
Abstract
Human bodies collectively turn over about 200 billion to 300 billion cells every day. Such turnover is an integral part of embryonic and postnatal development, as well as routine tissue homeostasis. This process involves the induction of programmed cell death in specific cells within the tissues and the specific recognition and removal of dying cells by a clearance 'crew' composed of professional, non-professional and specialized phagocytes. In the past few years, considerable progress has been made in identifying many features of apoptotic cell clearance. Some of these new observations challenge the way dying cells themselves are viewed, as well as how healthy cells interact with and respond to dying cells. Here we focus on the homeostatic removal of apoptotic cells in tissues.
Similar content being viewed by others
Main
Among the different forms of cell death, caspase-dependent apoptosis is thought to account for the majority of homeostatic cellular turnover1. Apoptosis is characterized by the rounding and shrinking of cells, chromatin condensation, and the formation of plasma membrane blebs or apoptotic bodies2. Apoptotic cell death helps to eliminate cells that are old or no longer needed without causing damage to the surrounding tissues or initiating an immune response. As part of routine homeostasis, different tissues turn over varying numbers of apoptotic cells, with some tissues undergoing an impressively high rate of renewal: hematopoiesis produces billions of cells daily, many with short lifespans (such as neutrophils); in the gastrointestinal tract, epithelial cells, which cover an area equivalent in size to a tennis court, are turned over every 4–5 days; in the thymus and the bone marrow, millions of thymocytes and immature B cells, respectively, are eliminated during maturation; in the brain, adult neurogenesis produces thousands of new neurons daily, but only a few survive; and in the testes, spermatogenesis produces millions of germ cells, of which many undergo apoptosis. In addition, there is an increase in homeostatic turnover under certain conditions, such as during involution of the mammary gland after lactation and weaning3. In some situations, such as during neuronal pruning, pieces of cells (rather than whole cells) undergo phagocytosis. Finally, there are situations, such as infection or acute tissue injury, in which the number of apoptotic cells increases beyond the normal rate within a given tissue.
In such contexts, apoptotic cells need to be disposed of quickly and without elicitation of inflammation in the local tissue milieu2,4. Under homeostatic conditions, tissue-resident phagocytes mediate removal of the cellular 'corpse'. In cases of increased cell death due to infection (apoptosis of epithelial cells during lung infection) or sustained 'sterile' inflammation (atherosclerotic plaques), such clearance is mediated both by resident phagocytes and by phagocytes recruited from the circulation. Failure in the clearance of apoptotic cells at early stages of death and progression to a secondary necrotic state can induce tissue inflammation due to the release of cellular contents or exposure of otherwise sequestered intracellular moieties2. The critical 'decision' of whether to initiate an immune response to the dying cell or not is made by the cell-clearance machinery, in response to molecules released by and/or exposed on the dying cells. The phagocyte ultimately responds by actively suppressing or eliciting inflammation2,4.
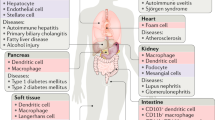
Phagocyte types and the tissue contexts
Homeostatic removal of cellular 'corpses' within a tissue is determined by the composition of the local 'clearance crew'. Phagocytes that ingest apoptotic cells have been categorized as professional and non-professional phagocytes. On the basis of existing evidence, we suggest a third category: specialized phagocytes (Fig. 1).

In many tissues of the body, clearance of apoptotic cells is performed by the professional phagocytes (P), among which are tissue-resident macrophages and immature dendritic cells. Many non-hematopoietic cells also have phagocytic functions in ex vivo or in vitro systems. Among these non-professional phagocytes (NP) are epithelial cells, hepatocytes and endothelial cells of the liver11, astrocytes, oligodendrocytes and neuronal progenitor cells of the central nervous system108,109,110,111, or the Muller's glia of the eye112. Satellite cells of the skeletal muscle have also been reported to engulf apoptotic myoblasts37. Specialized phagocytes (SP) are multifunctional cells that engulf apoptotic cells; this subset includes RPE cells113 and Sertoli cells in the testes16.
The subset of professional phagocytes includes macrophages and immature dendritic cells. Macrophages have long been known as professional engulfers of apoptotic cells due to their high capacity for engulfment in vitro and in vivo5,6. Although early studies used macrophages of various sources (native, thioglycollate elicited, bone marrow derived, etc.), understanding of macrophage types has grown substantially since then7,8. Elegant series of studies now suggest that embryonic yolk sac–derived stem cells colonize most tissues and contribute to the resident macrophage pool9,10. This self-renewing population differentiates into specific types of tissue-resident macrophages, such as peritoneal macrophages, Kupffer cells in the liver, alveolar macrophages in the lung, and microglia in the brain. These resident macrophage-like cells clear dying cells and debris: Kupffer cells clear aged red blood cells11, while microglia clear dying neurons and prune mature neurons12. In addition to resident phagocytes, circulating monocytes can also be recruited during infection or injury. Recruited phagocytes can act in cooperation with (or compete with) the resident phagocytes and thereby influence the immune response13.
Non-professional phagocytes, such as epithelial cells and fibroblasts, have gained greater appreciation for their ability to clear apoptotic cells under routine homeostatic conditions (Fig. 1). Although they are called 'non-professional' due to their lower phagocytosis efficiency than that of professional phagocytes, non-professional phagocytes have a major role in tissues in which macrophages are scarce or where the access of macrophages to apoptotic cells is not readily achieved, such as in the alveoli of the lungs or in the intestinal epithelium. The importance of non-professional phagocytes in the clearance of cellular 'corpses' has been revealed in several contexts. In macrophage-deficient animals, apoptotic cells generated during development continue to be cleared, albeit with lower efficiency14. Similarly, airway epithelial cells engulf dying apoptotic airway epithelial cells, a process that requires the small GTPase Rac1 (which functions downstream of several engulfment receptors)15. Epithelial cell–specific deletion of Rac1 results in increased susceptibility to allergen-induced airway inflammation and decreased production of anti-inflammatory mediators15. Similarly, intestinal epithelial cells can also engulf their neighbors in vivo and thus contribute to the regulation of inflammatory sequelae (Lee et al., personal communication). Moreover, during involution of the mammary tissue after lactation, the epithelial cells of the mammary gland (rather than macrophages) function as the main engulfers3. Because epithelial cells vastly outnumber professional phagocytes and are probably the first to contact a dying adjacent epithelial cell, engulfment by neighboring cells might help maintain the tissue barrier while providing the benefits of anti-inflammatory cytokine production15.
Specialized phagocytes are hybrid, multi-functional phagocytes that are increasingly recognized for their importance in specific tissue contexts. The best examples are Sertoli cells of the testes and retinal pigment epithelial (RPE) cells of the eye (Fig. 1). Sertoli cells, which are non-hematopoietic and post-mitotic, line the epithelium of seminiferous tubules and make up the blood-testes barrier. Sertoli cells clear millions of apoptotic germ cells that arise during spermatogenesis. Their hybrid function is exemplified by the fact that a single Sertoli cell is often in contact with 30–40 germ cells in various stages of differentiation. In addition to serving as nurse cells for the developing spermatocytes, the Sertoli cells phagocytose those germ cells that display improper meiosis or other developmental abnormalities, and disruption of either apoptosis or engulfment can affect spermatogenesis16,17. Another example of specialized phagocytes is the RPE cell. RPE cells are long-lived cells with a critical role in the homeostatic removal of the photoreceptor outer segment that occurs daily in a circadian fashion (with uptake of RPE cells triggered by the onset of light)18. Each RPE cell is estimated to engulf thousands of outer segment discs over its lifetime. Failures in RPE cell–mediated removal of outer segments can severely affect the integrity of retinal layers and contribute to a predisposition to adverse conditions, such as retinitis pigmentosa18.
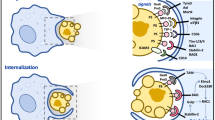
Accessing and identifying apoptotic cells
On the basis of studies by several groups, engulfment of apoptotic cells includes distinguishable steps (Fig. 2). First, the dying cell releases 'find-me' signals to attract and/or activate the phagocytes. The phagocytes then distinguish the apoptotic cell from healthy living cells via specific engulfment receptors, which recognize 'eat-me' signals on the dying cell. Next, the phagocyte undergoes extensive cytoskeletal rearrangement to internalize cellular 'corpses' that are often the same size (for example, an epithelial cell ingesting its neighbor). The final step is processing of the ingested cargo and elicitation of specific phagocyte responses, mainly the secretion of anti-inflammatory mediators that help dampen the local immune response.

When a cell initiates the apoptotic program, it releases soluble 'find-me' signals that attract phagocytes. The apoptotic cell is distinguished from the nearby living cell via the exposure of 'eat-me' signals, the most prominent of which is PtdSer. The 'eat-me' signals are recognized by different engulfment receptors on the phagocytes, which results in signaling events that facilitate uptake of the apoptotic cellular 'corpse'. Engulfment also elicits transcriptional upregulation of the cholesterol-efflux transporter ABCA1 and increased expression of engulfment receptors. Within the mitochondria, the levels of the uncoupling protein UCP2 are increased, which enables the continued uptake of apoptotic cellular 'corpses'. Anti-inflammatory mediators are expressed and secreted, which contributes to tissue homeostasis and inhibition of local inflammation. CX3CR, chemokine CX3CL1 receptor; G2A, G protein–coupled receptor; S1P1, sphingosine 1-phosphate (S1P) receptor; LPC, lysophosphatydilcholine; RAGE, receptor for advanced glycation end products; LRP1, low-density lipoprotein receptor–related protein; TSP1, thrombospondin; CRT, calreticulin.
The release of 'find-me' signals is a critical first step in many tissues, as it recruits a potentially distant phagocyte to the dying cell. In some tissues, such as the developing thymus, this is particularly important; a dying thymocyte will probably not be ingested by its neighbor, as lymphocytes generally lack the ability to engulf apoptotic cells. Therefore, motile resident phagocytes must be recruited to the proximity of apoptotic thymocytes. This is achieved through the release of 'find-me' signals from the dying cell, including nucleotides (ATP and UTP), the chemokine fractalkine (CX3CL1), and the lipids lysophosphatidylcholine and sphingosine 1-phosphate19,20,21,22,23. Among those, only nucleotides and a nucleotide receptor on the phagocyte (P2Y2) are linked to the clearance of apoptotic thymocytes in vivo19. It is possible that 'find-me' signals might serve other functions, such as during the removal of dying epithelial cells by a viable neighbor, when recruitment is not required. Since apoptotic epithelial cells also release 'find-me' signals19, perhaps these signals influence and/or enhance the engulfment capacity of the neighbor(s). For example, CX3CL1 stimulates the expression of MFG-E8 ('milk fat globule–epidermal growth factor 8′) by phagocytes, which bridges apoptotic cells to the phagocytes to facilitate engulfment24.
Next is the recognition of specific 'eat-me' signals on apoptotic cells by engulfment receptors on the phagocytes. So far, the best-studied 'eat-me' signal on apoptotic cells is exposure of the lipid phosphatidylserine (PtdSer), which is evolutionarily conserved from Caenorhabditis elegans to humans25,26. In living cells, PtdSer is actively restricted to the inner leaflet of the plasma membrane27, and elegant studies have identified apoptosis-mediated modes as well as calcium-induced modes of PtdSer exposure28,29,30. In addition to PtdSer, other moieties that are variably exposed on apoptotic cells include a modified form of the intracellular adhesion molecule ICAM-3, oxidized low-density lipoprotein, calreticulin, annexin I, cell suface–bound thrombospondin and complement C1q; other alterations on the surface include changes in the surface protein charge and glycosylation status31. Conversely, viable cells avoid their removal by displaying the 'don't-eat-me' signals CD47 and CD31 or by binding to the receptor CD300a on the phagocyte and suppressing phagocyte functions32,33,34.
Engulfment receptors linked to homeostatic clearance
Many apoptotic cell–recognition and engulfment receptors have been identified in inflammatory and/or homeostatic contexts. These receptors are of different 'flavors', such as members of the scavenger receptor family, immunoglobulin-containing proteins, seven-transmembrane proteins, tyrosine kinases, etc.31. Why there are many different types of engulfment receptors and how they provide specificity is still unclear. In some ways, the diversity of engulfment receptors is similar to that of accessory proteins linked to the interaction of T cells with antigen-presenting cells. While the exposed PtdSer could be viewed as having a function loosely analogous to that of a major histocompatibility complex molecule on an antigen-presenting cell, the distinction between the phagocyte–apoptotic cell interaction and the T cell–antigen-presenting cell interface lies in the lack of an equivalent to the T cell antigen receptor on phagocytes. Instead, the role of that receptor seems to be distributed among the various engulfment receptors. What has been shown so far by studies of animals (primarily mice) with specific deletion of individual receptors for PtdSer is that while there is redundancy in function, at least in some cases there are specific needs for particular engulfment receptors. Since not all engulfment receptors are expressed on all phagocyte types, the differences between professional phagocytes and non-professional phagocytes in their expression of such receptors might influence the homeostatic turnover of dying cells. In fact, a diverse set of phenotypes have been reported in mice with alterations in various molecules linked to the recognition of PtdSer (Table 1).
We discuss below three specific receptors that engage PtdSer: BAI1, TIM-4 and MerTK (Fig. 3). BAI1 is a seven-transmembrane protein that can directly engage PtdSer and also relay intracellular signaling to mediate engulfment, while TIM-4 can bind PtdSer directly but does not have signaling ability on its own (i.e., it is a tethering receptor). MerTK, however, is a membrane tyrosine kinase that cannot engage PtdSer directly but uses bridging molecules that bind PtdSer on apoptotic cells. We have chosen to focus on these receptors because they highlight some of the complexities in the recognition of PtdSer and are linked to homeostatic cell turnover.

Binding of the apoptotic cell to the phagocyte triggers signaling pathways. The seven-transmembrane receptor BAI1 directly binds the PtdSer on the surface of an apoptotic cell, which results in the recruitment of the ELMO-DOCK ('engulfment and cell motility–downstream of Crk') complex, which functions as a guanine-exchange factor for the small GTPase Rac39. The activation of Rac promotes remodeling of the actin cytoskeleton required for engulfment of the apoptotic cellular 'corpse'. Integrins αVβ3 or αVβ5 and members of the TAM family of receptors bind apoptotic cells indirectly, via PtdSer-bound bridging molecules MFG-E8, Gas-6 or protein S, which results in activation of the kinase FAK and contributes to the activation of Rac114. TAM receptors are tyrosine kinases that also activate cell-signaling pathways involving the kinases Src and PI(3)K and the phospholipase PLC114,115. TIM-4 functions as a tethering receptor that brings the apoptotic cell in contact with signaling engulfment receptors, and it signals through co-receptors. The extent of the connection between the signals elicited by different engulfment receptors awaits further characterization.
BAI1, along with its homologs BAI2 and BAI3, belongs to the adhesion subfamily of G protein–coupled receptors35. Originally identified as an inhibitor of angiogenesis, BAI1 serves functions in diverse biological processes, including the phagocytosis of apoptotic cells, myoblast fusion, synaptogenesis, and tumor growth36,37,38,39,40. Via its thrombospondin repeats, BAI1 can directly bind PtdSer39. Upon recognizing PtdSer, BAI1 interacts with a cytoplasmic signaling module composed of ELMO1 and DOCK, which function as guanine-exchange factors for Rac1, and thereby induces rearrangements of the actin cytoskeleton and facilitates the uptake of apoptotic cells39.
Although BAI1-deficient mice are grossly normal, they have several key homeostatic defects. Adult mice with global deletion of BAI1 have smaller skeletal muscle fibers and display delayed healing after muscle injury relative to wild-type mice; since myoblast fusion also seems to involve exposure of PtdSer, these results probably reflect an interesting additional BAI1 function37. In peritoneal macrophages, binding of apoptotic cells to BAI1 triggers signaling that promotes cholesterol efflux41 and contributes to the maintenance of lipid homeostasis (discussed further below). It has also been independently reported that mice that lack BAI1 have deficits in spatial learning and memory. This could be due to a function of BAI1 in regulating postsynaptic density40. BAI1 expression is particularly high in the brain, testes and certain hematopoietic compartments39. Although BAI1 mRNA levels in macrophages are lower than those of TIM-4 or MerTK mRNA (unpublished observations), macrophages from BAI1- and TIM-4 deficient mice have comparable deficiencies in the uptake of apoptotic cells41. However, direct comparisons of the levels of BAI1 mRNA and BAI1 protein have not been reported so far. BAI1 expression might also be regulated post-transcriptionally, or BAI1 might influence engulfment via mechanisms that do not require high expression.
TIM-4 belongs to a family of cell-surface glycoproteins originally identified as regulators of T cell function42. The discovery of TIM-4 as a PtdSer-recognition receptor was closely followed by the recognition of other members of the TIM family (such as TIM-1 and TIM-3) as receptors for PtdSer43,44. However, unlike BAI1, TIM-4 does not activate direct downstream signaling but instead acts as a tethering receptor45. Although integrins can function cooperatively with TIM-4 for signaling in vitro46, the co-signaling receptor(s) for TIM-4 under endogenous expression conditions is (are) unclear. An elegant study of zebrafish has shown that BAI1 and TIM-4 may act at distinct stages of engulfment, with possible cooperation between the receptors, whereby BAI1 contributes to phagosome formation, while TIM-4 contributes to phagosome stabilization47.
In mice, TIM-4 expression is high on tissue-resident macrophages, dendritic cells and, particularly, peritoneal macrophages44. Macrophages that lack TIM-4 show diminished engulfment of apoptotic cells48,49. Mice with global TIM-4 deficiency also variably develop signs of autoimmunity48,49,50, whereas mice with overexpression of TIM-4 display diminished secondary immune responses51. Such data suggest that the homeostatic clearance of apoptotic cells can be influenced by TIM-4, with potential links to immunotolerance. Conditional deletion of Tim-4 in specific cell types is needed for better characterization of its function in immune responses.
The tyrosine kinase MerTK is a member of the TAM receptor family, which includes Tyro, Axl and Mer receptor tyrosine kinases52. TAM receptors have immunoglobulin-like domains and fibronectin repeats in the extracellular region and a cytoplasmic tyrosine kinase domain. TAM receptors engage PtdSer on apoptotic cells indirectly via the soluble ligands protein S and Gas-6 (ref. 52). There are differential requirements for protein S and Gas-6 in mediating the ligation of TAM receptors and downstream signaling53,54. Although MerTK has been reported to be a specific marker of macrophages55, we note that many epithelial cells have high expression of MerTK.
TAM receptors are linked to the homeostatic clearance of apoptotic cells in several contexts. Single or combined deletion of members of the TAM family leads to the accumulation of apoptotic germ cells in the testes, with complete lack of mature sperm in mice lacking all three TAM receptors56. Also, mice lacking MerTK develop progressive blindness (by 8–12 weeks of age) due to deficiency in the circadian RPE cell–dependent removal of rod outer segments in the retina; such findings indicate the specific and critical requirement for MerTK in the function of retinal epithelial cells57. Moreover, while loss of all three TAM receptors does not affect embryonic development, adult mice lacking these receptors show decreased clearance of apoptotic cells and develop severe systemic autoimmunity56. The latter phenotype has been linked to the function of TAM receptors as powerful inhibitors of the immune response58.
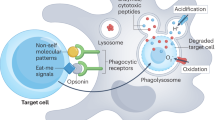
Processing the apoptotic cargo
A fascinating but understudied area of the clearance of apoptotic cells is how phagocytes process the ingested cargo. When a phagocyte engulfs an apoptotic cell, it may double its protein, lipid and carbohydrate content, yet professional phagocytes manage to rapidly engulf multiple cellular 'corpses'. In tissues that turn over a large number of cells, such as the thymus, the number of macrophages is much lower than that of thymocytes undergoing death. Therefore, a single phagocyte must ingest more than one cellular 'corpse', probably in succession. Several studies have suggested that the process of engulfment itself influences the capacity of the phagocyte to engulf additional corpses, linked to the increased expression of engulfment receptors via nuclear receptors (LXR, PPARδ, PPARγ and RXR)59,60,61 (Fig. 2). Continued clearance of apoptotic cells by the phagocyte is also positively regulated by increased expression of UCP2, a mitochondrial uncoupler of oxidative phosphorylation from ATP synthesis62. Whether the expression and induction of LXR, PPARδ, PPARγ, RXR and UCP2 differ among professional and non-professional phagocytes under homeostatic and inflammatory conditions remains to be established.
Among the ingested components degraded in the phagocytic lysosomes, the degradation of DNA is of particular importance, as 'escaped' DNA can induce breaks in self-tolerance and lead to the development of autoimmunity4. A key situation in which this happens in homeostasis is during erythropoiesis. During the definitive stage of erythropoiesis, DNA from erythroblasts is extruded in structures called 'pyrenocytes' (nuclei surrounded by membrane decorated with PtdSer63). Pyrenocytes are engulfed by neighboring macrophages in a MerTK-dependent fashion64, which allows erythropoiesis to proceed65. DNase II is the enzyme that degrades DNA in the lysosomes65. Macrophages have high expression of DNase II, and macrophages that lack DNase II cannot digest the DNA from engulfed apoptotic cells and cannot support erythropoiesis65. In fact, failed digestion of DNA leads to activation of the cyclic cGAS–STING nucleic acid–sensing pathway, with production of type I interferon and lethal anemia66. Although such mice (with failed digestion of DNA) are rescued from anemia by the added deletion of the type I interferon receptor, they develop arthritis due to excessive production of tumor-necrosis factor, which suggests that undigested DNA from apoptotic cells can induce inflammatory disease67.
Certain components of the ingested apoptotic cell, such as cholesterol, can also be disposed of in other ways. In macrophages, transporters of the ATP-binding cassette (ABC) family, ABCA1 and ABCG1, aid in the efflux of intracellular cholesterol to lipid-rich high-density lipoprotein, which is then taken up by the liver and excreted in the bile68. Impairments in cholesterol efflux are linked to dyslipidemia and atherosclerosis69. When macrophages engage apoptotic cells, they rapidly increase their ABCA1 expression and cholesterol efflux in a PtdSer-dependent manner70. Surprisingly, this early induction of ABCA1 does not require the canonical LXR-mediated pathway (although LXR can be relevant after prolonged exposure of apoptotic cells60). Instead, the BAI1-ELMO1-Dock180-Rac1 signaling module mediates the upregulation of ABCA1 and cholesterol efflux41. Furthermore, in atherosclerosis-prone mice fed a high-fat diet, deletion of BAI1 results in lower serum concentrations of high-density lipoprotein, a risk factor for cardiovascular disease, whereas overexpression of BAI1 results in a higher ratio of high-density lipoprotein to cholesterol and low-density lipoprotein in serum41, which suggests that BAI1 regulates normal lipidemia.
Cell clearance and anti-inflammatory responses
The clearance of cellular 'corpses' commences at the earliest stages of apoptosis, before the loss of plasma membrane integrity, which prevents the release of cellular contents. In homeostatic conditions, this occurs rather efficiently, and there is hardly any recruitment of inflammatory cells even in tissues with high cellular turnover. However, once the integrity of the plasma membrane is lost due to the secondary necrosis of late-stage apoptotic cells, the released cellular contents can engage receptors for damage-associated molecular patterns and contribute to immune responses to self antigens71. The mechanisms by which late apoptotic and necrotic cells are cleared include opsonization with lectins, properdin, pentraxins, thrombospondin and heparan sulfate proteoglycans. Interestingly, many of the opsonins that facilitate clearance of these cells also facilitate pathogen clearance72. Perhaps the concurrent recognition of the late-stage dying cell and the infectious pathogen contributes to faster recovery from infectious injury and resolution of inflammation. Treatment with recombinant human MFG-E8 has been shown to reduce disease in two mouse models of colitis73, which suggests that enhancing the clearance of all PtdSer-exposing cells could be of benefit in inflammation. Although delayed or impaired clearance of dying cells (Table 1) can aggravate inflammatory disease, the administration of early-stage apoptotic cells has been shown to help reduce disease severity in inflammation models, probably via elicitation of anti-inflammatory mediators74. This suggests that the benefit versus inflammatory potential of apoptotic cells is in a delicate balance and is probably critical for the design of apoptotic cell–based therapies for inflammatory diseases.
Should homeostasis be breached by tissue inflammation with infiltrating cells, the dying cell populations can include bystander cells and short-lived cells of the immune system (such as neutrophils) that need to be removed during resolution of inflammation. In addition to apoptosis, other forms of cell death may also be involved, including primary and secondary necrosis, pyroptosis and necroptosis75. Neutrophils recruited to sites of bacterial infection can also die via the formation of neutrophil extracellular traps76, with the release of nuclear chromatin and histones to facilitate trapping and killing of bacteria. Due to the release of cellular contents, the formation of such traps is generally thought to incite inflammation, although certain types of neutrophil extracellular traps can contribute to its resolution77. Necroptosis is a non-apoptotic cell death triggered by tumor-necrosis factor (a cytokine abundantly present at the sites of inflammation) or by other stimuli when apoptosis is blocked75. The clearance of necroptotic cells is not yet fully defined. In fact, fascinating but unexplored topics are the relative contributions of different forms of cell death to maintaining homeostasis in any given tissue, how the cells that die by different mechanisms within the same tissue are removed (by the same phagocytes?), and how 'decisions' are made about the phagocyte responses.
Rethinking apoptosis and PtdSer exposure
Apoptosis is closely linked to regenerative processes, as a dying cell can stimulate proliferation in the surrounding viable cells through apoptosis-induced compensatory proliferation78. This is observed even in the simple metazoan Hydra, in which a caspase-dependent apoptotic response caused by injury induces proliferation of the surrounding cells79 (Fig. 4a). Similarly, apoptosis is a requirement for regenerative processes in Xenopus80, planaria (flatworm)81 and newts82 and even the mammalian liver83.

(a) Regeneration: in the metazoan Hydra, tissue injury can lead to the apoptosis of cells, which stimulates regenerative processes in nearby viable tissues via a process called 'apoptosis-induced compensatory proliferation'. Apoptosis is required for re-growth of the new Hydra head. (b) Caspase-dependent inhibition of interferon production: in the context of viral infection, apoptosis leads to the activation of caspases that is linked to inhibition of the production of interferon-α and interferon-β (IFN-α/β) induced by mitochondrial DNA (mtDNA)-mediated activation of the cGAS-STING pathway. (c) Myoblast fusion: during the development and regeneration of muscle after muscle injury, apoptosis of myoblasts triggers signaling by BAI1 or BAL3 through the ELMO-DOCK complex, which leads to activation of Rac. This pathway contributes to the fusion of healthy myoblasts with the nascent myotube and promotes the development and regeneration of muscle. (d) Exploitation of engulfment receptors by pathogens: bacteria, parasites and even viruses have evolved to utilize the apoptotic cell–engulfment receptors for their entry into host cells and for the induction of anti-inflammatory signaling in the host cell (phagocyte), which aids in the establishment and persistence of infection.
Interestingly, caspases that are activated during cell death can also regulate subsequent induction of inflammation84. When apoptotic caspases are missing, viral infection causes permeabilization of the mitochondrial membrane dependent on the apoptosis promoters Bax and Bak, which leads to the release of mitochondrial DNA, activation of the cGAS-STING pathway via the recognition of cytosolic DNA, and the induction of type I interferons84 (Fig. 4b). This suggests that the caspase-dependent death that occurs during most homeostatic conditions might have evolved to dampen local inflammation that might have been adapted by viruses that induce cell lysis.
PtdSer can also be transiently exposed on viable cells. Since such transient PtdSer exposure does not lead to engulfment, PtdSer exposure alone may not be sufficient for stimulating phagocytosis. It is likely that 'eat-me' signals in addition to PtdSer, perhaps in combination with a lack of 'don't-eat-me' markers, might be needed to confirm the impending cell death to the phagocyte85. In T cells, exposure of PtdSer is triggered by stimulation of the T cell antigen receptor or the ATP receptor P2X7 (ref. 86). PtdSer on activated T cells contributes to the downregulation of immune responses by engaging protein S and triggering TAM receptor–mediated anti-inflammatory signaling in antigen-presenting cells87. Therefore, PtdSer exposure in this context acts as a 'rheostat' of the immune response, instead of acting as an 'eat-me' signal. Transient PtdSer exposure is also observed upon activation of neutrophils and mast cells88,89. The distinction between apoptotic PtdSer exposure and non-apoptotic PtdSer exposure is that the latter is reversible and generally lasts only minutes or even seconds. Exposure of PtdSer has been noted during the fusion of myoblasts into skeletal muscle myotubes in vitro90 and, subsequently, fusion-inducing cues have been shown to cause the death of some myoblasts, and the caspase-dependent exposure of PtdSer is required for the fusion to occur37. Furthermore, the PtdSer receptor BAI1 and its homolog BAI3 act as promoters of myoblast fusion, as mice deficient in BAI1 and BAI3 develop smaller myofibers and show delayed healing after muscle injury37,91 (Fig. 4c).
PtdSer exposure is also exploited by several microorganisms due to the anti-inflammatory nature of PtdSer-dependent clearance of apoptotic cells (Fig. 4d). This was first reported in Leishmania, which exposes PtdSer on the cell surface during the amastigote stage of the life cycle92. PtdSer promotes internalization of amastigotes by the macrophage while also inhibiting the immune response via induction of transforming growth factor-β. Similar mechanisms of evasion have been reported for Toxoplasma gondii93 and Trypanosoma cruzi94. Enveloped viruses also use PtdSer for entry into the cell via a process called 'apoptotic mimicry'95. The list of viruses that utilize this mechanism is growing rapidly, including human immunodeficiency virus96, vaccinia virus95, Ebola virus97,98, dengue virus99 and Pichinde viruses100. Remarkably, it has been suggested that even non-enveloped viruses, conventionally thought to require cell lysis for viral transmission, use PtdSer-decorated vesicles for packaging of multiple virions for transfer into the new host cell101. Similarly, many PtdSer receptors are linked to viral entry100. Finally, certain PtdSer receptors, including BAI1 (ref. 102), TREM-2 (ref. 103), stabilin-2 (ref. 104), CD36 and the scavenger receptor SCARF-1 (SREC-1)105, can also bind bacteria and fungi. Thus, rethinking of the role of PtdSer receptors is warranted, both in the context of the clearance of apoptotic cells and in non-apoptotic homeostatic functions and pathogen encounters.
Impending challenges
In terms of how the clearance of apoptotic cells regulates homeostasis in tissues, many interesting questions remain to be addressed. The first challenge is understanding the role of specific receptors. It is unclear whether there is 'preference' for the use of particular engulfment receptors or clearance mechanisms to achieve the distinction between homeostatic turnover of apoptotic cells versus inflammatory turnover of apoptotic cells. The second challenge is defining the anti-inflammatory responses. The difference between the phagocytosis of apoptotic cells and that of other targets (such as bacteria or other pathogens) is that routine uptake of apoptotic cells is generally not immunogenic; furthermore, it elicits the production of mediators that actively suppress inflammation in the local tissue milieu. However, the phagocyte molecular events that lead to specific downstream consequences are just beginning to be defined41,106. Engagement of apoptotic cells is well known to induce transforming growth factor-β, which is linked to the differentiation of immunosuppressive regulatory T cells. Whether routine clearance of apoptotic cells has a role in generating regulatory T cells specific for self antigens not expressed in the thymus remains to be explored. The third major challenge is in understanding the 'labor distribution' between professional phagocytes and non-professional phagocytes. An intriguing question is whether there is crosstalk between professional phagocytes and non-professional phagocytes under homeostatic conditions and whether this might influence the phagocytic capacity of either. Furthermore, in many inflammatory conditions, different phagocytes are present (resident macrophages, non-professional phagocytes and recruited phagocytes). It is not known whether professional phagocytes redirect non-professional phagocytes (such as epithelial cells) to shift their efforts toward proliferation or matrix production for tissue recovery. Moreover, non-professional phagocytes can also produce anti-inflammatory cytokines15, but there may be differences in the spectrum of factors produced and their contribution to the maintenance of the local anti-inflammatory state. Such knowledge could be useful for therapeutic targeting and accelerating tissue recovery after injury. The fourth challenge is delineating the 'metabolomics' of apoptotic cargo processing. Relatively little is known about how target-derived metabolites are processed and used by the phagocyte or in the phagocyte neighborhood. Release of some of these metabolites might also provide a means for communication between cells in a tissue. In this context, the secretion of lactate from tumor cells regulates macrophage phenotypes in a tumor environment107; perhaps similar strategies exist whereby a non-professional phagocyte engulfing an apoptotic cell secretes metabolites that regulate the activation status of macrophages in the local environment. Comprehensive determination of the metabolomics of engulfment could be of relevance to human diseases such as obesity and diabetes. Thus, better definition of the homeostatic clearance of apoptotic cells could have important implications for the understanding of basic physiology, immunotolerance and responses to infection.
References
Nagata, S., Hanayama, R. & Kawane, K. Autoimmunity and the clearance of dead cells. Cell 140, 619–630 (2010).
Poon, I.K., Lucas, C.D., Rossi, A.G. & Ravichandran, K.S. Apoptotic cell clearance: basic biology and therapeutic potential. Nat. Rev. Immunol. 14, 166–180 (2014).
Monks, J. et al. Epithelial cells as phagocytes: apoptotic epithelial cells are engulfed by mammary alveolar epithelial cells and repress inflammatory mediator release. Cell Death Differ. 12, 107–114 (2005).
Nagata, S. Autoimmune diseases caused by defects in clearing dead cells and nuclei expelled from erythroid precursors. Immunol. Rev. 220, 237–250 (2007).
Metchnikoff, E. in Lectures on the Comparative Pathology of Inflammation (Dover, New York, 1968).
van Furth, R. et al. The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bull. World Health Organ. 46, 845–852 (1972).
Lavin, Y. & Merad, M. Macrophages: gatekeepers of tissue integrity. Cancer Immunol. Res. 1, 201–209 (2013).
Epelman, S., Lavine, K.J. & Randolph, G.J. Origin and functions of tissue macrophages. Immunity 41, 21–35 (2014).
Gomez Perdiguero, E. et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518, 547–551 (2015).
Hoeffel, G. et al. C-myb+ erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 42, 665–678 (2015).
Dini, L., Pagliara, P. & Carla, E.C. Phagocytosis of apoptotic cells by liver: a morphological study. Microsc. Res. Tech. 57, 530–540 (2002).
Bilimoria, P.M. & Stevens, B. Microglia function during brain development: New insights from animal models. Brain Res. 1617, 7–17 (2015).
Uderhardt, S. et al. 12/15-lipoxygenase orchestrates the clearance of apoptotic cells and maintains immunologic tolerance. Immunity 36, 834–846 (2012).
Wood, W. et al. Mesenchymal cells engulf and clear apoptotic footplate cells in macrophageless PU.1 null mouse embryos. Development 127, 5245–5252 (2000).
Juncadella, I.J. et al. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature 493, 547–551 (2013).
Elliott, M.R. et al. Unexpected requirement for ELMO1 in clearance of apoptotic germ cells in vivo. Nature 467, 333–337 (2010).
Lysiak, J.J., Turner, S.D. & Turner, T.T. Molecular pathway of germ cell apoptosis following ischemia/reperfusion of the rat testis. Biol. Reprod. 63, 1465–1472 (2000).
Burstyn-Cohen, T. et al. Genetic dissection of TAM receptor-ligand interaction in retinal pigment epithelial cell phagocytosis. Neuron 76, 1123–1132 (2012).
Elliott, M.R. et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286 (2009).
Gude, D.R. et al. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 22, 2629–2638 (2008).
Lauber, K. et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 113, 717–730 (2003).
Peter, C., Wesselborg, S., Herrmann, M. & Lauber, K. Dangerous attraction: phagocyte recruitment and danger signals of apoptotic and necrotic cells. Apoptosis 15, 1007–1028 (2010).
Truman, L.A. et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 112, 5026–5036 (2008).
Miksa, M., Amin, D., Wu, R., Ravikumar, T.S. & Wang, P. Fractalkine-induced MFG-E8 leads to enhanced apoptotic cell clearance by macrophages. Mol. Med. 13, 553–560 (2007).
Darland-Ransom, M. et al. Role of C. elegans TAT-1 protein in maintaining plasma membrane phosphatidylserine asymmetry. Science 320, 528–531 (2008).
Fadok, V.A. et al. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148, 2207–2216 (1992).
Balasubramanian, K. & Schroit, A.J. Aminophospholipid asymmetry: A matter of life and death. Annu. Rev. Physiol. 65, 701–734 (2003).
Suzuki, J., Denning, D.P., Imanishi, E., Horvitz, H.R. & Nagata, S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 341, 403–406 (2013).
Segawa, K. et al. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344, 1164–1168 (2014).
Suzuki, J., Umeda, M., Sims, P.J. & Nagata, S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468, 834–838 (2010).
Lauber, K., Blumenthal, S.G., Waibel, M. & Wesselborg, S. Clearance of apoptotic cells: getting rid of the corpses. Mol. Cell 14, 277–287 (2004).
Brown, S. et al. Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature 418, 200–203 (2002).
Simhadri, V.R. et al. Human CD300a binds to phosphatidylethanolamine and phosphatidylserine, and modulates the phagocytosis of dead cells. Blood 119, 2799–2809 (2012).
Barclay, A.N. & Van den Berg, T.K. The interaction between signal regulatory protein α (SIRP α) and CD47: structure, function, and therapeutic target. Annu. Rev. Immunol. 32, 25–50 (2014).
Hamann, J. et al. International Union of Basic and Clinical Pharmacology. XCIV. Adhesion G protein-coupled receptors. Pharmacol. Rev. 67, 338–367 (2015).
Duman, J.G. et al. The adhesion-GPCR BAI1 regulates synaptogenesis by controlling the recruitment of the Par3/Tiam1 polarity complex to synaptic sites. J Neurosci. 33, 6964–6978 (2013).
Hochreiter-Hufford, A.E. et al. Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature 497, 263–267 (2013).
Kaur, B., Brat, D.J., Devi, N.S. & Van Meir, E.G. Vasculostatin, a proteolytic fragment of brain angiogenesis inhibitor 1, is an antiangiogenic and antitumorigenic factor. Oncogene 24, 3632–3642 (2005).
Park, D. et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450, 430–434 (2007).
Zhu, D. et al. BAI1 regulates spatial learning and synaptic plasticity in the hippocampus. J. Clin. Invest. 125, 1497–1508 (2015).
Fond, A.M., Lee, C.S., Schulman, I.G., Kiss, R.S. & Ravichandran, K.S. Apoptotic cells trigger a membrane-initiated pathway to increase ABCA1. J. Clin. Invest. 125, 2748–2758 (2015).
Freeman, G.J., Casasnovas, J.M., Umetsu, D.T. & DeKruyff, R.H. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol. Rev. 235, 172–189 (2010).
Miyanishi, M. et al. Identification of Tim4 as a phosphatidylserine receptor. Nature 450, 435–439 (2007).
Kobayashi, N. et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity 27, 927–940 (2007).
Park, D., Hochreiter-Hufford, A. & Ravichandran, K.S. The phosphatidylserine receptor TIM-4 does not mediate direct signaling. Curr. Biol. 19, 346–351 (2009).
Flannagan, R.S., Canton, J., Furuya, W., Glogauer, M. & Grinstein, S. The phosphatidylserine receptor TIM4 utilizes integrins as coreceptors to effect phagocytosis. Mol. Biol. Cell 25, 1511–1522 (2014).
Mazaheri, F. et al. Distinct roles for BAI1 and TIM-4 in the engulfment of dying neurons by microglia. Nat. Commun. 5, 4046 (2014).
Rodriguez-Manzanet, R. et al. T and B cell hyperactivity and autoimmunity associated with niche-specific defects in apoptotic body clearance in TIM-4-deficient mice. Proc. Natl. Acad. Sci. USA 107, 8706–8711 (2010).
Wong, K. et al. Phosphatidylserine receptor Tim-4 is essential for the maintenance of the homeostatic state of resident peritoneal macrophages. Proc. Natl. Acad. Sci. USA 107, 8712–8717 (2010).
Miyanishi, M., Segawa, K. & Nagata, S. Synergistic effect of Tim4 and MFG-E8 null mutations on the development of autoimmunity. Int. Immunol. 24, 551–559 (2012).
Albacker, L.A. et al. TIM-4, a receptor for phosphatidylserine, controls adaptive immunity by regulating the removal of antigen-specific T cells. J. Immunol. 185, 6839–6849 (2010).
Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 5, a009076 (2013).
Lew, E.D. et al. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. eLife 10, 7554 (2014).
Zagórska, A., Traves, P.G., Lew, E.D., Dransfield, I. & Lemke, G. Diversification of TAM receptor tyrosine kinase function. Nat. Immunol. 15, 920–928 (2014).
Gautier, E.L. et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 13, 1118–1128 (2012).
Lu, Q. et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature 398, 723–728 (1999).
Duncan, J.L. et al. An RCS-like retinal dystrophy phenotype in mer knockout mice. Invest. Ophthalmol. Vis. Sci. 44, 826–838 (2003).
Rothlin, C.V., Ghosh, S., Zuniga, E.I., Oldstone, M.B. & Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 131, 1124–1136 (2007).
Mukundan, L. et al. PPAR-δ senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat. Med. 15, 1266–1272 (2009).
N, A.G. et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 31, 245–258 (2009).
Roszer, T. et al. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor α deficiency. J. Immunol. 186, 621–631 (2011).
Park, D. et al. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature 477, 220–224 (2011).
Yoshida, H. et al. Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature 437, 754–758 (2005).
Toda, S., Segawa, K. & Nagata, S. MerTK-mediated engulfment of pyrenocytes by central macrophages in erythroblastic islands. Blood 123, 3963–3971 (2014).
Kawane, K. et al. Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science 292, 1546–1549 (2001).
Yoshida, H., Okabe, Y., Kawane, K., Fukuyama, H. & Nagata, S. Lethal anemia caused by interferon-β produced in mouse embryos carrying undigested DNA. Nat. Immunol. 6, 49–56 (2005).
Kawane, K. et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 443, 998–1002 (2006).
Marcel, Y.L., Ouimet, M. & Wang, M.D. Regulation of cholesterol efflux from macrophages. Curr. Opin. Lipidol. 19, 455–461 (2008).
Oram, J.F. & Heinecke, J.W. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol. Rev. 85, 1343–1372 (2005).
Kiss, R.S., Elliott, M.R., Ma, Z., Marcel, Y.L. & Ravichandran, K.S. Apoptotic cells induce a phosphatidylserine-dependent homeostatic response from phagocytes. Curr. Biol. 16, 2252–2258 (2006).
Janko, C. et al. CRP/anti-CRP antibodies assembly on the surfaces of cell remnants switches their phagocytic clearance toward inflammation. Frontiers in immunology 2, 70 (2011).
Poon, I.K., Hulett, M.D. & Parish, C.R. Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Differ. 17, 381–397 (2010).
Zhang, Y., Brenner, M., Yang, W.L. & Wang, P. Recombinant human MFG-E8 ameliorates colon damage in DSS- and TNBS-induced colitis in mice. Lab. Invest. 95, 480–490 (2015).
Gatza, E. et al. Extracorporeal photopheresis reverses experimental graft-versus-host disease through regulatory T cells. Blood 112, 1515–1521 (2008).
Pasparakis, M. & Vandenabeele, P. Necroptosis and its role in inflammation. Nature 517, 311–320 (2015).
Remijsen, Q. et al. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 18, 581–588 (2011).
Schauer, C. et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 20, 511–517 (2014).
Fan, Y. & Bergmann, A. Apoptosis-induced compensatory proliferation. The Cell is dead. Long live the Cell!. Trends Cell Biol. 18, 467–473 (2008).
Chera, S. et al. Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev. Cell 17, 279–289 (2009).
Tseng, A.S., Adams, D.S., Qiu, D., Koustubhan, P. & Levin, M. Apoptosis is required during early stages of tail regeneration in Xenopus laevis. Dev. Biol. 301, 62–69 (2007).
Fuchs, Y. & Steller, H. Programmed cell death in animal development and disease. Cell 147, 742–758 (2011).
Vlaskalin, T., Wong, C.J. & Tsilfidis, C. Growth and apoptosis during larval forelimb development and adult forelimb regeneration in the newt (Notophthalmus viridescens). Dev. Genes Evol. 214, 423–431 (2004).
Li, F. et al. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci. Signal. 3, ra13 (2010).
Rongvaux, A. et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159, 1563–1577 (2014).
Ravichandran, K.S. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J. Exp. Med. 207, 1807–1817 (2010).
Elliott, J.I. et al. Membrane phosphatidylserine distribution as a non-apoptotic signalling mechanism in lymphocytes. Nat. Cell Biol. 7, 808–816 (2005).
Carrera Silva, E.A. et al. T cell-derived protein S engages TAM receptor signaling in dendritic cells to control the magnitude of the immune response. Immunity 39, 160–170 (2013).
Martin, S. et al. Immunologic stimulation of mast cells leads to the reversible exposure of phosphatidylserine in the absence of apoptosis. Int. Arch. Allergy Immunol. 123, 249–258 (2000).
Frasch, S.C. et al. Phospholipid flip-flop and phospholipid scramblase 1 (PLSCR1) co-localize to uropod rafts in formylated Met-Leu-Phe-stimulated neutrophils. J. Biol. Chem. 279, 17625–17633 (2004).
van den Eijnde, S.M. et al. Transient expression of phosphatidylserine at cell-cell contact areas is required for myotube formation. J. Cell Sci. 114, 3631–3642 (2001).
Hamoud, N., Tran, V., Croteau, L.P., Kania, A. & Cote, J.F. G-protein coupled receptor BAI3 promotes myoblast fusion in vertebrates. Proc. Natl. Acad. Sci. USA 111, 3745–3750 (2014).
Wanderley, J.L., Moreira, M.E., Benjamin, A., Bonomo, A.C. & Barcinski, M.A. Mimicry of apoptotic cells by exposing phosphatidylserine participates in the establishment of amastigotes of Leishmania (L) amazonensis in mammalian hosts. J. Immunol. 176, 1834–1839 (2006).
Seabra, S.H., de Souza, W. & Damatta, R.A. Toxoplasma gondii exposes phosphatidylserine inducing a TGF-β1 autocrine effect orchestrating macrophage evasion. Biochem. Biophys. Res. Commun. 324, 744–752 (2004).
Damatta, R.A. et al. Trypanosoma cruzi exposes phosphatidylserine as an evasion mechanism. FEMS Microbiol. Lett. 266, 29–33 (2007).
Mercer, J. & Helenius, A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320, 531–535 (2008).
Callahan, M.K. et al. Phosphatidylserine on HIV envelope is a cofactor for infection of monocytic cells. J. Immunol. 170, 4840–4845 (2003).
Brindley, M.A. et al. Tyrosine kinase receptor Axl enhances entry of Zaire ebolavirus without direct interactions with the viral glycoprotein. Virology 415, 83–94 (2011).
Hunt, C.L., Kolokoltsov, A.A., Davey, R.A. & Maury, W. The Tyro3 receptor kinase Axl enhances macropinocytosis of Zaire ebolavirus. J. Virol. 85, 334–347 (2011).
Meertens, L. et al. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 12, 544–557 (2012).
Morizono, K. & Chen, I.S. Role of phosphatidylserine receptors in enveloped virus infection. J. Virol. 88, 4275–4290 (2014).
Chen, Y.H. et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 160, 619–630 (2015).
Das, S. et al. Brain angiogenesis inhibitor 1 (BAI1) is a pattern recognition receptor that mediates macrophage binding and engulfment of Gram-negative bacteria. Proc. Natl. Acad. Sci. USA 108, 2136–2141 (2011).
N'Diaye, E.N. et al. TREM-2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J. Cell Biol. 184, 215–223 (2009).
Adachi, H. & Tsujimoto, M. FEEL-1, a novel scavenger receptor with in vitro bacteria-binding and angiogenesis-modulating activities. J. Biol. Chem. 277, 34264–34270 (2002).
Means, T.K. et al. Evolutionarily conserved recognition and innate immunity to fungal pathogens by the scavenger receptors SCARF1 and CD36. J. Exp. Med. 206, 637–653 (2009).
Henson, P.M. Dampening inflammation. Nat. Immunol. 6, 1179–1181 (2005).
Colegio, O.R. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 (2014).
Chung, W.S. et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400 (2013).
Gaultier, A. et al. Low-density lipoprotein receptor-related protein 1 is an essential receptor for myelin phagocytosis. J. Cell Sci. 122, 1155–1162 (2009).
Tasdemir-Yilmaz, O.E. & Freeman, M.R. Astrocytes engage unique molecular programs to engulf pruned neuronal debris from distinct subsets of neurons. Genes Dev. 28, 20–33 (2014).
Lu, Z. et al. Phagocytic activity of neuronal progenitors regulates adult neurogenesis. Nat. Cell Biol. 13, 1076–1083 (2011).
García, M. & Vecino, E. Role of Muller glia in neuroprotection and regeneration in the retina. Histol. Histopathol. 18, 1205–1218 (2003).
Kevany, B.M. & Palczewski, K. Phagocytosis of retinal rod and cone photoreceptors. Physiology (Bethesda) 25, 8–15 (2010).
Wu, Y., Singh, S., Georgescu, M.M. & Birge, R.B. A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J. Cell Sci. 118, 539–553 (2005).
Todt, J.C., Hu, B. & Curtis, J.L. The receptor tyrosine kinase MerTK activates phospholipase C gamma2 during recognition of apoptotic thymocytes by murine macrophages. J. Leukoc. Biol. 75, 705–713 (2004).
Ji, H. et al. T-cell immunoglobulin and mucin domain 4 (TIM-4) signaling in innate immune-mediated liver ischemia-reperfusion injury. Hepatology 60, 2052–2064 (2014).
Karikoski, M. et al. Clever-1/stabilin-1 controls cancer growth and metastasis. Clin. Cancer Res. 20, 6452–6464 (2014).
Hirose, Y. et al. Inhibition of Stabilin-2 elevates circulating hyaluronic acid levels and prevents tumor metastasis. Proc. Natl. Acad. Sci. USA 109, 4263–4268 (2012).
Schledzewski, K. et al. Deficiency of liver sinusoidal scavenger receptors stabilin-1 and -2 in mice causes glomerulofibrotic nephropathy via impaired hepatic clearance of noxious blood factors. J. Clin. Invest. 121, 703–714 (2011).
Englert, J.M. et al. A role for the receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Am. J. Pathol. 172, 583–591 (2008).
Englert, J.M. et al. Paradoxical function for the receptor for advanced glycation end products in mouse models of pulmonary fibrosis. Int. J. Clin. Exp. Pathol. 4, 241–254 (2011).
He, M. et al. The role of the receptor for advanced glycation end-products in lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L1427–L1436 (2007).
He, M. et al. Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep. 12, 358–364 (2011).
Liliensiek, B. et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J. Clin. Invest. 113, 1641–1650 (2004).
Tian, L. et al. p85α recruitment by the CD300f phosphatidylserine receptor mediates apoptotic cell clearance required for autoimmunity suppression. Nat. Commun. 5, 3146 (2014).
Cantoni, C. et al. TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol. 129, 429–447 (2015).
Jay, T.R. et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J. Exp. Med. 212, 287–295 (2015).
Poliani, P.L. et al. TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Invest. 125, 2161–2170 (2015).
Wang, Y. et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160, 1061–1071 (2015).
Bosurgi, L. et al. Paradoxical role of the proto-oncogene Axl and Mer receptor tyrosine kinases in colon cancer. Proc. Natl. Acad. Sci. USA 110, 13091–13096 (2013).
Camenisch, T.D., Koller, B.H., Earp, H.S. & Matsushima, G.K. A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J. Immunol. 162, 3498–3503 (1999).
D'Cruz, P.M. et al. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum. Mol. Genet. 9, 645–651 (2000).
Lu, Q. & Lemke, G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 293, 306–311 (2001).
Neher, J.J. et al. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc. Natl. Acad. Sci. USA 110, E4098–E4107 (2013).
Prasad, D. et al. TAM receptor function in the retinal pigment epithelium. Mol. Cell. Neurosci. 33, 96–108 (2006).
Scott, R.S. et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411, 207–211 (2001).
Weinger, J.G. et al. Loss of the receptor tyrosine kinase Axl leads to enhanced inflammation in the CNS and delayed removal of myelin debris during experimental autoimmune encephalomyelitis. J. Neuroinflammation 8, 49 (2011).
Acharya, M. et al. αv Integrin expression by DCs is required for Th17 cell differentiation and development of experimental autoimmune encephalomyelitis in mice. J. Clin. Invest. 120, 4445–4452 (2010).
Lacy-Hulbert, A. et al. Ulcerative colitis and autoimmunity induced by loss of myeloid alphav integrins. Proc. Natl. Acad. Sci. USA 104, 15823–15828 (2007).
McCarty, J.H. et al. Selective ablation of αV integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development 132, 165–176 (2005).
McCarty, J.H. et al. Genetic ablation of αV integrins in epithelial cells of the eyelid skin and conjunctiva leads to squamous cell carcinoma. Am. J. Pathol. 172, 1740–1747 (2008).
Nandrot, E.F. & Finnemann, S.C. Lack of αVβ5 integrin receptor or its ligand MFG-E8: distinct effects on retinal function. Ophthalmic Res. 40, 120–123 (2008).
Aziz, M., Matsuda, A., Yang, W.L., Jacob, A. & Wang, P. Milk fat globule-epidermal growth factor-factor 8 attenuates neutrophil infiltration in acute lung injury via modulation of CXCR2. J. Immunol. 189, 393–402 (2012).
Fricker, M. et al. MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. J. Neurosci. 32, 2657–2666 (2012).
Hanayama, R. et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science 304, 1147–1150 (2004).
Kusunoki, R. et al. Role of milk fat globule-epidermal growth factor 8 in colonic inflammation and carcinogenesis. J. Gastroenterol. http://dx.doi.org/10.1007/s00535-014-1036-x (2015).
Neher, J.J., Neniskyte, U. & Brown, G.C. Primary phagocytosis of neurons by inflamed microglia: potential roles in neurodegeneration. Front. Pharmacol. 3, 27 (2012).
Peng, Y. & Elkon, K.B. Autoimmunity in MFG-E8-deficient mice is associated with altered trafficking and enhanced cross-presentation of apoptotic cell antigens. J. Clin. Invest. 121, 2221–2241 (2011).
Ait-Oufella, H. et al. Lactadherin deficiency leads to apoptotic cell accumulation and accelerated atherosclerosis in mice. Circulation 115, 2168–2177 (2007).
Akitake-Kawano, R. et al. Inhibitory role of Gas6 in intestinal tumorigenesis. Carcinogenesis 34, 1567–1574 (2013).
Angelillo-Scherrer, A. et al. Deficiency or inhibition of Gas6 causes platelet dysfunction and protects mice against thrombosis. Nat. Med. 7, 215–221 (2001).
Binder, M.D. et al. Gas6 deficiency increases oligodendrocyte loss and microglial activation in response to cuprizone-induced demyelination. J. Neurosci. 28, 5195–5206 (2008).
Binder, M.D. et al. Gas6 increases myelination by oligodendrocytes and its deficiency delays recovery following cuprizone-induced demyelination. PLoS ONE 6, e17727 (2011).
Burnier, L. et al. Gas6 deficiency in recipient mice of allogeneic transplantation alleviates hepatic graft-versus-host disease. Blood 115, 3390–3397 (2010).
Llacuna, L. et al. Growth arrest-specific protein 6 is hepatoprotective against murine ischemia/reperfusion injury. Hepatology 52, 1371–1379 (2010).
Yanagita, M. et al. Essential role of Gas6 for glomerular injury in nephrotoxic nephritis. J. Clin. Invest. 110, 239–246 (2002).
Cai, L., Wang, Z., Ji, A., Meyer, J.M. & van der Westhuyzen, D.R. Scavenger receptor CD36 expression contributes to adipose tissue inflammation and cell death in diet-induced obesity. PLoS ONE 7, e36785 (2012).
Greenberg, M.E. et al. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J. Exp. Med. 203, 2613–2625 (2006).
Kennedy, D.J. et al. Dietary cholesterol plays a role in CD36-mediated atherogenesis in LDLR-knockout mice. Arterioscler. Thromb. Vasc. Biol. 29, 1481–1487 (2009).
Parks, B.W. et al. CD36, but not G2A, modulates efferocytosis, inflammation, and fibrosis following bleomycin-induced lung injury. J. Lipid Res. 54, 1114–1123 (2013).
Overton, C.D., Yancey, P.G., Major, A.S., Linton, M.F. & Fazio, S. Deletion of macrophage LDL receptor-related protein increases atherogenesis in the mouse. Circ. Res. 100, 670–677 (2007).
Yancey, P.G. et al. Macrophage LRP-1 controls plaque cellularity by regulating efferocytosis and Akt activation. Arterioscler. Thromb. Vasc. Biol. 30, 787–795 (2010).
Yancey, P.G. et al. Low-density lipoprotein receptor-related protein 1 prevents early atherosclerosis by limiting lesional apoptosis and inflammatory Ly-6Chigh monocytosis: evidence that the effects are not apolipoprotein E dependent. Circulation 124, 454–464 (2011).
Subramanian, M. et al. An AXL/LRP-1/RANBP9 complex mediates DC efferocytosis and antigen cross-presentation in vivo. J. Clin. Invest. 124, 1296–1308 (2014).
Ramirez-Ortiz, Z.G. et al. The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat. Immunol. 14, 917–926 (2013).
Bhatia, V.K. et al. Complement C1q reduces early atherosclerosis in low-density lipoprotein receptor-deficient mice. Am. J. Pathol. 170, 416–426 (2007).
Bossi, F. et al. C1q as a unique player in angiogenesis with therapeutic implication in wound healing. Proc. Natl. Acad. Sci. USA 111, 4209–4214 (2014).
Botto, M. et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat. Genet. 19, 56–59 (1998).
Chu, Y. et al. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc. Natl. Acad. Sci. USA 107, 7975–7980 (2010).
Stevens, B. et al. The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178 (2007).
Acknowledgements
We thank the members of the Ravichandran laboratory, as well as colleagues in the field, for comments and discussions. Supported by the US National Institutes of Health (NIGMS GM064709, HD074981, GM107848, HL120840 and MH096484).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Arandjelovic, S., Ravichandran, K. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol 16, 907–917 (2015). https://doi.org/10.1038/ni.3253
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ni.3253
