
Abstract
Disruption of human neural precursor proliferation can give rise to a small brain (microcephaly), and failure of neurons to migrate properly can lead to an abnormal arrest of cerebral cortical neurons in proliferative zones near the lateral ventricles (periventricular heterotopia). Here we show that an autosomal recessive condition characterized by microcephaly and periventricular heterotopia1 maps to chromosome 20 and is caused by mutations in the gene ADP-ribosylation factor guanine nucleotide-exchange factor-2 (ARFGEF2). By northern-blot analysis, we found that mouse Arfgef2 mRNA levels are highest during embryonic periods of ongoing neuronal proliferation and migration, and by in situ hybridization, we found that the mRNA is widely distributed throughout the embryonic central nervous system (CNS). ARFGEF2 encodes the large (>200 kDa) brefeldin A (BFA)-inhibited GEF2 protein (BIG2), which is required for vesicle and membrane trafficking from the trans-Golgi network (TGN). Inhibition of BIG2 by BFA, or by a dominant negative ARFGEF2 cDNA, decreases cell proliferation in vitro, suggesting a cell-autonomous regulation of neural expansion. Inhibition of BIG2 also disturbed the intracellular localization of such molecules as E-cadherin and β-catenin by preventing their transport from the Golgi apparatus to the cell surface. Our findings show that vesicle trafficking is an important regulator of proliferation and migration during human cerebral cortical development.
Similar content being viewed by others


Main
Autosomal recessive periventricular heterotopia with microcephaly (ARPHM) is a severe malformation of the cerebral cortex, characterized by severe developmental delay and recurrent infections1. No anomalies extrinsic to the CNS, such as dysmorphic features or grossly abnormal endocrine or other conditions, have been observed in individuals with ARPHM. Magnetic resonance imaging of the brains of affected individuals shows notable periventricular heterotopia, resulting from the failure of neurons to migrate normally from the lateral ventricular proliferative zone, where they are formed, to the cerebral cortex (Fig. 1a–d). Abnormal magnetic resonance image signals in subcortical white matter and elsewhere also suggest a delay in the normal myelination by glial cells.

(a) Axial T1-weighted image from the affected female in pedigree 2 at 6 months of age shows bilateral, continuous nodular heterotopia along the margin of the lateral ventricles and small cerebral cortex. The ventricles are prominent or enlarged. (b) Coronal T1-weighted image from the same female. (c) Sagittal flair sequence from the same female shows a slightly small corpus callosum, especially posteriorly, and relative preservation of the cerebellum and brainstem. (d) Axial T2-weighted image from the affected male sibling (in pedigree 2) at ∼18 months of age shows the characteristic radiographic findings of periventricular heterotopia alone with mild hydrocephalus. Scale bars in a–d, 3 cm, subdivided into subintervals of 1 cm. (e,f) Pedigrees of families with ARPHM showing genotypes for microsatellite markers on chromosome 20q11.21–q13.2. The marker order is centromere, D20S195, D20S839, telomere. The boxed regions indicate regions of homozygosity in each affected individual. The crossover regions are indicated in the shaded boxes adjacent to the alleles. (g) Combined multipoint lod score analysis of ARPHM in chromosome 20q11.21–13.2 for both families generated a maximal multipoint lod score of 4.41 between microsatellite markers D20S197 and D20S845. Analysis of pedigree 1 gave a maximal lod score of 2.23 between microsatellite markers D20S107 and D20S845. Analysis of pedigree 2 gave a maximal lod score of 2.18 between microsatellite markers D20S197 and D20S845. The horizontal axis extends over 9 cM. (h) Two-point analyses of each pedigree shows that the maximal lod scores also fall between microsatellite markers D20S197 and D20S845 with lod scores >3.0 at D20S197, D20S109 and D20S857. infini, infinity; Ped1, pedigree 1; Ped2, pedigree 2.
An initial genome-wide screen at 10-cM intervals of two Turkish families with ARPHM identified shared homozygosity at a single locus on chromosome 20q11.21–13.2, which we refined by further marker analysis (Fig. 1e,f). Because the two families had indistinguishable clinical and radiographic features, we assumed that they had an allelic disorder and summed their linkage results. The ARPHM region (66.16–77.75 cM on chromosome 20) that was identical by descent in each of the two families yielded a summed multipoint lod score of 4.41 (Fig. 1g). Two-point analyses gave combined maximal lod scores of 3.28 (pedigree 1 = 1.53 and pedigree 2 = 1.75) at marker D20S109 (Fig. 1h).
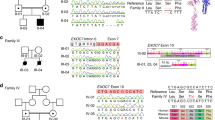
We sequenced several candidate genes in the minimal linked region and found a different deleterious mutation in ARFGEF2 (encoding BIG2) in each pedigree (Fig. 2a). In pedigree 1, exon 6 contained the nucleotide change 625G→A, leading to the predicted nonconservative amino acid substitution E209K (Fig. 2b). This residue is identical between BFA-inhibited GEF1 (BIG1) and BIG2, is embedded in a larger area of 79% amino acid identity between the two proteins at the N terminus and is identical in mouse and humans (Fig. 2d). Pedigree 2 has a complex mutation in exon 3 with the nucleotide substitutions 242C→A and 247G→T and the deletion 249delA. These nucleotide changes predict amino acid substitutions P81Q and V83L and a translational frameshift at amino acid residue 84, resulting in predicted premature termination of translation after 30 additional abnormal amino acids (Fig. 2b). The mutated allele in pedigree 2 was present in the mRNA of an affected individual but not in that of an unaffected sibling (Fig. 2c). The mutations segregated with the disease manifestations in each family and were not found in 220 unrelated control individuals of European descent or 110 unrelated control individuals of Middle Eastern descent (data not shown). These data are consistent with the conclusion that mutations in ARFGEF2 cause ARPHM.

(a) Candidate genes in and adjacent to the region of common haplotype. (b) In pedigree 1, 625G→A predicts the amino acid substitution E209K, with a negative-to-positive polarity shift at physiological pH and predicted disruption of protein conformation. In pedigree 2, 242C→A;247G→T;249delA results in a predicted premature protein truncation. Similarly shaded arrowheads point to respective mutations. (c) The mutated allele in pedigree 2 is identified at the mRNA level in the affected individual and is not seen in the unaffected sibling. (d) Diagram of the BIG2 molecule (the protein product of ARFGEF2) shows position numbers of amino acids above and percentage of identity of sequence with that of BIG1 in the Sec7 domain and other regions as indicated. Alignment of BIG1 and BIG2 showed 72% amino acid identity overall with 90% identity in the Sec7 domain. I, identity; +, conservative differences as determined by alignment in the National Center for Biotechnology Information database.
ADP-ribosylation factors (ARFs) are required to assemble coat proteins around intracellular vesicles in a GTP-dependent fashion, and thereby regulate vesicle cycling between the TGN, endosome and cell surface2,3. ARFGEFs, such as BIG2 (the protein product of ARFGEF2), initiate this process by accelerating the exchange of GDP for GTP through a Sec7 domain and thus activating the ARF2,3,4,5,6. This regulatory role is essential in embryonic pattern formation, apical-basal transport of proteins, cell division, elongation and adhesion in plants and yeast7,8,9.
To investigate how BIG2 might regulate neural proliferation and neuronal migration, we first carried out northern-blot analyses of whole-mouse Arfgef2 mRNA. Expression gradually increased from embryonic day (E) 11 to E17, during the period of cerebral cortical neurogenesis (Fig. 3a). In situ hybridization analysis of E17 mice (Fig. 3b–d) showed that there was somewhat more Arfgef2 mRNA in the cerebral cortical and diencephalic ventricular zones than in other CNS structures. This pattern of expression, though widespread, is consistent with a potential role in early neural precursor proliferation and neuronal migration, which might explain the microcephaly and periventricular heterotopia. Furthermore, BIG2 immunoreactivity was restricted to the Golgi apparatus in mouse neuroepithelial cells, consistent with its known role in vesicle trafficking (Fig. 3e).

(a) Northern-blot analysis of mouse whole embryos at various ages shows a gradual increase in Arfgef2 mRNA expression during late gestation. Actb was used as a loading control. (b) Sagittal section of E17 mouse shows widespread Arfgef2 mRNA expression with preferential expression in the CNS, liver, gut and kidneys by in situ hybridization. (c,d) Coronal sections of the rostral and caudal forebrain of E17 mouse show widespread expression throughout the CNS, with increased expression in the cerebral cortical ventricular zone, the proliferative region of the medial ganglionic eminence and lateral ganglionic eminence and the ventricular zone of the dorsal diencephalon as compared to other CNS structures. Elevated expression is also observed in the neocortex, hippocampus and olfactory bulbs. This pattern of expression was identical in three separate antisense probes and was not seen with control sense probes (data not shown). (e) Neuroepithelial cell from mouse primary neuronal culture shows colocalization of BIG2 (ARFGEF2) and the Golgi marker GM130 by immunostaining. Scale bars: 50 μm in b and d; 25 μm in e.
As ARPHM is probably a disorder of neural proliferation and migration, and as disruption of plant, yeast or mammalian ARFGEF homologs impairs proliferation, adhesion or neurite extension7,8,9,10,11,12, we investigated the effects of GEF inhibition on neural precursor proliferation and neuronal migration. Neural stem cells, derived from the subventricular zone (the region responsible for neural proliferation and initial migration into cortex) of adult mice, undergo continual expansion and glial-independent chain migration in vitro13,14. We treated cultured neural progenitors with BFA, a fungal metabolite that specifically inhibits the Sec7 domains of the high-molecular-weight (>200 kDa) ARFGEFs (BIG1 and BIG2 but not GBF1; ref. 8). Inhibition of BIG1 and BIG2 by BFA specifically disrupts the Golgi complex and TGN with preservation of other cytoplasmic structures15 (Fig. 4a). Progenitors treated with BFA had less proliferation, manifested as less uptake of bromodeoxyuridine (BrdU; Fig. 4b), with no increase in cell death as measured by TUNEL staining (Supplementary Fig. 1 online). To analyze the potential role of BIG2 specifically in proliferation, we expressed a dominant negative mutant BIG2 protein in HEK293 cells. The amino acid substitution E738K (in the Sec7 domain) interferes with BIG2 activity and membrane trafficking from the TGN5. Proliferation of HEK293 cells overexpressing hemagglutinin (HA)-tagged mutant BIG2 (E738K) was significantly less than that of cells expressing wild-type BIG2 (Fig. 4c). The results of treating cerebellar granule cells and neural stem cells with BFA suggest that BIG2 functions in neural migration. Treatment with BFA inhibited migration of cerebellar granule cells across a filter and inhibited spreading of neural precursors derived from the subventricular zone, suggesting that ARFGEF function is required for neuronal migration (Fig. 4d,e).

(a) Electron micrograph of mouse neurospheres 3 h after exposure to 4 μM BFA shows specific disruption of the Golgi apparatus (arrowheads) with relative preservation of the nuclei (Nu), mitochondria and other subcellular constituents. Higher magnification of the boxed region in a shows the extent of disruption of the Golgi (Go, concave arrowheads) and preservation of other organelles, such as the mitochondria (Mt, arrowheads) and the endoplasmic reticulum (ER, arrows). (b) Fluorescence photomicrograph of neural precursors immunostained for BrdU (with rhodamine) 24 h after addition of the indicated concentration of BFA and incubation in BrdU. The level of BrdU incorporation is graphically summarized to the right (n = 6–8 samples per concentration of BFA). **P < 0.0002. (c) Fluorescence photomicrographs of HEK293 cells expressing HA-tagged BIG2 (E738K) or wild-type BIG2 (WT) after immunostaining with an antibody to HA (rhodamine), an antibody to BrdU (fluorescein) and a Hoechst nuclear stain. Proliferation of cells containing HA-tagged dominant negative BIG2 (E738K) 24 h after transfection and 3 h after BrdU addition was less than that of cells transfected with wild-type BIG2. Cells containing HA-tagged protein (rhodamine) are marked with arrows or arrowheads, respectively, to indicate that they have or have not incorporated BrdU. Results are summarized to the right. *χ2 = 21.2, P < 0.02. (d) Inhibition of BIG2 by BFA impairs cerebellar granule cell migration. Less migration of cerebellar granule cells through transwell membrane can be observed after pretreatment with 4 μM BFA. *P < 0.0004 by paired t-test analysis. (e) Fluorescence photomicrograph of Hoechst-stained nuclei from neural precursors 24 h after plating of neurospheres and addition of the indicated concentration of BFA. Less dispersion and migration of precursors can be observed with increasing concentrations of the BIG2 inhibitor. The degree of neural precursor migration from the center of the neurosphere is graphically summarized to the right (n = 3–6 samples per concentration). *P < 0.003 and **P < 0.008 compared to controls. Scale bars: 2 μm (low magnification) and 500 nm (high magnification) in a; 100 μm in b and e; 25 μm in c.
Inhibition of the ARFGEFs by BFA can impair the expression of many proteins on the cell surface16,17, and this disruption is a probable mechanism of action for BIG2 in neural proliferation and migration. We therefore examined the effect of BFA and of dominant negative BIG2 (E738K) on the expression pattern of a few representative proteins in MDCK cells. Inhibition of BIG2 disrupted the normal localization of E-cadherin and the normal colocalization of β-catenin, which binds the cytoplasmic tail of cadherins at the cell surface18. In contrast, such markers as the atypical protein kinase C λ (PKCλ) and early endosome antigen 1 (EEA-1) were not apparently affected (Figs. 5 and 6). The mutant BIG2 (E738K) protein colocalized with E-cadherin, β-catenin and the Golgi marker GM130, suggesting that E-cadherin and β-catenin were arrested in the Golgi (Fig. 6). The exact target proteins whose trafficking is regulated by BIG2 in dividing and migrating cells are not known, though components of the Wnt signaling pathway, such as β-catenin, are good candidates. For example, β-catenin is implicated in regulation of cerebral cortical size19, and so redistribution of this protein might impair proliferation in a cell-autonomous fashion. Likewise, blockade of the transport of E-cadherin, β-catenin or other proteins from the Golgi to the cell surface could also disrupt cell-cell adhesion and prevent postmitotic neurons from exiting the ventricular zone.

Disruption of E-cadherin–catenin complexes is apparent 3 h after exposure to 4 μM BFA, with redistribution of the proteins from the adherens junction to the cytoplasm after 7 h. Perinuclear cytoplasmic staining for E-cadherin and β-catenin suggest that the transport of these proteins is arrested in the Golgi, blocking membrane association of both. No significant change is observed in expression pattern of EEA-1 and PKCλ at the same time points. Scale bar, 50 μm.

MDCK cells expressing HA-tagged BIG2 (E738K) or wild-type BIG2 (WT) were stained with antibody to HA, antibody to E-cadherin, β-catenin, EEA-1 or GM130, and a Hoechst nuclear stain. The perinuclear redistribution of both E-cadherin or β-catenin in cells transfected with BIG2 (E738K) but not with wild-type BIG2 overlaps with distribution of BIG2, suggesting that their transport from the Golgi to the cell membrane is arrested in the Golgi. Colocalization of both the wild-type and dominant negative BIG2 (E738K) constructs with the Golgi marker GM130 confirms this observation.
The essential requirement for vesicle trafficking in neuronal proliferation and migration shifts our view of both processes in the developing human brain. For instance, mutations in ASPM are the most common cause of recessive microcephaly in humans20, and ASPM is the human ortholog of the Drosophila melanogaster gene called abnormal spindle, which is essential for mitotic spindle function21. Other genes associated with microcephaly are involved in DNA repair, for example, in Nijmegen breakage syndrome (NBS1; ref. 22). On the other hand, mutations in FLNA are the only other identified cause of periventricular heterotopia, but unlike ARFGEF2, FLNA is not associated with microcephaly, and it encodes an actin-binding protein23. Thus, the current findings emphasize the crucial, if not yet widely recognized, role of vesicular trafficking in human embryonic development and raise the possibility that the enlarging superfamily of ARFGEF proteins may have evolved more specialized and overlapping functions than suspected2,3,4,5,6.
Methods
Subjects.
The clinical features of pedigree 1 with periventricular heterotopia and an autosomal recessive mode of inheritance were previously reported1. The affected Turkish children in pedigree 2 share the clinical features of severe developmental delay (no postural acquisition, quadriparesis, poor eye contact), microcephaly (head circumference >2 s.d. below the mean), early-onset refractory epilepsy (electroencephalogram with hypsarrhythmia), bilateral nodular periventricular heterotopia and no extraneurological anomalies. The male child died from pneumonia at 13 years of age.
The study was approved by the Institutional Review Board of the Beth Israel Deaconess Medical Center, Children's Hospital Boston and the National Institutes of Health Office of Human Research Protection. We obtained informed consent from all subjects and, for those under 18 years of age, also from their parents. All animal procedures were reviewed and approved by the Harvard Medical School Standing Committee on Animals and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Linkage analysis.
We carried out a genome-wide screen on each of the pedigrees, using human MapPairs with fluorescent labels and separated at intervals of 10 cM on average (ABI 10cM Linkage Mapping Set Version 2.5, Applied Biosystems). We isolated DNA from the peripheral whole blood of individuals from each pedigree using previously described protocols (Qiagen). We carried out PCR using the selected markers, analyzed the samples on an ABI Prism 3100 genetic analyzer and determined alleles using a standard software package (Genotyper Analysis) with size standards provided by the manufacturer. We further refined the map using custom markers D20S195, D20S107, D20S119, D20S481, D20S836, D20S886, D20S891, D20S197, D20S178, D20S176, D20S887, D20S108, D20S868, D20S196, D20S857, D20S845 and D20S838 (Research Genetics, Invitrogen). We calculated multipoint lod scores using the GeneHunter program with five-point runs. Serial runs were concatenated to generate the graph24. For multipoint analysis of linkage, we assumed the disorder to be autosomal recessive with a penetrance of 90% in males and females. In all families, we assumed a disease allele frequency of 1 in 10,000 and eight alleles per marker at an equal allele frequency (12.5%). Variation in disease-allele frequency did not significantly alter the lod score analysis (Supplementary Table 1 online). To ensure that variations in marker allele frequencies did not significantly alter the lod scores, we also carried out two-point analyses assuming equal allele frequency and repeated them under the assumption that alleles in the region of linkage and shared by affected individuals were either rare (having lower frequency in the population) or common (having higher frequency in the population). Maximal two-point lod scores ranged between 2.56 and 4.07 after analyses with variations in allele frequency (Supplementary Table 1 online).
Candidate gene analysis.
We identified candidate genes in the region of homozygosity using available databases (National Center for Biotechnology Information, Celera and University of California at Santa Cruz). We designed exon-specific primers using standard software (Primer 3), purified the amplified PCR products (PSIΨClone PCR 96) and sequenced the products through the Dana-Farber/Harvard Cancer Center High-Throughput DNA Sequencing Facility. We then compared the exon and intron-exon sequences to consensus sequences obtained from the available databases listed above using standard software for DNA sequencing (Sequencher, version 3.1.1). We confirmed the mutations by bidirectional sequencing of genomic DNA. Normal control DNA represented samples (n = 220) from random individuals of European descent. We sequenced an additional 110 control DNA samples from individuals of Middle Eastern descent. A search of the single-nucleotide polymorphism databases of both National Center for Biotechnology Information and Celera did not detect either variant found in these two families.
Human RNA isolation.
We isolated RNA from transformed leukocytes of individuals in pedigree 2 using the TRIzol reagent protocol (GibcoBRL) and obtained cDNA by reverse transcription with random priming. We amplified the ARFGEF2 region of exon 3, which contains the identified mutation, by PCR using primers intrinsic to exons 2 and 4.
Mouse in situ hybridization.
We carried out in situ hybridization using previously published methods25. We obtained the probes from PCR products using Arfgef2 expressed-sequence tag (EST) templates from mouse EST clones BF181048 and AI413956. The Arfgef2 primers contained flanking T7 and T3 promoters. Two additional probes, one generated from PCR primer pairs and the second from a PstI and HindIII fragment from the mouse EST clone, yielded similar results. We also examined sense control probes, which gave no specific hybridization (data not shown). Primer sequences are available on request.
Mouse antibody to BIG2.
We generated an antibody against mouse BIG2 in a rabbit using a previously described epitope and confirmed its activity by western-blot analysis3 (Sigma Genosys).
Mouse northern-blot analysis.
We carried out northern-blot analysis of Arfgef2 mRNA from total mouse embryos using a filter purchased from Clontech Laboratories, following the manufacturer's instructions. We used the same probes as in the mouse in situ hybridization. We used Actb mRNA as a loading control.
Migration assay.
We cultured mouse neurospheres, derived from adult mice and clonally propagated, using suggested procedures (Clonetics, Biowhittaker). We grew cells in neurosphere medium (Clonetics bullet kit 200 ml, CC-3209), containing 0.4 ml epidermal growth factor, 0.4 ml fibroblast growth factor, 200 μl leukemia inhibitory factor (Sigma), 4 ml neural survival factor-1 and 0.4 ml gentamicin/amphotericin. We passaged the cells on tissue-culture dishes (Corning) in a 37 °C, 5% CO2 incubator, exposed the mouse neurospheres to varying concentrations of BFA (0.04–4 μM) in neurosphere medium and seeded the neurospheres onto poly-L-lysine-coated slides. After 24 h, we fixed the cultures in 4% paraformaldehyde and stained them with Hoechst 33342. We imaged random fields containing individual neurospheres (n = 3–6 fields per concentration) using an Olympus fluorescence microscope and digital camera and counted the number of Hoechst-stained nuclei in a 150-μm radius of the center of the neurosphere. We used a two-tailed t-test to determine statistical significance of differences between control samples and samples exposed to BFA.
We prepared and analyzed cerebellar granule cell assays using previously described methods26. Briefly, we isolated granule cells from postnatal day (P) 7 mice using a Percoll gradient, placed them in a migration chamber, exposed them to 4 μM BFA and induced them to migrate using brain-derived neurotrophic factor as a chemotactic factor. We assessed migration induced by brain-derived neurotrophic factor by determining the number of cells that had migrated through the transwell membrane 12 h after exposure.
Proliferation assay.
We incubated mouse neurospheres in neurosphere medium with 100 μM BrdU and the indicated concentrations of BFA (similar to those used by Shinotsuka et al.5,6) for 6 h, washed them and incubated them in neurosphere medium with BrdU but no BFA for 18 h before fixing them in 4% paraformaldehyde. We carried out immunocytochemical analysis for BrdU (Boerhinger Mannheim) using previously described protocols27. We imaged random fields containing individual neurospheres (n = 6–8 per concentration) using an Olympus fluorescence microscope with a digital camera. We counted the number of BrdU-positive cells per unit area arising from a single neurosphere using standard imaging software (NIH image version 1.62).
We incubated HEK293 cells with either the mutant BIG2 (E738K) or the wild-type construct for 24 h, then incubated them for 3 h with 100 μM BrdU and counted the number of HA-tagged cells with and without BrdU incorporation after immunocytochemical analysis of HA and BrdU. We evaluated the significance of differences in proliferative rates using a standard χ2 statistical test.
Electron microscopy.
We exposed cultured mouse neurospheres to 4 μM BFA or normal neurosphere medium for 3 h, immersed them in glutaraldehyde fixative and processed them for electron microscopy according to standard techniques28.
Immunocytochemistry.
We cultured MDCK cells using previously described methods29,30. We exposed confluent cultures to 4 μM BFA for the indicated time before fixating them in 4% paraformaldehyde and preparing them for immunofluorescence microscopy. We observed expression of HA (Covance), E-cadherin, β-catenin, atypical PKCλ, EEA-1 (all from BD Transduction Laboratories) or FLNA (Novacastra) using immunofluorescence from conjugated secondary antibodies.
BIG2 transfection.
The BIG2 (E738K) mutant and wild-type BIG2 constructs, subcloned into pcDNA4-HAN with an N-terminal HA tag, were previously described5,6. We transfected MDCK or HEK293 cells, grown in eight-well Biocoat chamber slides (Becton Dickinson), with expression vectors encoding HA-tagged BIG2 (E738K) or wild-type BIG2 (refs. 5,6) using the FuGene6 reagent. After 24 h, the cells were fixed, permeabilized and processed for immunocytochemistry.
Cell death.
We quantified the percentage of cells undergoing cell death using a tetramethylrhodamine (TMR) red in situ cell-death detection kit (Roche) with slight modifications to the manufacturer's suggested methods.
URLs.
The mouse and human genome databases are available at http://www.celera.com, http://genome.ucsc.edu and http://www.ncbi.nlm.nih.gov.
GenBank accession numbers.
ARFGEF2 genomic sequence, NT_011362; ARFGEF2 mRNA, NM_006420; BIG2 protein sequence, NP_006411. Arfgef2 genomic and mRNA sequences and Big2 protein sequences are available in the Celera database.
Note: Supplementary information is available on the Nature Genetics website.
References
Sheen, V.L. et al. Autosomal recessive form of periventricular heterotopia. Neurology 60, 1108–1112 (2003).
Yamaji, R. et al. Identification and localization of two brefeldin A-inhibited guanine nucleotide-exchange proteins for ADP-ribosylation factors in a macromolecular complex. Proc. Natl. Acad. Sci. USA 97, 2567–2572 (2000).
Pacheco-Rodriguez, G., Moss, J. & Vaughan, M. BIG1 and BIG2: brefeldin A-inhibited guanine nucleotide-exchange proteins for ADP-ribosylation factors. Methods Enzymol. 345, 397–404 (2002).
Togawa, A., Morinaga, N., Ogasawara, M., Moss, J. & Vaughan, M. Purification and cloning of a brefeldin A-inhibited guanine nucleotide-exchange protein for ADP-ribosylation factors. J. Biol. Chem. 274, 12308–12315 (1999).
Shinotsuka, C., Waguri, S., Wakasugi, M., Uchiyama, Y. & Nakayama, K. Dominant-negative mutant of BIG2, an ARF-guanine nucleotide exchange factor, specifically affects membrane trafficking from the trans-Golgi network through inhibiting membrane association of AP-1 and GGA coat proteins. Biochem. Biophys. Res. Commun. 294, 254–260 (2002).
Shinotsuka, C., Yoshida, Y., Kawamoto, K., Takatsu, H. & Nakayama, K. Overexpression of an ADP-ribosylation factor-guanine nucleotide exchange factor, BIG2, uncouples brefeldin A-induced adaptor protein-1 coat dissociation and membrane tubulation. J. Biol. Chem. 277, 9468–9473 (2002).
Shevell, D.E. et al. EMB30 is essential for normal cell division, cell expansion, and cell adhesion in Arabidopsis and encodes a protein that has similarity to Sec7. Cell 77, 1051–1062 (1994).
Bonifacino, J.S. & Jackson, C.L. Endosome-specific localization and function of the ARF activator GNOM. Cell 112, 141–142 (2003).
Geldner, N. et al. The Arabidopsis GNOM ARF-GEF mediates endosomal recycling, auxin transport, and auxin-dependent plant growth. Cell 112, 219–230 (2003).
Poon, P.P., Nothwehr, S.F., Singer, R.A. & Johnston, G.C. The Gcs1 and Age2 ArfGAP proteins provide overlapping essential function for transport from the yeast trans-Golgi network. J. Cell Biol. 155, 1239–1250 (2001).
Ruthel, G. & Banker, G. Role of moving growth cone-like “wave” structures in the outgrowth of cultured hippocampal axons and dendrites. J. Neurobiol. 39, 97–106 (1999).
Jareb, M. & Banker, G. Inhibition of axonal growth by brefeldin A in hippocampal neurons in culture. J. Neurosci. 17, 8955–8963 (1997).
Reynolds, B.A. & Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255, 1707–1710 (1992).
Wichterle, H., Garcia-Verdugo, J.M. & Alvarez-Buylla, A. Direct evidence for homotypic, glia-independent neuronal migration. Neuron 18, 779–791 (1997).
Doms, R.W., Russ, G. & Yewdell, J.W. Brefeldin A redistributes resident and itinerant Golgi proteins to the endoplasmic reticulum. J. Cell Biol. 109, 61–72 (1989).
Schroeter, E.H., Kisslinger, J.A. & Kopan, R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 393, 382–386 (1998).
Peng, C.Y., Manning, L., Albertson, R. & Doe, C.Q. The tumour-suppressor genes lgl and dlg regulate basal protein targeting in Drosophila neuroblasts. Nature 408, 596–600 (2000).
Klymkowsky, M.W. & Parr, B. The body language of cells: the intimate connection between cell adhesion and behavior. Cell 83, 5–8 (1995).
Chenn, A. & Walsh, C.A. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297, 365–369 (2002).
Roberts, E. et al. Autosomal recessive primary microcephaly: an analysis of locus heterogeneity and phenotypic variation. J. Med. Genet. 39, 718–721 (2002).
Bond, J. et al. ASPM is a major determinant of cerebral cortical size. Nat. Genet. 32, 316–320 (2002).
Sheen, V.L. & Walsh, C.A. Developmental genetic malformations of the cerebral cortex. Curr. Neurol. Neurosci. Rep. 3, 433–441 (2003).
Fox, J.W. et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron 21, 1315–1325 (1998).
Kruglyak, L., Daly, M.J., Reeve-Daly, M.P. & Lander, E.S. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am. J. Hum. Genet. 58, 1347–1363 (1996).
Berger, U.V. & Hediger, M.A. Differential distribution of the glutamate transporters GLT-1 and GLAST in tanycytes of the third ventricle. J. Comp. Neurol. 433, 101–114 (2001).
Borghesani, P.R. et al. BDNF stimulates migration of cerebellar granule cells. Development 129, 1435–1442 (2002).
Sheen, V.L., Arnold, M.W., Wang, Y. & Macklis, J.D. Neural precursor differentiation following transplantation into neocortex is dependent on intrinsic developmental state and receptor competence. Exp. Neurol. 158, 47–62 (1999).
Sheen, V.L. & Macklis, J.D. Apoptotic mechanisms in targeted neuronal cell death by chromophore-activated photolysis. Exp. Neurol. 130, 67–81 (1994).
Lu, Q. et al. delta-catenin, an adhesive junction-associated protein which promotes cell scattering. J. Cell Biol. 144, 519–532 (1999).
Sheen, V.L. et al. Filamin A and Filamin B are co-expressed within neurons during periods of neuronal migration and can physically interact. Hum. Mol. Genet. 11, 2845–2854 (2002).
Acknowledgements
We thank K. Nakayama and H.-W. Shin for providing the BIG-2 dominant-negative and wild-type constructs. This work was supported by grants to C.A.W. from the US National Institute of Neurological Disorders and Stroke, the March of Dimes and the McKnight Foundation. C.A.W. is an Investigator of the Howard Hughes Medical Institute. V.L.S. is supported by a grant from the US National Institute of Mental Health and is a Charles A. Dana fellow and Clinical Investigators Training Program fellow for the Beth Israel Deaconess Medical Center and the Harvard/Massachusetts Institute of Technology Division of Health Sciences and Technology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Sheen, V., Ganesh, V., Topcu, M. et al. Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat Genet 36, 69–76 (2004). https://doi.org/10.1038/ng1276
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1276
