
Abstract
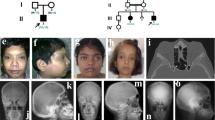
Schimke immuno-osseous dysplasia (SIOD, MIM 242900) is an autosomal-recessive pleiotropic disorder with the diagnostic features of spondyloepiphyseal dysplasia, renal dysfunction and T-cell immunodeficiency1,2,3. Using genome-wide linkage mapping and a positional candidate approach, we determined that mutations in SMARCAL1 (SWI/SNF2-related, matrix-associated, actin-dependent regulator of chromatin, subfamily a–like 1), are responsible for SIOD. Through analysis of data from persons with SIOD in 26 unrelated families, we observed that affected individuals from 13 of 23 families with severe disease had two alleles with nonsense, frameshift or splicing mutations, whereas affected individuals from 3 of 3 families with milder disease had a missense mutation on each allele. These observations indicate that some missense mutations allow retention of partial SMARCAL1 function and thus cause milder disease.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


References
Boerkoel, C.F. et al. Manifestations and treatment of Schimke immuno-osseous dysplasia: 14 new cases and a review of the literature. Eur. J. Pediatr. 159, 1–7 (2000).
Spranger, J. et al. Schimke immuno-osseous dysplasia: a newly recognized multisystem disease. J. Pediatr. 119, 64–72 (1991).
Schimke, R.N., Horton, W.A. & King, C.R. Chondroitin-6-sulfaturia, defective cellular immunity, and nephrotic syndrome. Lancet 2, 1088–1089 (1971).
Saraiva, J.M. et al. Schimke immuno-osseous dysplasia: case report and review of 25 patients. J. Med. Genet. 36, 786–789 (1999).
Coleman, M.A., Eisen, J.A. & Mohrenweiser, H.W. Cloning and characterization of HARP/SMARCAL1: a prokaryotic HepA-related SNF2 helicase protein from human and mouse. Genomics 65, 274–282 (2000).
Havas, K., Whitehouse, I. & Owen-Hughes, T. ATP-dependent chromatin remodeling activities. Cell. Mol. Life Sci. 58, 673–682 (2001).
Pazin, M.J. & Kadonaga, J.T. SWI2/SNF2 and related proteins: ATP-driven motors that disrupt protein–DNA interactions? Cell 88, 737–740 (1997).
Koonin, E.V. A common set of conserved motifs in a vast variety of putative nucleic acid–dependent ATPases including MCM proteins involved in the initiation of eukaryotic DNA replication. Nucleic Acids Res. 21, 2541–2547 (1993).
Gorbalenya, A.E., Koonin, E.V., Donchenko, A.P. & Blinov, V.M. Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res. 17, 4713–4730 (1989).
Hall, M.C. & Matson, S.W. Helicase motifs: the engine that powers DNA unwinding. Mol. Microbiol. 34, 867–877 (1999).
Troelstra, C. et al. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne's syndrome and preferential repair of active genes. Cell 71, 939–953 (1992).
Gibbons, R.J., Picketts, D.J., Villard, L. & Higgs, D.R. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with α-thalassemia (ATR-X syndrome). Cell 80, 837–845 (1995).
Sévenet, N. et al. Spectrum of hSNF5/INI1 somatic mutations in human cancer and genotype–phenotype correlations. Hum. Mol. Genet. 8, 2359–2368 (1999).
Bochar, D.A. et al. BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell 102, 257–265 (2000).
Schäffer, A.A., Gupta, S.K., Shriram, K. & Cottingham, R.W. Jr. Avoiding recomputation in linkage analysis. Hum. Hered. 44, 225–237 (1994).
Gudbjartsson, D.F., Jonasson, K., Frigge, M.L. & Kong, A. Allegro, a new computer program for multipoint linkage analysis. Nature Genet. 25, 12–13 (2000).
den Dunnen, J.T. & Antonarakis, S.E. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum. Mutat. 15, 7–12 (2000).
Shaffer, L.G., Kennedy, G.M., Spikes, A.S. & Lupski, J.R. Diagnosis of CMT1A duplications and HNPP deletions by interphase FISH: implications for testing in the cytogenetics laboratory. Am. J. Med. Genet. 69, 325–331 (1997).
Walker, J.E., Saraste, M., Runswick, M.J. & Gay, N.J. Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1, 945–951 (1982).
Korolev, S., Hsieh, J., Gauss, G.H., Lohman, T.M. & Waksman, G. Major domain swiveling revealed by the crystal structures of complexes of E. coli Rep helicase bound to single-stranded DNA and ADP. Cell 90, 635–647 (1997).
Subramanya, H.S., Bird, L.E., Brannigan, J.A. & Wigley, D.B. Crystal structure of a DExx box DNA helicase. Nature 384, 379–383 (1996).
Yao, N. et al. Structure of the hepatitis C virus RNA helicase domain. Nature Struct. Biol. 4, 463–467 (1997).
Kim, J.L. et al. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure 6, 89–100 (1998).
Hall, M.C., Özsoy, A.Z. & Matson, S.W. Site-directed mutations in motif VI of Escherichia coli DNA helicase II result in multiple biochemical defects: evidence for the involvement of motif VI in the coupling of ATPase and DNA binding activities via conformational changes. J. Mol. Biol. 277, 257–271 (1998).
Hall, M.C. & Matson, S.W. Mutation of a highly conserved arginine in motif IV of Escherichia coli DNA helicase II results in an ATP-binding defect. J. Biol. Chem. 272, 18614–18620 (1997).
Acknowledgements
We thank the families described for their cooperation and H. Bellen for critical review of this manuscript. H.T. is a recipient of a postdoctoral fellowship from the Charcot–Marie–Tooth Association. This study was supported in part by the Kleberg Foundation and by grants from the US National Institute of Diabetes, Digestive, and Kidney Diseases (to C.F.B.), from the US National Institute of Eye Diseases (to D.W.S.) and from the US National Institute of Neurological Disorders and Stroke (to J.R.L.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Boerkoel, C., Takashima, H., John, J. et al. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet 30, 215–220 (2002). https://doi.org/10.1038/ng821
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng821
This article is cited by
-
Schimke immunoosseous dysplasia: an ultra-rare disease. a 20-year case series from the tertiary hospital in the Czech Republic
Italian Journal of Pediatrics (2023)
-
Pathological consequences of DNA damage in the kidney
Nature Reviews Nephrology (2023)
-
Hidden genetics behind glomerular scars: an opportunity to understand the heterogeneity of focal segmental glomerulosclerosis?
Pediatric Nephrology (2023)
-
CHR721, interacting with OsRPA1a, is essential for both male and female reproductive development in rice
Plant Molecular Biology (2020)
-
Genetic testing in steroid-resistant nephrotic syndrome: why, who, when and how?
Pediatric Nephrology (2019)
