
Abstract
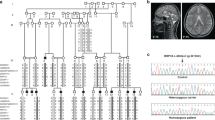
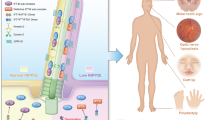
Cerebello-oculo-renal syndrome (CORS), also called Joubert syndrome type B, and Meckel (MKS) syndrome belong to the group of developmental autosomal recessive disorders that are associated with primary cilium dysfunction. Using SNP mapping, we identified missense and truncating mutations in RPGRIP1L (KIAA1005) in both CORS and MKS, and we show that inactivation of the mouse ortholog Rpgrip1l (Ftm) recapitulates the cerebral, renal and hepatic defects of CORS and MKS. In addition, we show that RPGRIP1L colocalizes at the basal body and centrosomes with the protein products of both NPHP6 and NPHP4, known genes associated with MKS, CORS and nephronophthisis (a related renal disorder and ciliopathy). In addition, the RPGRIP1L missense mutations found in CORS individuals diminishes the interaction between RPGRIP1L and nephrocystin-4. Our findings show that mutations in RPGRIP1L can cause the multiorgan phenotypic abnormalities found in CORS or MKS, which therefore represent a continuum of the same underlying disorder.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

References
Joubert, M., Eisenring, J.J., Robb, J.P. & Andermann, F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology 19, 813–825 (1969).
Patel, S. & Barkovich, A.J. Analysis and classification of cerebellar malformations. AJNR Am. J. Neuroradiol. 23, 1074–1087 (2002).
Saunier, S., Salomon, R. & Antignac, C. Nephronophthisis. Curr. Opin. Genet. Dev. 15, 324–331 (2005).
Valente, E.M. et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet. 38, 623–625 (2006).
Sayer, J.A. et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet. 38, 674–681 (2006).
Ferland, R.J. et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat. Genet. 36, 1008–1013 (2004).
Parisi, M.A. et al. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am. J. Hum. Genet. 75, 82–91 (2004).
Caridi, G. et al. Nephronophthisis type 1 deletion syndrome with neurological symptoms: prevalence and significance of the association. Kidney Int. 70, 1342–1347 (2006).
Betz, R. et al. Children with ocular motor apraxia type Cogan carry deletions in the gene (NPHP1) for juvenile nephronophthisis. J. Pediatr. 136, 828–831 (2000).
Tory, K. et al. High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J. Am. Soc. Nephrol. 18, 1566–1575 (2007).
Saar, K. et al. Homozygosity mapping in families with Joubert syndrome identifies a locus on chromosome 9q34.3 and evidence for genetic heterogeneity. Am. J. Hum. Genet. 65, 1666–1671 (1999).
Valente, E.M. et al. Description, nomenclature and mapping of a novel cerebello-renal syndrome with the molar tooth malformation. Am. J. Hum. Genet. 73, 663–670 (2003).
Kyttala, M. et al. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat. Genet. 38, 155–157 (2006).
Smith, U.M. et al. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat. Genet. 38, 191–196 (2006).
Baala, L. et al. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am. J. Hum. Genet. 80, 186–194 (2007).
Baala, L. et al. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel-Gruber syndrome. Am. J. Hum. Genet. (in the press).
Dawe, H.R. et al. The Meckel-Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum. Mol. Genet. 16, 173–186 (2007).
van der Hoeven, F. et al. Programmed cell death is affected in the novel mouse mutant Fused toes (Ft). Development 120, 2601–2607 (1994).
Anselme, I., Laclef, C., Lanaud, M., Rüther, U. & Schneider-Maunoury, S. Defects in brain patterning and head morphogenesis in the mouse mutant Fused-Toes. Dev. Biol. 304, 208–220 (2007).
Roepman, R. et al. Interaction of nephrocystin-4 and RPGRIP1 is disrupted by nephronophthisis or Leber congenital amaurosis-associated mutations. Proc. Natl. Acad. Sci. USA 102, 18520–18525 (2005).
Mollet, G. et al. The gene mutated in juvenile nephronophthisis type 4 encodes a novel protein that interacts with nephrocystin. Nat. Genet. 32, 300–305 (2002).
Mollet, G. et al. Characterization of the nephrocystin/nephrocystin-4 complex and subcellular localization of nephrocystin-4 to primary cilia and centrosomes. Hum. Mol. Genet. 14, 645–656 (2005).
Blacque, O.E. et al. Functional genomics of the cilium, a sensory organelle. Curr. Biol. 15, 935–941 (2005).
Dryja, T.P. et al. Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am. J. Hum. Genet. 68, 1295–1298 (2001).
Chen, D. & Shou, C. Molecular cloning of a tumor-associated antigen recognized by monoclonal antibody 3H11. Biochem. Biophys. Res. Commun. 280, 99–103 (2001).
Watnick, T. & Germino, G. From cilia to cyst. Nat. Genet. 34, 355–356 (2003).
Otto, E.A. et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat. Genet. 34, 413–420 (2003).
Huangfu, D. & Anderson, K.V. Cilia and Hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. USA 102, 11325–11330 (2005).
Kruglyak, L., Daly, M.J., Reeve-Daly, M.P. & Lander, E.S. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am. J. Hum. Genet. 58, 1347–1363 (1996).
Espinosa-Parrilla, Y. et al. Expression of the SMADIP1 gene during early human development. Mech. Dev. 114, 187–191 (2002).
Acknowledgements
We thank the patients and their families for participation. We thank N. Spassky, R. Benkirane, G. Guedu, S. Audollent, C. Gazengel, S. Delahaye and C. Esculpavit for technical assistance and the SOFFOET for clinical data and material support. We thank M. Teboul and the staff of the Centre d'Orthogénie at the Broussais hospital in Paris for their contributions. This work was supported by grants from GIS-Maladies Rares (AAE05017KSA), the Association pour l'Utilisation du Rein Artificiel, the Association pour la Recherche sur le Cancer (ARC) (to S.S.M.) and the Deutsche Forschungsgemeinschaft (DFG) through SFB 590 and 612 (to U.R.). M.D. is the recipient of a fellowship from the Ministère de la Recherche, K.T. and N.E.H. were supported from a grant from the Fondation Recherche Médicale, K.T. benefits from an ERA-EDTA fellowship, N.E.H. is the recipient of a Fulbright grant and L.B. is supported by the CANAM. F.H. is a Doris Duke Distinguished Clinical Scientist and supported by US National Institutes of Health grant DK068306. M.T.F.W. was supported by grants from the German Kidney Fund (Deutsche Nierenstiftung) and the German Research Foundation (DFG WO 1229/2-1).
Author information
Authors and Affiliations
Contributions
The senior authors T.A.-B. and S.S. contributed equally to this work. T.A.-B. and S.S. designed this study; M.D. and C.G. performed expression studies in humans; M.D., F.S. and I.M. performed functional analyses (interaction and localization studies); L.B., T.L., K.T. and C.O. contributed to mutation screening; S.S.-M., C.L., L.B., I.A. and C.V. performed mouse phenotype analyses; U.R., J.V. and C.G. generated mice and antibodies against Rpgrip1l; C.A., R.S., M.C.G., F.E.-R., M.V. and N.B. performed phenotype assessment, C.A.J., F.M., E.R., F.H. and M.T.F.W. contributed to the SNP genotyping; C.A., N.E.H, R.S. and S.S.-M. contributed to the writing of this paper and M.G., M.A.M., H.N., G.C., J.P.B. and P.N. assessed patients in the study. R.S. and S.S.-M. initiated the collaboration.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Fig. 1
RPGRIP1L mutations in individuals with CORS and fetuses with MKS. (PDF 292 kb)
Supplementary Fig. 2
Anatomical and pathological data from individuals with CORS and fetuses with MKS. (PDF 471 kb)
Supplementary Fig. 3
RPGRIP1L localization during cell division. (PDF 413 kb)
Supplementary Fig. 4
Characterization of polyclonal antibody to nephrocystin-6. (PDF 143 kb)
Supplementary Fig. 5
Characterization of polyclonal antibodies to RPGRIP1L. (PDF 178 kb)
Supplementary Table 1
Exon-flanking oligonucleotide PCR primers for the human RPGRIP1L gene. (PDF 12 kb)
Rights and permissions
About this article
Cite this article
Delous, M., Baala, L., Salomon, R. et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet 39, 875–881 (2007). https://doi.org/10.1038/ng2039
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng2039
