
Abstract
The shift from outcrossing to selfing is common in flowering plants1,2, but the genomic consequences and the speed at which they emerge remain poorly understood. An excellent model for understanding the evolution of self fertilization is provided by Capsella rubella, which became self compatible <200,000 years ago. We report a C. rubella reference genome sequence and compare RNA expression and polymorphism patterns between C. rubella and its outcrossing progenitor Capsella grandiflora. We found a clear shift in the expression of genes associated with flowering phenotypes, similar to that seen in Arabidopsis, in which self fertilization evolved about 1 million years ago. Comparisons of the two Capsella species showed evidence of rapid genome-wide relaxation of purifying selection in C. rubella without a concomitant change in transposable element abundance. Overall we document that the transition to selfing may be typified by parallel shifts in gene expression, along with a measurable reduction of purifying selection.
Similar content being viewed by others


Main
The switch from obligatory outcrossing to predominant self fertilization in plants is one of the most striking and repeated examples of convergent evolution1,2. Selfing is thought to be favored because of its inherent transmission advantage, as well as the advantage of assured reproduction when mates, pollinators or both are scarce. Selfing should evolve whenever these advantages outweigh the costs associated with inbreeding depression3. In contrast to the immediate benefits of selfing, reduced effective recombination rates, greater population subdivision and more frequent genetic bottlenecks may incur longer-term costs as a result of reductions in effective population size and selective interference among linked sites4, all of which are potential contributors to the high rates of extinction of selfing lineages5. A key problem in understanding the causes and consequences of the evolution of selfing has been partitioning the changes that occurred after the mating system evolution, as many species diverged before the evolution of selfing. As an example, Arabidopsis thaliana probably became selfing only several million years after it was established as a separate species6,7.
A unique opportunity to understand the evolution of selfing is offered by the genus Capsella, which is from the same family as Arabidopsis. The highly selfing species C. rubella, found throughout much of southern and western Europe, separated less than 200,000 years ago from the self-incompatible, obligate outcrosser C. grandiflora, which is restricted primarily to the northwest of Greece8,9. In contrast to Arabidopsis, the breakdown of self incompatibility in Capsella was concurrent with species divergence8,9,10.
We shotgun sequenced the genome of the C. rubella reference line Monte Gargano (Italy) to 22× coverage using a combination of platforms (Online Methods, Supplementary Table 1 and Supplementary Note). For the final assembly of 134.8 Mb, covering all eight chromosomes, we used a genetic map with 768 markers11 (Supplementary Figs. 1 and 2, Supplementary Tables 2,3,4,5 and Supplementary Note), Arabidopsis lyrata synteny (Supplementary Fig. 3) and BAC and fosmid paired-end link support (Supplementary Note and Supplementary Fig. 4). We predicted 28,447 transcripts from 26,521 protein-coding genes and 86 microRNA loci (Table 1 and Online Methods). We also conducted de novo genome assemblies from Illumina libraries for an outbred C. grandiflora accession and the close outgroup species Neslia paniculata (Supplementary Note, Supplementary Fig. 5 and Supplementary Tables 6,7,8,9).
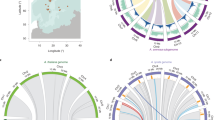
Although the C. rubella genome assembly is ∼40% shorter than the nuclear DNA content estimated from flow cytometry, 219-Mb, k-mer analysis and remapping of Illumina reads indicated that the assembly encompasses most of the euchromatin (Supplementary Note, Supplementary Table 10 and Supplementary Fig. 6). Almost half of the 219-Mb genome seems to be repetitive, including centromeric satellite repeats (Supplementary Fig. 7). Apart from the centromeres, fluorescence in situ hybridization (FISH) identified the 45S ribosomal DNA (rDNA) arrays on chromosomes 2, 3 and 7, the 5S rDNA locus on chromosome 5 and an interstitial telomeric sequence in the pericentromeric region of chromosome 7 as notable genomic locations of repeats (Fig. 1a,b, Supplementary Note and Supplementary Fig. 8).

(a) Comparative genome structure and major chromosome landmarks in C. rubella (CR). The 24 ancestral genomic blocks are indicated by uppercase letters (A–X) and are colored according to their position on the eight chromosomes of the ancestral crucifer karyotype (ACK12). (b) Comparative chromosome painting of CR1 and CR2. Differentially labeled A. thaliana BAC contigs corresponding to the genomic blocks A, B, C, D and E were used as painting probes on the pachytene bivalents of CR1 and CR2. The true fluorescence signals were pseudocolored according to the color code used in a. The arrowheads indicate unpainted pericentromeric heterochromatin. Scale bars, 10 μm. (c,d) Comparative genome mapping of C. rubella with A. lyrata (AL; c) and A. thaliana (AT; d). The outer ring shows the percentage of the genomic window that comprises transposable elements, with a maximum at 60% coverage, the second ring shows gene density, and the inner ring shows orthologous regions between species on the basis of whole-genome alignment and orthologous chaining. Note that the A. lyrata, but not the C. rubella, assembly includes gaps for inferred centromeric heterochromatin. From synteny analyses of the three species, the approximate gene intervals contained within each block include: A/B, AT1G01010–AT1G36980; C, AT1G41830–AT1G56200; D, AT1G56210–AT1G64720; E, AT1G64790–AT1G80950; F, AT3G01070–AT3G25530; G, AT2G04039–AT2G07050; H, AT2G10870–AT2G20900; I, AT2G20920–AT2G26430; J, AT2G26670–AT2G48160; K, AT2G01060–AT2G04038; L, AT3G25545–AT3G32980; M/N, AT3G42170–AT3G63490; O, AT4G00026–AT4G05530; P, AT4G06534–AT4G12590; Q/R, AT5G01010–AT5G30510; S, AT5G32440–AT5G42110; T/U, AT4G12640–AT4G40100; V, AT5G42140–AT5G47760; W/X, AT5G47800–AT5G67640.
Consistent with previous findings12, we found that the large-scale synteny between C. rubella and A. lyrata is almost complete (Fig. 1c and Supplementary Note). Comparisons with Schrenkiella parvula (Supplementary Fig. 9) indicated that all three major differences between C. rubella and A. lyrata are either specific to the A. lyrata genome or errors in the A. lyrata assembly (Supplementary Note). Further comparisons delimited the breakpoints of major rearrangements in A. thaliana (Fig. 1d)12. Overall, we conclude that C. rubella, despite having gone through an extreme genetic bottleneck, retains a largely ancestral genome structure.
To investigate the functional consequences of the mating system change, we compared flower transcriptomes from four C. rubella and four C. grandiflora accessions. Many C. rubella alleles are found in C. grandiflora8,9; thus, DNA sequence variation should not confound RNA sequencing (RNA-seq) comparisons. RNA expression levels were more highly correlated within (average Pearson correlation coefficients, 0.95 for C. grandiflora and 0.94 for C. rubella) than between species (average Pearson correlation coefficient, 0.82; Supplementary Fig. 10). We identified 246 genes that were expressed more strongly in C. rubella relative to C. grandiflora and 373 that were expressed more weakly relative to C. grandiflora, with a minimum fold change of 1.5, false discovery and significance thresholds of 0.5% and a minimum normalized expression of 19 (Supplementary Note). The set was enriched for Gene Ontology terms related to floral development and growth functions, which is consistent with changes in reproductive organ size and development between species (Supplementary Table 11). One-hundred fifty-eight differentially expressed genes colocalized with interspecific quantitative trait loci that are responsible for differences in petal size and pollen number11 (Supplementary Table 12), with 17 found within 2 Mb of petal size quantitative trait loci peaks. Thus, some of the expression changes may be due to cis-regulatory changes that had a role in floral evolution. Pathway analyses (Supplementary Note) identified a reduction in brassinosteroid signaling, which is involved in hormone-triggered pollen maturation13, in C. rubella (Supplementary Table 13).
To investigate whether the C. rubella and C. grandiflora pair is representative of the expression differences in flowers of closely related selfers and outcrossers, we compared the selfer A. thaliana and the predominantly outcrossing A. lyrata. We found that the overlap in expression changes between the two species pairs was much higher than that expected by chance. For example, of 373 genes that were expressed more strongly in C. rubella, 75 orthologs were also expressed more strongly in A. thaliana compared to A. lyrata, whereas only 16 showed higher expression in A. lyrata. In contrast, of 246 genes that were expressed more weakly in C. rubella, 46 orthologs were also expressed more weakly in A. thaliana, whereas only 12 showed lower expression in A. lyrata (Fisher's exact test P < 1 × 10−13; Fig. 2). These results suggest that parallel floral evolution in selfers may be associated with parallel changes in gene expression. A caveat is that some of these changes could reflect the altered abundance of specific tissue types because of changes in flower morphology.

Distribution of fold changes in gene expression in C. grandiflora relative to C. rubella (x axis) and A. lyrata relative to A. thaliana (y axis) at genes showing significant downregulation or significant upregulation in C. rubella.
Population genetic theory predicts that selfers should accumulate slightly deleterious mutations because of a reduced effective population size3. To test this hypothesis, we characterized genome-wide patterns of coding sequence polymorphisms discovered in RNA-seq data from five outbred C. grandiflora (ten haploid chromosomes) and six C. rubella individuals (Supplementary Note and Supplementary Table 14). We identified 48,518 high-quality SNPs in 4,225 genes. The vast majority (81%) segregated only in C. grandiflora. Of the remainder, 7% segregated in both species, 8% segregated only in C. rubella and only 4% were fixed between the two species. On average, diversity at synonymous sites was 0.02 in C. grandiflora, whereas it was sixfold lower (0.003) in C. rubella (Fig. 3a), which is similar to previously documented differences in smaller gene sets10,11,12.

(a) Average pairwise differences (π) at nonsynonymous (nonsyn) and synonymous (syn) sites. Error bars indicate standard errors across all loci. Cr, C. rubella; Cg, C. grandiflora. (b) Ratio of nonsynonymous to synonymous polymorphisms at each derived frequency class using data subsampled to six chromosomes per species. N. paniculata was used as an outgroup to infer derived status. (c) Proportion of synonymous and nonsynonymous polymorphisms unique to each species, as well as shared and fixed differences. Simulated (sim) values are from forward computer simulations using the inferred demographic model and strength of selection on nonsynonymous sites (see main text). Obs, observed.
To test for a change in the efficacy of selection, we examined the ratios of nonsynonymous to synonymous polymorphisms. The ratio was much higher for C. rubella–specific (0.68) than C. grandiflora–specific SNPs (0.35; two-tailed Fisher's exact test P < 0.0001). This was true for all site frequency classes when subsampling the data to equivalent haploid sample sizes (Fig. 3b), suggesting that relaxed selection is not simply due to rare variants in C. rubella having experienced less purifying selection after the recent genetic bottleneck that purged the majority of variation. In contrast, fixed SNPs behaved similarly to C. grandiflora–only SNPs, with a nonsynonymous-to-synonymous ratio of 0.38, whereas SNPs segregating in both species had the lowest ratio (0.22), consistent with these being the oldest on average and thus having experienced the most selection.
There are at least three potential explanations for the elevated nonsynonymous-to-synonymous ratio in C. rubella. The first is experimental error: because diversity is generally lower in C. rubella, SNP errors relative to true polymorphisms may inflate the relative measure of nonsynonymous variation. However, dideoxy sequencing indicated that the false positive rate was less than 2 × 10−3 per SNP, which is much lower than the C. rubella polymorphism density (Supplementary Note). Another explanation could be that the distribution of selection coefficients is altered because the evolution of selfing in C. rubella is associated with changes in morphology, life history, habitat and range. However, extensive population genetic modeling and computer simulations indicated that such a scenario is not required to explain the data (Fig. 3c, Supplementary Note, Supplementary Fig. 11 and Supplementary Tables 15 and 16). The elevated nonsynonymous-to-synonymous ratio in C. rubella could also be due to a reduced efficacy of purifying selection caused by the demographic and selective effects associated with the shift to selfing, as is predicted by theory4. Our results seem to be the most consistent with this third hypothesis. Most previous work has found little or no evidence for relaxed selection on nonsynonymous sites in selfing lineages14,15,16; our use of genome-wide polymorphism data to quantify current selection pressures in a recently derived selfing lineage provides a more powerful test of this hypothesis, suggesting that selfing lineages may in fact experience considerable accumulation of deleterious mutations even over short timescales.
Apart from genome-wide changes in the nonsynonymous-to-synonymous ratio, a potential consequence of the transition to selfing is a change in the abundance and distribution of transposable elements. Not only are self-replicating transposable elements expected to spread more efficiently through the genomes of highly outcrossing species, but self-regulated transposition is also more probable in selfers17,18. In agreement, many transposable element classes have fewer members in the A. thaliana than in the A. lyrata genome19. Despite its nuclear DNA content being in the same range as that of A. lyrata, the C. rubella genome is more similar to that of A. thaliana in several features related to transposable element frequency and density (Fig. 4). First, intergenic distances are more similar in C. rubella and A. thaliana (Fig. 4a). Across all chromosomal blocks, the A. lyrata–to–C. rubella ratios were positive, with a mean of 1.6, whereas the mean for A. thaliana compared to C. rubella was 0.95. Thus, intergenic space has either shrunk in A. thaliana and C. rubella or has expanded in A. lyrata. Similarly, transposable element density is low in the C. rubella assembly (Fig. 4b), particularly in gene-rich regions (Fig. 1), and is more comparable to that in A. thaliana (Fig. 4c). This suggests that genome expansion and contraction have occurred in different regions, with C. rubella having a compact euchromatic region comparable to A. thaliana, whereas A. lyrata has experienced greater recent transposable element activity near genes.

(a) Slope of the physical positions in orthologous blocks between C. rubella and A. thaliana and A. lyrata. Ancestral orthologous blocks are labeled on the x axis (see Fig. 1). (b) Transposable element genomic coverage in the three species. LINE, long interspersed nucleotide repetitive elements; SINE, short interspersed nucleotide repetitive elements. (c) Distribution of the distances between transposable elements and their nearest protein-coding genes. (d) Age distribution of full-length LTR retrotransposons estimated using the rate of substitution between LTRs of individual insertions and assuming a substitution rate of 7 × 10−9 per bp per generation. MYA, million years ago.
Given that the structure of gene-rich regions in the C. rubella genome is similar to that in A. thaliana, we tested whether the shift to selfing was associated with a rapid loss of transposable element abundance, activity or both. The age distribution of long terminal repeat (LTR) retrotransposons in C. rubella seemed to be similar to that in A. thaliana, with no evidence for the high rate of recent transposition seen in A. lyrata (Fig. 4d). Only 5% of the full-length LTR retrotransposons seemed to be younger than 100,000 years, which is close to the estimated speciation time (Supplementary Table 10), suggesting that the vast majority of transposition occurred before the shift to selfing. However, identification of transposable element insertions using paired-end genomic Illumina sequencing revealed no clear evidence for consistent copy number differences between C. rubella and C. grandiflora (Fig. 5a and Supplementary Note). Similarly, although expression comparisons indicate a possible higher variance in transposable element expression in C. rubella, there is no evidence for consistently higher transposable element expression in C. grandiflora (Wilcoxon rank sum test P = 0.4857; Fig. 5b). In addition, transposable element density along the chromosomes is very similar in the two species (Fig. 5c). Given that species divergence is low relative to the coalescent history of C. grandiflora (that is, most of the C. rubella alleles are shared with C. grandiflora), it is probable that longer timescales are required for transposable element copy number to diverge noticeably. Thus, our analyses suggest little evidence for large-scale changes in transposable element abundance, as the evolution of selfing in Capsella occurred about 100,000 years ago, and imply that transposable element activity may be specifically elevated in A. lyrata20.

(a) Numbers of insertion sites identified using read mapping of paired-end Illumina genomic data (using Popoolation TE) in two C. rubella accessions and two C. grandiflora accessions. SINE/LINE, SINE and LINE. (b) Mean and standard error of the proportion of RNA-seq transcripts mapping to transposable elements. (c) Distribution of transposable element insertions along chromosome 1 in two C. rubella accessions and two C. grandiflora accessions.
C. rubella is a young species with an origin that is probably associated with a severe founder event and a shift to a highly selfing mating system. These recent and correlated events have had genome-wide consequences that range from divergence in gene expression for a suite of reproductively related genes to a genome-wide decline in the efficacy of natural selection on amino acid polymorphisms. Moreover, our comparisons among three closely related species, A. thaliana, A. lyrata and C. rubella, highlight the fluidity of large-scale genome structure, typified by differential expansion of centromeric repeats and changes in transposable element activity. The factors driving such contrasting modes of genome expansion and shrinkage are far from resolved, and it will be important to broaden future comparisons to larger phylogenetic scales to better understand the processes driving genome structure evolution.
Methods
Genome assembly and annotation.
Sequencing. Whole-genome shotgun sequencing of C. rubella was conducted using the Monte Gargano (Italy) reference strain. The majority of the sequencing reads were collected with standard Sanger sequencing protocols and Roche 454 XLR and Illumina GAIIx machines at the DoE JGI in Walnut Creek, California (JGI sequencing protocols; see URLs). One linear Roche 454 library (ten runs, 2.78 Gb), three 2.5-kb insert size paired libraries (three runs, 713.1 Mb), two 4-kb insert size paired libraries (four runs, 434.5 Mb), one 8-kb insert size library (three runs, 935.4 Mb) and one 10-kb insert size library (two runs, 515.4 Mb) were sequenced with standard XLR protocols.
Genome assembly and construction of pseudomolecule chromosomes. The sequence reads were assembled using our modified version of Arachne v.20071016 (ref. 21). This produced 1,859 scaffold sequences, with a scaffold L50 value of 6.7 Mb, 71 scaffolds larger than 100 kb and a total genome size of 137.1 Mb. Scaffolds were screened against bacterial proteins, organelle sequences and the GenBank nucleotide repository (nr) and removed if they were found to be contaminants. Additional scaffolds were removed if they (i) consisted of >95% 24-mers that occurred four other times in scaffolds larger than 50 kb, (ii) contained only unanchored RNA sequences or (iii) were less than 1 kb in length.
The combination of 768 markers in an Illumina-based genetic map (Supplementary Note), A. lyrata synteny derived from whole-genome alignments (Online Methods) and BAC and fosmid paired-end link support was used to identify misjoins in the assembly. Scaffolds were broken if they contained syntenic and linkage group discontiguity coincident with an area of low BAC and fosmid coverage. To avoid bias, discontiguity on the basis of synteny alone was not deemed sufficient for breaking scaffolds. A total of 16 breaks were executed, and 45 broken scaffolds were oriented, ordered and joined using 37 joins to form the final assembly containing eight pseudomolecule chromosomes. The final assembly contained 853 scaffolds (9,675 contigs) that covered 134.8 Mb of the genome with a contig L50 value of 134.1 kb and a scaffold L50 value of 15.1 Mb. Except for a single potential misplacement of a small chromosome 7 contig on chromosome 4, the assembly was supported by an independent map with 999 markers within C. rubella, with 194 F2 individuals from the cross of 1408 and Monte Gargano22.
Completeness of the euchromatic portion of the genome assembly was assessed using 1,167 full-length cDNAs from C. rubella along with an 108-bp Illumina EST library from C. grandiflora leaf material (plants grown at the University of Toronto and library construction and sequencing conducted at the Genome Quebec Innovation Centre in Montreal). The aim of this analysis was to obtain a measure of the completeness of the assembly rather than a comprehensive examination of gene space. The screened alignments indicated that 1,166 of 1,167 (99.74%) of the full-length cDNAs aligned to the assembly, and 27,876 of 32,766 (86.29%) of the Illumina ESTs aligned. The ESTs that did not align were checked against the NCBI nr, and a large fraction was found to be prokaryotic rDNA.
Annotation. Protein-coding genes were predicted with a pipeline that combines ESTs, homology and de novo prediction methods23. Three-hundred twenty-nine million 108-bp-long single-ended Illumina reads from ten libraries of C. grandiflora cDNAs were assembled with tophat 1.3.0 and cufflinks 1.0.3, aligned to the Capsella genome and assembled with PASA24 (alignments required 95% identity and 50% length), generating 28,322 EST assemblies with a median length of 1,426 bp. These were aligned to the Capsella genome (requiring 95% sequence identity and 50% coverage of the input sequence) and further assembled with PASA24 to generate 27,399 EST assemblies with a median length of 1,432 bp. Predicted protein sequences from Arabidopsis (v. TAIR10), papaya (ASGPB v0.4 Dec2) and grapevine (Genoscope 12 × 05/10/10) to the softmasked Capsella v1.0 assembly with gapped blastx25 were aligned to generate putative protein-coding gene loci from regions with EST assemblies, protein homology or both, extending to include overlap where necessary. Gene predictions were generated from putative loci with FGenesH+26, exonerate27 (with setting –model protein2genome) and GenomeScan28. The gene prediction at each locus with the highest amount of support from EST assemblies and protein homology was chosen to be improved using evidence from the EST assemblies with a second round of PASA. Gene models with homology to repeats were removed. This produced an annotation at each of 26,521 protein-coding loci, with 1,926 alternative splice forms predicted to produce a total of 28,447 transcripts.
Whole-genome alignment. Two approaches were used to conduct whole-genome alignments and identify orthologous gene positions. First, CoGe29 was used, using the quota align algorithm in Synmap, to identify orthologous gene positions across the genomes of C. rubella, A. thaliana, A. lyrata and S. parvula.
Additionally, to gain more comprehensive synteny information for gene-poor regions, whole-genome orthologous alignments were generated for C. rubella, A. thaliana and A. lyrata. To generate three-species whole-genome alignments, LASTZ30 alignments and orthologous chaining31 were conducted using Capsella as the reference, retaining only a single chain per species per region. Version 0.62-1 of the circos software32 was used to create the circular plots. For comparative mapping in the circos plots, orthologous chains were filtered to a minimum length of 100 kb.
RNA extraction, sequencing and read mapping. Total RNA was harvested from mixed flower buds flash frozen in liquid nitrogen from five C. grandiflora genotypes from Greece and six C. rubella genotypes sampled from different geographic locations (Supplementary Table 2) using the RNAeasy plant mini kit (Qiagen) with minor modifications to obtain the required yield (∼5 μg) for RNA sequencing. After extraction, mRNA isolation, library preparation and paired-end 38-bp read sequencing were conducted on three flow cells of an Illumina Genome Analyzer (GAII) at the Centre for Analysis of Genome Evolution and Function (CAGEF) at the University of Toronto.
To compare our expression patterns in Capsella with those in Arabidopsis, we prepared duplicate RNA from stage-12 floral buds of A. thaliana (Col-0 reference accession) and A. lyrata (accession MN47 (ref. 19)). Barcoded RNA-seq libraries were prepared from each sample with an adaptation of the standard Illumina mRNA-seq method, and single-end sequencing (one flowcell lane worth) was performed to generate 78-bp reads on an Illumina Genome Analyzer (GAII). Methods for plant growth, floral bud collection, RNA extraction, library construction and sequencing for the Arabidopsis samples are as reported in Gan et al.33.
We used TopHat34 to map sequence reads to the reference genome of C. rubella. A. thaliana and A. lyrata RNA sequence reads were mapped to their respective reference genomes using the same parameters described above.
Genomic resequencing and analysis.
Genomic extraction of leaf material was conducted for two C. grandiflora accessions and three C. rubella accessions, as well as one accession of N. paniculata, using a modified CTAB protocol; 108-bp paired-end genomic sequencing was conducted at the Genome Quebec Innovation Centre, and 150-bp sequencing was performed at the Max Planck Institute for Developmental Biology. To infer the ancestral state of segregating polymorphisms in Capsella, we mapped Neslia Illumina reads onto the Capsella reference genome using Stampy35, which is optimized for mapping divergent sequences. Additionally, we conducted 5 kb–insert mate-pair sequencing of one of the C. grandiflora accessions and the N. paniculata accession and produced Illumina-only de novo assemblies as described in the Supplementary Note.
Transposable element annotation.
The TEdenovo pipeline from the REPET v2.0 package36 was used for de novo identification of repeated sequences in the following genomes: A. thaliana (ecotypes Col, Ler, Kro, Bur and C24 from the 1001 Arabidopsis genomes project; see URLs), A. lyrata, A. alpina (courtesy of G. Coupland), Brassica rapa, C. rubella, Eutrema halophila and S. parvula. The selected sequences from all species were combined into the Brassicaceae repeat library, to which we also appended the A. thaliana repeat library from the Repbase database. TEannot from the REPET v2.0 package was run against the C. rubella, A. thaliana and A. lyrata genomes using the Brassicaceae library.
LTR retrotransposons were identified de novo for each genome using LTRharvest from the Genome Tools v1.3.9 package37 with default parameters. MUSCLE v3.8.31 (ref. 38) was used to align LTRs from annotated elements, alignment ends were trimmed at each end to have three consecutive matching nucleotides, and the Kimura two-parameter distance was calculated for each alignment using the EMBOSS v6.4.0 dismat function39. Insertion time was then calculated using the methods described in Hu et al.19.
FISH and comparative chromosome painting.
Cytogenetic analyses were conducted using meiotic chromosomes at the stage of pachytene or diakinesis prepared from young flower buds. See Supplementary Figure 8 for details of rDNA and telomere repeat probes and A. thaliana BAC contigs used as chromosome-specific probes for comparative chromosome painting in C. rubella. All DNA probes were labeled with biotin–deoxyuridine triphosphate (dUTP), digoxigenin-dUTP or Cy3-dUTP by nick translation and hybridized to suitable chromosome spreads. Fluorescence signals were analyzed with an Olympus BX-61 epifluorescence microscope and a CoolCube camera (MetaSystems) and pseudocolored using Adobe Photoshop CS2 software (Adobe Systems). See Supplementary Figure 8 and ref. 40 for a detailed description of all protocols used.
URLs.
JGI sequencing protocols, http://www.jgi.doe.gov/sequencing/protocols/prots_production.html; 1001 Arabidopsis Genomes project, http://www.1001genomes.org/; PHYTOZOME portal, http://www.phytozome.net/capsella.php.
Accession codes.
The assembly and annotation (Entrez BioProject ID PRJNA13878) are available from GenBank (accession number ANNY00000000) and from the JGI PHYTOZOME portal (see URLs). RNA-seq data sets are available from GenBank (GEO SuperSeries GSE45687 and SRA accession PRJNA194469). Seeds from the reference C. rubella strain Monte Gargano are available from the Arabidopsis Biological Resource Center (ABRC) under accession number CS22697 and the Nottingham Arabidopsis Stock Centre (NASC) under accession number N9609.
Accession codes
Primary accessions
BioProject
Gene Expression Omnibus
NCBI Reference Sequence
Sequence Read Archive
References
Barrett, S.C.H. The evolution of plant sexual diversity. Nat. Rev. Genet. 3, 274–284 (2002).
Stebbins, G.L. Self fertilization and population variability in the higher plants. Am. Nat. 91, 337–354 (1957).
Charlesworth, D. & Willis, J.H. The genetics of inbreeding depression. Nat. Rev. Genet. 10, 783–796 (2009).
Charlesworth, D. & Wright, S.I. Breeding systems and genome evolution. Curr. Opin. Genet. Dev. 11, 685–690 (2001).
Lynch, M., Conery, J. & Burger, R. Mutational meltdowns in sexual populations. Evolution 49, 1067–1080 (1995).
Ossowski, S. et al. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327, 92–94 (2010).
Tang, C. et al. The evolution of selfing in Arabidopsis thaliana. Science 317, 1070–1072 (2007).
Guo, Y.L. et al. Recent speciation of Capsella rubella from Capsella grandiflora, associated with loss of self-incompatibility and an extreme bottleneck. Proc. Natl. Acad. Sci. USA 106, 5246–5251 (2009).
Foxe, J.P. et al. Recent speciation associated with the evolution of selfing in Capsella. Proc. Natl. Acad. Sci. USA 106, 5241–5245 (2009).
St Onge, K.R., Kallman, T., Slotte, T., Lascoux, M. & Palme, A.E. Contrasting demographic history and population structure in Capsella rubella and Capsella grandiflora, two closely related species with different mating systems. Mol. Ecol. 20, 3306–3320 (2011).
Slotte, T., Hazzouri, K.M., Stern, D., Andolfatto, P. & Wright, S.I. Genetic architecture and adaptive significance of the selfing syndrome in Capsella. Evolution 66, 1360–1374 (2012).
Schranz, M.E., Lysak, M.A. & Mitchell-Olds, T. The ABC's of comparative genomics in the Brassicaceae: building blocks of crucifer genomes. Trends Plant Sci. 11, 535–542 (2006).
Ye, Q. et al. Brassinosteroids control male fertility by regulating the expression of key genes involved in Arabidopsis anther and pollen development. Proc. Natl. Acad. Sci. USA 107, 6100–6105 (2010).
Escobar, J.S. et al. An integrative test of the dead-end hypothesis of selfing evolution in Triticeae (Poaceae). Evolution 64, 2855–2872 (2010).
Haudry, A. et al. Mating system and recombination affect molecular evolution in four Triticeae species. Genet. Res. (Camb.) 90, 97–109 (2008).
Wright, S.I., Lauga, B. & Charlesworth, D. Rates and patterns of molecular evolution in inbred and outbred Arabidopsis. Mol. Biol. Evol. 19, 1407–1420 (2002).
Wright, S.I. & Schoen, D.J. Transposon dynamics and the breeding system. Genetica 107, 139–148 (1999).
Charlesworth, B. & Langley, C.H. The evolution of self-regulated transposition of transposable elements. Genetics 112, 359–383 (1986).
Hu, T.T. et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 43, 476–481 (2011).
Hollister, J.D. et al. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 108, 2322–2327 (2011).
Jaffe, D.B. et al. Whole-genome sequence assembly for mammalian genomes: Arachne 2. Genome Res. 13, 91–96 (2003).
Guo, Y.L., Todesco, M., Hagmann, J., Das, S. & Weigel, D. Independent FLC mutations as causes of flowering time variation in Arabidopsis thaliana and Capsella rubella. Genetics 192, 729–739 (2012).
Goodstein, D.M. et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186 (2012).
Haas, B.J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Altschul, S.F., Gish, W., Miller, W., Myers, E.W. & Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Solovyev, V., Kosarev, P., Seledsov, I. & Vorobyev, D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 7 (suppl. 1), S10.1–S10.12 (2006).
Slater, G.S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005).
Yeh, R.F., Lim, L.P. & Burge, C.B. Computational inference of homologous gene structures in the human genome. Genome Res. 11, 803–816 (2001).
Lyons, E. & Freeling, M. How to usefully compare homologous plant genes and chromosomes as DNA sequences. Plant J. 53, 661–673 (2008).
Harris, R.S. Improved Pairwise Alignment of Genomic DNA. PhD thesis, Penn. State Univ. (2007).
Kent, W.J., Baertsch, R., Hinrichs, A., Miller, W. & Haussler, D. Evolution's cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl. Acad. Sci. USA 100, 11484–11489 (2003).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Gan, X. et al. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 477, 419–423 (2011).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Lunter, G. & Goodson, M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 21, 936–939 (2011).
Flutre, T., Duprat, E., Feuillet, C. & Quesneville, H. Considering transposable element diversification in de novo annotation approaches. PLoS ONE 6, e16526 (2011).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18 (2008).
Edgar, R.C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 (2000).
Lysak, M.A. & Mandáková, T. Analysis of plant meiotic chromosomes by chromosome painting. Methods Mol. Biol 990, 13–24 (2013).
Acknowledgements
The work conducted by the DoE JGI is supported by the Office of Science of the DoE under contract number DE-AC02-05CH11231. We thank J. Bergelson, J. Borevitz, A. Hall, C. Langley, K. Mayer, J. Nasrallah, B. Neuffer, Y. Van de Peer and O. Savolainen for contributing to the initial sequencing proposal submitted to the Community Sequencing Program at JGI. We also thank T. Bureau, D. Schoen, P. Harrison, J. Stinchcombe, A. Moses and E. Harmsen for their contributions to the Value-directed Evolutionary Genomics Initiative (VEGI) grant (Genome Quebec/Genome Canada), which funded C. grandiflora genomic and mRNA sequencing, and G. Coupland (Max Planck Institute for Plant Breeding Research) and colleagues for information on Arabis alpina repeats. The work was supported by the Max Planck Society (D.W.), the Genome Quebec and Genome Canada VEGI grant (S.I.W. and M.B.), the Natural Sciences and Engineering Research Council of Canada (NSERC) (S.I.W.), the Swedish Research Council (T.S.), the Carl Trygger and Erik Philip-Sörensen foundations (T.S.), National Science Foundation (NSF) grant 0929262 (J. Steffen and R.M.C.), the French National Research Agency (ANR-08-KBBE-012-02 to H.Q.), the Czech Science Foundation (excellence cluster P501/12/G090 to M.A.L.) and the European Regional Development Fund (CZ.1.05/1.1.00/02.0068 to M.A.L.). D.K. was supported by a Human Frontiers in Science Program Long-Term Fellowship, and G.C. was supported by the Alfred P. Sloan Foundation. Population genetics analyses were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) through the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) under project b2012122. L.M.S. was supported by a European Community FP7 Marie Curie Fellowship (PIEF-GA-2008-221553) and an EMBO Long-Term fellowship.
Author information
Authors and Affiliations
Contributions
B.S.G., M.N., J. Schmutz, T.S., D.R., D.W. and S.I.W. conceived and designed the study. Y.-L.G., K.M.H., D.K., J. Steffen and T.S. prepared samples for sequencing. J.J. and J. Schmutz conducted de novo assembly of C. rubella. Y.-L.G., S.R.H., S.P., D.R. and S.S. performed genome annotation. J.G. led the data collection of BAC-end and clone sequencing. P.A., K.M.H., T.T.H., T.S. and S.I.W. conducted genetic mapping analysis. J.A.Å., Y.-L.G., D.K., F.M., H.Q. and W.W. performed the transposon analysis. Y.B., G.C., J.S.E., K.M.H., L.K.N., K.S., T.S. and S.I.W. conducted population genetic analysis. R.M.C., J. Steffen, L.M.S., K.M.H. and A.E.P. conducted the RNA sequence and expression analysis. M.B. and A.E.P. conducted de novo assembly of N. paniculata and whole-genome alignments. J.C. led the de novo assembly of C. grandiflora. A.E.P., S.T. and E.V. conducted comparative genomic analysis. T.M. and M.A.L. conducted FISH and chromosome painting. B.S.G., M.N., T.S., D.W. and S.I.W. wrote the paper with contributions from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–11, Supplementary Tables 1–16 and Supplementary Note (PDF 2506 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article
Cite this article
Slotte, T., Hazzouri, K., Ågren, J. et al. The Capsella rubella genome and the genomic consequences of rapid mating system evolution. Nat Genet 45, 831–835 (2013). https://doi.org/10.1038/ng.2669
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.2669
