge CTL responses in the colon were higher in the intrarectally immunized group than the s.c. group. Because the control-infected animals had almost no specific colonic CTLs, such CTLs likely represent in part a secondary response induced by infection but dependent on priming by the vaccine. Thus, these data provide further evidence for more effective priming of gut CTLs by mucosal immunization.
It is becoming clear that the outcome of SIV/HIV infection depends on the interplay of the CD4+ T-helper function and the preservation of CD8+ CTL function33. With regard to the need for helper T-cell induction by vaccines, we and others have demonstrated the importance of a linked helper epitope for CTL induction in mice34,35,36, but the requirement for help to induce CTLs is not known in primates. Thus, our results are important in showing that SIV-specific CTL activity correlated with the HIV helper epitope-specific T-cell proliferative activity, not previously reported in a primate.
We are not convinced that we have optimized mucosal immunization. We used mutant E. coli labile toxin LT(R1926)22 as an alternative adjuvant to the more usual CT, because it is less toxic and does not inhibit endogenous interleukin-12 production as does CT (refs. 7,37). It is also apparent that we had to vary the adjuvant at the same time we varied the immunization route. A recent study by Allen et al.38 showed that CTLs specific for a single TAT epitope can select for new viral variants within two months of infection. Our data show that even a synthetic vaccine with only two helper and three CTL epitope peptides, given mucosally, can impact the course of pathogenic SHIV infection. A mucosal vaccine that incorporates more epitopes and immuno-enhancing molecules may be even more effective at controlling virus infection by reducing the viral reservoir in the gut, independent of the route of exposure.
Methods
Macaques and vaccines.
Indian rhesus macaques (Macaca mulatta) were matched for genetic origin, source, and comparable age and weight, and maintained in accordance with guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care International and under the approval of the Applied BioScience Labs Animal Center Review Committee. All were seronegative for SIV, simian retroviruses 1, 2 and 5, and simian T-cell leukemia/lymphotropic virus type 1 (STLV-1) prior to the study, and were Mamu-A*01-positive MHC type as determined by PCR-sequence–specific primers and direct sequencing39. The peptides in Fig. 1 each contain an HIV-envelope helper epitope10 and an SIV Gag19 or Pol20 CTL epitope.
SHIV-Ku2 virus stock.
SHIV-Ku2 is a chimeric virus containing the HIV-1 IIIB strain (HXBc2) envelope gene and SIVmac239 gag and pol genes, and is pathogenic in rhesus macaques23,24. SHIV-Ku2 stock was isolated from PBMC of SHIV-Ku2–infected macaques and titered in vivo intrarectally using 10-fold serial dilutions. Inoculation with 1 ml undiluted virus infected 8/8 (100%), as shown by the SIV plasma RNA within 2 weeks after virus challenge. Virus stock diluted 1:10 infected 2/4 macaques (50%), and none was infected with dilutions ≥ 1:100. Thus, the undiluted virus challenge dose contained ∼10 AID50 for intrarectal administration.
Immunization and challenge of macaques.
Each intrarectal peptide vaccine dose contained 0.5 mg of each peptide (total 2 mg peptide) mixed with mutant Escherichia coli labile toxin LT(R192G) as mucosal adjuvant (50 μg per immunization)22. Each s.c. vaccine dose was administered in the upper arm with the same peptide mixture emulsified 1:1 with Montanide ISA 51 adjuvant (Seppic, Fairfield, New Jersey). Control macaques received intrarectal LT(R192G) alone (50 μg per dose) by the same schedule (Fig. 1). For intrarectal inoculations, animals were sedated with ketamine hydrochloride (10 mg/kg i.m.) and placed within a biosafety cabinet in ventral recumbency with the hindquarters elevated. The tail was elevated dorsally and a 3-cc slip-tip syringe was atraumatically inserted into the rectum. Vaccine or virus was administered and the tail lowered assuring complete delivery.
Before challenge, one animal, #699L, that had received the intrarectal vaccine (group C), developed peritonitis following laparatomy for the final colonic biopsy, requiring urgent surgery to remove a large segment of devitalized bowel, and nearly died. Thus, shortly prior to challenge with SHIV-Ku2 the immune system of this macaque was under considerable stress, as corroborated by greatly decreased numbers of circulating lymphocytes (including CD8+ T cells), monocytes and neutrophils immediately prior to challenge. Although challenged, this animal could not be included in the analysis because of the apparent immune deficiency related to the severe peritonitis.
Biopsies and isolation of lamina propria lymphocytes.
MLN and intestinal tissue were obtained by laparotomy. MLN were chosen as an interface between intestinal and systemic immune systems, and para-aortic and para-iliac nodes were not accessible in this procedure. Intestinal tissue was obtained either from colonic-wedge resection (∼4 cm2) at laparotomy, or at the time of necropsy (∼100 cm2). Lymphocytes were prepared as previously established for human tissue40.
Lymphocyte proliferation assay.
Lymphocytes from blood and lymph nodes were freshly isolated by density-gradient centrifugation on Ficoll, resuspended in complete T-cell medium (CTM)6, and cultured at 3 × 105 cells per well in 96-well plates for 3 d in the absence or presence of T-helper peptides (PCLUS3 or PCLUS6.1)10,41, using PHA as a positive control, and then pulsed for 8 h with 1 mCi [3H]thymidine.
CTL and tetramer assays.
Immune cells were cultured 7 d at 5 × 106 per ml in 12-well plates with separate synthetic SIV CTL epitope peptides in CTM. On day 3 rhIL-2 (Boehringer) (20 international units per ml) was added. Autologous B-lymphoblastoid cell line (B-LCL) target cells were pulsed with 1 μM peptide for 2 h during 51Cr labeling, and cultured with effectors at 37 °C for 4 h. Specific release was calculated as described6,32. Spontaneous release was < 15% of maximal release in all assays. Soluble tetrameric Mamu-A*01/CM9 complex conjugated to PE-labeled streptavidin prepared as described19,42 was used to stain CD8+ cells gated on CD3+ cells (as CD8+-specific monoclonal antibodies can bind to natural killer cells of rhesus macaques43) and analyzed on FACScan (Becton-Dickinson).
Determination of viral load.
SIVmac251 mRNA in plasma was quantified by nucleic acid sequence-based amplification (ref. 44), with a detection limit of 2 × 103 RNA copies. For colon and jejunum, snap-frozen samples of 300–500 mg were homogenized and lysed in 1 ml of lysis buffer. 20-μl samples were analyzed, and the results normalized to 6 mg tissue.
For detection of virus in spleen cells at necropsy, thawed cells were washed in medium with 10% FCS, centrifuged at 10,000g for 15 min, lysed in DNA-STAT (Tel-Test, Friendswood, Texas), and processed according to the directions. The DNA was resuspended in 10 mM Tris and 0.1 mM EDTA, and amplified in 50 μl PCR buffer, 2 μM primers, and 200 μM NTPs, for 25 cycles of 95 °C (30 s), 50 °C (30 s) and 72 °C (3 min). The amplified product was run on a 2% agarose gel. The sensitivity was about 1000 copies of DNA per sample.
Software for fitting and statistical analysis.
The kinetics of plasma viral mRNA during the 'decay stage' (21 d after challenge) were analyzed by an exponential function: V(t) = Vie−bt, where Vi is the maximum number of mRNAs extrapolated on day 0, b is the rate of mRNA decline in plasma, and t is time (days). Alternatively we used the model, V(t) = Vie−bt + ct⁁2 (where c is the non-negative constant), which assumes a 'virus-escape' mechanism. Curve fitting and statistical analysis were performed in MLAB (Civilized Software, Silver Spring, Maryland), and parameters estimated using the Marquardt–Levenberg method. Statistica-6 (StatSoft, Tulsa, Oklahoma) and SigmaPlot-2000 (SSPS, Chicago, Illinois) were also employed. Statistical comparisons were performed using the Mann–Whitney, Wilcoxon, Kruskal–Wallis and χ2 tests, as well as a non-parametric repeated measures ANOVA for plasma viral loads over time26,27 because the data could not be taken as normally distributed. We found no significant interaction between time and treatment by this repeated measures ANOVA, consistent with the initial assumption of lack of interaction between time and treatment. Software for the non-parametric repeated measures ANOVA is available from V. Kuznetsov (vk28u@nih.gov).
References
Veazey, R.S. et al. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science 280, 427–431 (1998).
Belyakov, I.M. et al. Mucosal immunization with HIV-1 peptide vaccine induces mucosal and systemic cytotoxic T lymphocytes and protective immunity in mice against intrarectal recombinant HIV-vaccinia challenge. Proc. Natl. Acad. Sci. USA 95, 1709–1714 (1998).
Berzofsky, J.A. et al. Approaches to improve engineered vaccines for human immunodeficiency virus (HIV) and other viruses that cause chronic infections. Immunol. Rev. 170, 151–172 (1999).
Lehner, T., Bergmeier, L., Wang, Y., Tao, L. & Mitchell, E. A rational basis for mucosal vaccination against HIV infection. Immunol. Rev. 170, 183–196 (1999).
Czerkinsky, C. et al. Mucosal immunity and tolerance: relavance to vaccine development. Immunol. Rev. 170, 197–222 (1999).
Belyakov, I.M. et al. The importance of local mucosal HIV-specific CD8+ cytotoxic T lymphocytes for resistance to mucosal–viral transmission in mice and enhancement of resistance by local administration of IL-12. J. Clin. Invest. 102, 2072–2081 (1998).
Belyakov, I.M., Ahlers, J.D., Clements, J.D., Strober, W. & Berzofsky, J.A. Interplay of cytokines and adjuvants in the regulation of mucosal and systemic HIV-specific cytotoxic T lymphocytes. J. Immunol. 165, 6454–6462 (2000).
Murphey-Corb, M. et al. Selective induction of protective MHC class I restricted CTL in the intestinal lamina propria of rhesus monkeys by transient SIV infection of the colonic mucosa. J. Immunol. 162, 540–549 (1999).
Kaul, R. et al. HIV-1-specific mucosal CD8+ lymphocyte responses in the cervix of HIV-1-resistant prostitutes in Nairobi. J. Immunol. 164, 1602–1611 (2000).
Berzofsky, J.A. et al. Construction of peptides encompassing multideterminant clusters of HIV envelope to induce in vitro T-cell responses in mice and humans of multiple MHC types. J. Clin. Invest. 88, 876–884 (1991).
Takahashi, H. et al. An immunodominant epitope of the HIV gp160 envelope glycoprotein recognized by class I MHC molecule-restricted murine cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 85, 3105–3109 (1988).
Ahlers, J.D. et al. Construction of an HIV-1 peptide vaccine containing a multideterminant helper peptide linked to a V3 loop peptide 18 inducing strong neutralizing antibody responses in mice of multiple MHC haplotypes after two immunizations. J. Immunol. 150, 5647–5665 (1993).
Ahlers, J.D. et al. Candidate HIV type 1 multideterminant cluster peptide-P18MN vaccine constructs elicit type 1 helper T cells, cytotoxic T cells, and neutralizing antibody, all using the same adjuvant immunization. AIDS Res. Hum. Retroviruses 12, 259–272 (1996).
Palker, T.J. et al. Polyvalent human immunodeficiency virus synthetic immunogen comprised of envelope gp120 T helper cell sites and B cell neutralization epitopes. J. Immunol. 142, 3612–3619 (1989).
Hart, M.K. et al. Priming of anti-human immunodeficiency virus (HIV) CD8+ cytotoxic T cells in vivo by carrier-free HIV synthetic peptides. Proc. Natl. Acad. Sci. USA 88, 9448–9452 (1991).
Yasutomi, Y., Palker, T.J., Gardner, M.B., Haynes, B.F. & Letvin, N.L. Synthetic peptide in mineral oil adjuvant elicits simian immunodeficiency virus-specific CD8+ cytotoxic T lymphocytes in Rhesus monkeys. J. Immunol. 151, 5096–5105 (1993).
Rose, J.R., Williams, M.B., Rott, L.S., Butcher, E.C. & Greenberg, H.B. Expression of the mucosal homing receptor α4β7 correlates with the ability of CD8+ memory T cells to clear rotavirus infection. J. Virol. 72, 726–730 (1998).
Miller, M.D., Yamamoto, H., Hughes, A.L., Watkins, D.I. & Letvin, N.L. Definition of an epitope and MHC class I molecule recognized by gag-specific cytotoxic T lymphocytes in SIVmac-infected rhesus monkeys. J. Immunol. 147, 320–329 (1991).
Kuroda, M.J. et al. Analysis of Gag-specific cytotoxic T lymphocytes in simian immunodeficiency virus-infected rhesus monkeys by cell staining with a tetrameric major histocompatibility complex class I-peptide complex. J. Exp. Med. 187, 1373–1381 (1998).
Allen, T.M. et al. CD8+ lymphocytes from simian immunodeficiency virus-infected rhesus macaques recognize 14 different epitopes bound by the major histocompatibility complex class I molecule mamu-A*01: implications for vaccine design and testing. J. Virol. 75, 738–749 (2000).
Dickinson, B.L. & Clements, J.D. Dissociation of Escherichia coli heat-labile enterotoxin adjuvanticity from ADP-ribosyltransferase activity. Infect. Immun. 63, 1617–1623 (1995).
Morris, C.B., Cheng, E., Thanawastien, A., Cardenas-Freytag, L. & Clements, J.D. Effectiveness of intranasal immunization with HIV-gp160 Env CTL epitope peptide (E7) in combination with the mucosal adjuvant LT(R192G). Vaccine 18, 1944–1951 (2000).
Joag, S.V. et al. Chimeric simian/human immunodeficiency virus that causes progressive loss of CD4+ T cells and AIDS in pig-tailed macaques. J. Virol. 70, 3189–3197 (1996).
Joag, S.V. et al. Chimeric SHIV that causes CD4+ T cell loss and AIDS in rhesus macaques. J. Med. Primatol. 27, 59–64 (1998).
Egan, M.A. et al. Use of major histocompatibility complex class I/peptide/β2M tetramers to quantitate CD8+ cytotoxic T lymphocytes specific for dominant and nondominant viral epitopes in simian-human immunodeficiency virus-infected rhesus monkeys. J. Virol. 73, 5466–5472 (1999).
Brunner, E., Munzel, U. & Puri, M.L. Ranks-score tests in factorial designs with repeated measures. J. Multivariate Anal. 70, 286–317 (1999).
Brunner, E. & Puri, M.L. Nonparametric methods in factorial designs. Statistical Papers 42, 1–52 (2001).
Walker, B.D. et al. HIV-specific cytotoxic T lymphocytes in seropositive individuals. Nature 328, 345–351 (1987).
Barouch, D.H. et al. Control of viremia and prevention of clinical AIDS in rhesus monkeys by cytokine-augmented DNA vaccination. Science 290, 486–492 (2000).
Cranage, M.P. et al. Intrarectal challenge of macaques vaccinated with formalin-inactivated simian immunodeficiency virus. Lancet 339, 273–274 (1992).
Amara, R.R. et al. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Science 292, 69–74 (2001).
Belyakov, I.M. et al. Induction of mucosal CTL response by intrarectal immunization with a replication-deficient recombinant vaccinia virus expressing HIV 89.6 envelope protein. J. Virol. 72, 8264–8272 (1998).
Rosenberg, E.S. et al. Vigorous HIV-1-specific CD4+ T cell responses associated with control of viremia. Science 278, 1447–1450 (1997).
Shirai, M. et al. Helper-CTL determinant linkage required for priming of anti-HIV CD8+ CTL in vivo with peptide vaccine constructs. J. Immunol. 152, 549–556 (1994).
Ahlers, J.D., Takeshita, T., Pendleton, C.D. & Berzofsky, J.A. Enhanced immunogenicity of HIV-1 vaccine construct by modification of the native peptide sequence. Proc. Natl. Acad. Sci. USA 94, 10856–10861 (1997).
Ossendorp, F., Mengede, E., Camps, M., Filius, R. & Melief, C.J. Specific T helper cell requirement for optimal induction of cytotoxic T lymphocytes against major histocompatibility complex class II negative tumors. J. Exp. Med. 187, 693–702 (1998).
Braun, M.C., He, J., Wu, C.-Y. & Kelsall, B.L. Cholera toxin suppresses interleukin (IL)-12 production and IL-12 receptor B1 and B2 chain expression. J. Exp. Med. 189, 541–552 (1999).
Allen, T.M. et al. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407, 386–90 (2000).
Knapp, L.A., Lehmann, E., Piekarczyk, M.S., Urvater, J.A. & Watkins, D.I. A high frequency of Mamu-A*01 in the rhesus macaque detected by polymerase chain reaction with sequence-specific primers and direct sequencing. Tissue Antigens 50, 657–661 (1997).
Bull, D.M. & Bookman, M.A. Isolation and functional characterization of human intestinal mucosal lymphoid cells. J. Clin. Invest. 59, 966–974 (1977).
Hosmalin, A. et al. Priming with helper T-cell epitope peptides enhances the antibody response to the envelope glycoprotein of HIV 1 in primates. J. Immunol. 146, 1667–1673 (1991).
Altman, J.D. et al. Phenotypic analysis of antigen-specific T lymphocytes. Science 274, 94–96 (1996).
Kuroda, M.J. et al. Emergence of CTL coincides with clearance of virus during primary simian immunodeficiency virus infection in rhesus monkeys. J. Immunol. 162, 5127–5133 (1999).
Romano, J.W. et al. Quantitative evaluation of simian immunodeficiency virus infection using NASBA technology. J. Virol. Methods 86, 61–70 (2000).
Acknowledgements
We thank M. Spano and the animal care staff at the ABL for their assistance; R. Pal for the titered stock of SHIV-Ku2; J.-P. Planchot for the Montanide ISA51; M. Lewis for the DNA PCR of provirus; V. Stepanov, Y. Tuyrin, S. Steinberg, D. Venzon and E. Brunner for assistance, advice and critical comments on statistical analysis; V. Hirsch, B. Moss, and J. Snyder for critical reading of the manuscript and helpful suggestions; and N. Miller for support of the animals. The work of A.S. was partially supported by NIH contract N01-AI-95362 and grant R24 RR 15731.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Belyakov, I., Hel, Z., Kelsall, B. et al. Mucosal AIDS vaccine reduces disease and viral load in gut reservoir and blood after mucosal infection of macaques. Nat Med 7, 1320–1326 (2001). https://doi.org/10.1038/nm1201-1320
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nm1201-1320
