
Abstract
Here we present the first diploid genome sequence of an Asian individual. The genome was sequenced to 36-fold average coverage using massively parallel sequencing technology. We aligned the short reads onto the NCBI human reference genome to 99.97% coverage, and guided by the reference genome, we used uniquely mapped reads to assemble a high-quality consensus sequence for 92% of the Asian individual’s genome. We identified approximately 3 million single-nucleotide polymorphisms (SNPs) inside this region, of which 13.6% were not in the dbSNP database. Genotyping analysis showed that SNP identification had high accuracy and consistency, indicating the high sequence quality of this assembly. We also carried out heterozygote phasing and haplotype prediction against HapMap CHB and JPT haplotypes (Chinese and Japanese, respectively), sequence comparison with the two available individual genomes (J. D. Watson and J. C. Venter), and structural variation identification. These variations were considered for their potential biological impact. Our sequence data and analyses demonstrate the potential usefulness of next-generation sequencing technologies for personal genomics.
Similar content being viewed by others

Main
The completion of a highly refined, encyclopaedic human genome sequence1,2 was a major scientific development. Such reference sequences have accelerated human genetic analyses and contributed to advances in biomedical research. Given the growth of information on genetic risk factors, researchers are developing new tools and analyses for deciphering the genetic composition of a single person to refine medical intervention at a level tailored to the individual. The announcements that J. Craig Venter and James D. Watson have had their genomes sequenced3,4, along with the announcement of the Personal Genome Project5, highlight the growth of personal genomics.
Using a massively parallel DNA sequencing method, we have generated the first diploid genome sequence of a Han Chinese individual, a representative of an East Asian population that accounts for nearly 30% of the human population. The consensus sequence of the donor, assembled as pseudo-chromosomes, serves as one of the first sequences available from a non-European population and adds to the small number of publicly available individual genome sequences. This sequence and the analyses herein provide an initial step towards attaining information on population and individual genetic variation, and, given the use and analysis of next-generation sequencing technology, constitute advancement towards the goal of providing personalized medicine.
Data production and short read alignment
The genomic DNA used in this study came from an anonymous male Han Chinese individual who has no known genetic diseases. The donor gave written consent for public release of the genomic data for use in scientific research (see Supplementary Information for consent forms).
We carried out G-banded karyotyping to check the overall structural suitability of this DNA for use as a genomic standard for other genetic comparison and found no obvious chromosomal abnormalities (Supplementary Fig. 1). We then proceeded with whole-genome sequencing of the individual’s DNA (hereafter referred to as YH) using Illumina Genome Analysers (GA; see Methods for details). To minimize the likelihood of systematic biases in genome representation, multiple DNA libraries were prepared and data were generated from eight single-end and two paired-end libraries (Supplementary Table 1). The read lengths averaged 35 base pairs (bp), and the two paired-end libraries had a span size of 135 bp and 440 bp, respectively. We collected a total of 3.3 billion reads of high-quality data: approximately 117.7 gigabases (Gb) of sequence (72 Gb from single-end reads and 45.7 Gb from paired-end reads). The data have been deposited in the EBI/NCBI Short Read Archive (accession number ERA000005). (See Supplementary Information for details concerning the availability of all data.)
Using the Short Oligonucleotide Alignment Program (SOAP)6, 102.9 Gb of sequence (87.4% of all data) was properly aligned to the NCBI human reference genome (build 36.1; hereafter called NCBI36). This resulted in a 36-fold average coverage of NCBI36 (Table 1). The effective genome coverage of the single- and paired-end sequencing was 22.5-fold and 13.5-fold, respectively. In total, 99.97% of NCBI36 (excluding Ns, which are undetermined sequence of the reference genome) was covered by at least one uniquely or repeatedly aligned read (uniquely aligned reads had only one best hit on NBCI36; repeatedly aligned reads had multiple possible alignments; see Methods for details).
About 86.1% (83.6% of single-end and 90.2% of paired-end reads) of the mapped reads could be uniquely aligned and had an average per-nucleotide difference of 1.45% from the NCBI36 sequence. (See Supplementary Information for additional sequence alignment assessment.) We used the alignment of uniquely mapped single-end and paired-end reads to build the consensus YH genome sequence and to detect genetic variations: SNPs, insertions and deletions (indels), and structural variations.
SNP and indel identification
For SNP identification, we estimated the genotype and its accuracy for each nucleotide using Bayesian theory with probabilities based on previous observation of a SNP at that site. Each location was assigned a score value as a measure of SNP call accuracy (see Methods for details).
For SNP detection, we used a series of filtering criteria (see Methods) to remove unreliable portions of the consensus sequence from the analysis. The resulting calculated YH genome consensus sequence covered 92% of the NCBI36 sequence (92.6% of the autosomes; 83.1% of the sex chromosomes), in which we identified 3.07 million SNPs. The remaining 8% of the reference sequence was composed of either repetitive sequence (6.6%) that did not have any uniquely mapped reads or sequence that didn’t pass our filtering steps (1.4%).
For indel identification, we required at least three pairs of reads to define an indel. We only considered paired-end read-gapped alignments that had insertion or deletion sizes of 3 bp or less to avoid creating alignment errors. Confining indel size was necessary to obtain the best detection accuracy given our short-read sequencing strategy. From this analysis, we identified a total of 135,262 indels.
SNP and indel identification accuracy
We assessed our SNP calling accuracy by comparing the identified SNPs in the YH sequence with dbSNP7. We found that 2.26 million (73.5%) of the YH SNPs were present in dbSNP as validated SNPs, and 0.4 million (12.9%) were present as non-validated SNPs. The remaining 0.42 million SNPs were novel (Fig. 1a). Of the 135,262 small indels that we identified, the percentage that overlapped dbSNP indels was much lower than that of the YH SNPs (40.9% compared with 86.4%, respectively). Additionally, most (59.1%) of the indels were novel (Fig. 1b). This isn’t surprising given that dbSNP contains only 13,727 validated and 1,589,264 non-validated 1–3-bp indels.

The dbSNP alleles were separated into validated and non-validated SNPs, and the detected SNPs that were not present in dbSNP were classified as novel.
We also used the Illumina 1M BeadChip for genotyping. The YH consensus sequence covered 99.22% of the genotyped SNPs with a concordance rate at 99.90% (Table 2). We used polymerase chain reaction (PCR) amplification and traditional Sanger sequencing technology on a subset of the inconsistent SNPs and small indels to determine whether they conformed to the genotyping or GA sequencing results (Supplementary Table 2). Of the 50 SNPs examined, 82.0% (41 SNPs) were consistent with the GA sequencing, indicating that the YH genome has a 99.98% accuracy over these genotyped sites (Supplementary Table 3). We also validated 100% of the PCR-amplified YH genome non-coding-region indels and 90% of the frameshift indels (Supplementary Table 4).
Depth effect on genome sequencing
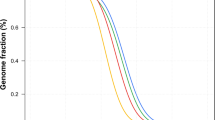
To determine what sequencing depth provides the best genome coverage and lowest SNP-calling error rates for a diploid human genome, we randomly extracted subsets of reads with different average depths from all the mapped reads on chromosome 12, which has a relatively moderate number of repeats. SNPs were identified using GA sequencing and then compared with the genotyping data. We applied the same filtering steps as used in SNP identification (see Methods).
At a depth greater than 10-fold, the assembled consensus covered 83.63% of the reference genome using single-end reads and 95.88% coverage using paired-end reads. Thus, greater sequencing depth provides only a small increase in genome coverage (Fig. 2).

Analyses were carried out on human chromosome 12, and subsets of reads from all mapped 22.5× single-end and 13.5× paired-end reads were randomly extracted from areas of different average depth. The same method and filtering threshold (Q20) was used for SNP detection over different sequencing depths. The error rate for SNP calling—the sum of ‘over call’, ‘under call’ and ‘misses’ rate (see Supplementary Information)—was separated into heterozygotes (HET) and homozygotes (HOM), and was validated against the Illumina 1M genotyping alleles.
The error rate of SNP calling, however, greatly decreases with increased sequencing depth. Additionally, the use of paired-end reads as opposed to single-end reads further reduces SNP calling errors. Of note, SNP calling errors of homozygous and heterozygous SNPs differ significantly.
Individual genome comparison
With the availability of the YH genome sequence, there are now three different individual genome sequences that can be compared. In looking at the SNPs of the three individual genomes, all share 1.2 million SNPs. Each also has a set of SNPs unique to their own genome: for YH, 978,370 (31.8%) SNPs; for Venter, 924,333 (30.1%); and for Watson, 1,096,873 (33.0%) (Supplementary Fig. 2).
The three individuals also have a similar fraction of non-synonymous SNPs (YH, 7,062 (0.23%); Venter, 6,889 (0.22%); Watson, 7,319 (0.20%)). There are 2,622 non-synonymous SNPs shared among the three individuals, accounting for 37.1% of non-synonymous SNPs in the YH genome.
Mutation and selection
To determine which are the ancestral versions of the small indels between the YH genome and the NCBI reference genome, we used the chimpanzee genome as an outgroup and assumed that the alleles on the chimpanzee genome were the ancestral type (Supplementary Table 5). Notably, the YH genome has the ancestral version of 66.2% of the homozygous insertions, whereas the NCBI reference genome contained the ancestral versions of 66.0% of the homozygous deletions. This suggests that during the process of mutation and selection of the human genome, small DNA deletions occur more frequently than do small DNA insertions. Among the heterozygous indels, the allele types that are identical to those in the NCBI reference were mostly comprised of the ancestral versions. This is probably because alleles that are identical between two random individuals are more likely to be the most common type of allele in the population, whereas the fraction of alleles that differ between individuals is likely to be those with a minor allele frequency in the population or genetic drift mutations. The same pattern was also observed with heterozygous indels, indicating that mutations may be biased to DNA loss.
Additional mutation and selection analyses done comparing the YH and NCBI36 genomes are available as Supplementary Information.
Structural variation identification
We used paired-end alignment information to identify structural variations. We identified structural variation boundaries between the YH and NCBI36 genomes by detecting abnormally aligned read pairs that have improper orientation relationships or span sizes (see Methods for details). We identified a total of 2,682 structural variations (Fig. 3a). Because our YH genome sequencing methodology generates paired-end reads with short but very accurate insert sizes, we could identify variations larger than 100 bp, about 6 times the insert size standard deviation. Identified structural variations had a median length of 492 bp, smaller than that of the database of genomic variants (DGV; 30.8 kb)8. This indicates that our methods were biased towards the detection of small structural variation events, but also indicates that it has an acceptable resolution as compared to current structural variation analyses9,10,11.

a, Abundance of each class of structural variation. The overlap with known structural variations in the DGV (http://projects.tcag.ca/variation/) and with transposons (transposable elements, TEs) was calculated. About 34% of our identified structural variations are novel (having less than 10% of a portion of the YH structural variations overlapping with structural variations in the DGV). Transposable elements are a major component of the identified deletions, with Alus and LINEs involved in 49% and 34% of the deletions, respectively. b, An example of a deletion of a transposon complex on YH chromosome 1. The sequencing depth by both single-end and paired-end reads are shown. Normally aligned paired-end reads are shown in green, whereas abnormally aligned paired-end reads, which have unexpected long insert sizes or an incorrect orientation relationship, are shown in red. c, An example of an inversion on YH chromosome 19. Local assembly showed that a 102,405-bp fragment was inverted and reinserted in the genome. There are three genes in this sequence fragment, and the last exon of gene CYP4F12 was destroyed by this inversion event.
Using paired-end methods, we identified more deletion (2,441) than duplication (33) events. Greater detection of deletions may be because they are identified by observing unexpectedly long insert sizes in paired-end clusters, whereas detection of insertions longer than our paired-end library span size will probably be missed.
We searched for candidate regions where larger insertions might have occurred by adopting a method based on the ratio of single-end to paired-end read depth and found 4,819 regions with a ratio significantly higher (P < 0.001) than the average ratio over the whole genome. Our data indicated that 4,377 (90.8%) of these candidate regions were likely to have insertions of repetitive elements, such as mammalian interspersed repeats (MIR; 2,067) and Alu elements (692) in the short interspersed nuclear elements (SINE) category, or L1 elements (1,601) in the long interspersed nuclear elements (LINE) category (see Methods for details).
Recent studies10,11 have shown that novel sequences (those not anchored to the NCBI reference genome) are a considerable source of structural variations. To search for sequences unique to the YH genome, we analysed 487 million unmapped short reads. Among these, 0.39% could be aligned on unanchored scaffolds of NCBI36, 1.09% on novel small contigs of the Venter genome, and 0.67% on novel sequences identified by ref. 10. Using the de novo assembler Velvet12, we could assemble only 1,731,355 (0.36%) reads into 20,949 contigs with lengths >100 bp. In total, 10,398 (49.6%) of these contigs aligned well with unplaced human clones in GenBank. Of the remaining short contigs, 961 (4.6%) aligned with chimpanzee and mouse genomes at greater than 90% identity. These may represent deletions present in populations of European descent or be regions missed in the assembly of both NCBI36 and the Venter genome.
Because most structural variations occur in transposable elements or repetitive sequences, they are unlikely to have any major impact on function. (See Fig. 3b for an example of a deletion of a transposable element complex.) In the YH genome, we did find structural variations that resulted in the complete or partial deletion of 33 genes, and 30.3% of these are homozygous deletions, increasing their likelihood of affecting gene function (Supplementary Table 6). An example of a gene disruption event is in the CYP4F12 gene on YH chromosome 19, where an inversion has broken the gene into two segments (Fig. 3c). We used PCR amplification and sequencing to validate the inversion breakpoints. This gene also had non-synonymous mutations in its obsolete exons, indicating that it may have been under neutral selection.
Haplotype analysis
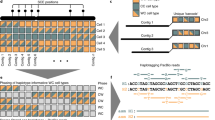
We used PHASE13 and the available phased genotypes of the HapMap CHB/JPT population to predict the YH genome haplotypes. The 700,320 YH genome heterozygotes that overlapped with HapMap loci were used to construct 4,399 haplotype blocks that averaged 587 kb in size (Fig. 4). Of these heterozygous SNPs, 3,039 (0.43%) showed an inconsistent phase in the two adjacent fragments, which may potentially break the haplotype blocks. Additional potential haplotype breakpoints were 1,021,953 heterozygous YH genome SNPs absent in the HapMap. We evaluated this by checking paired-end reads that simultaneously covered two of the heterozygotes used in phasing. A total of 43,902 heterozygous SNP pairs were covered by read pairs, among which 97.37% (42,746 pairs) were in agreement with haplotypes as the corresponding covered read pairs. In total, the 2,434 haplotypes that had sizes greater than 200 kb covered 2.38 Gb of the genome.

Haplotypes were constructed using PHASE software with the 700,300 autosomal heterozygous SNPs that overlapped with the CHB/JPT genotypes from the HapMap phase II data.
Genetic ancestry
To estimate the ancestral composition of the YH individual’s genome, we did a cluster analysis using an evenly sampled 87,614 loci with known alleles in all 270 HapMap individuals (Supplementary Fig. 3). The YH individual was estimated to share alleles14 (thus ancestry) at 94.12% with the Asian, 4.12% with the European and 1.76% with the African populations. Collection of more data from all representative worldwide populations and development of analytical models to provide better estimates of time since admixture will improve the ability to assess an individual’s personal genetic history.
Effective population size, Ne, is the number of breeding individuals in an idealized population that would show the same amount of allele frequency dispersion under random genetic drift or the same amount of inbreeding as the population under consideration15. Assuming an infinite-site model of neutral mutations and equilibrium of mutation and drift, and adopting the mutation rate used by ref. 16 with 2.63 × 10-8 per site per generation, we estimated that the effective Chinese population size is about 5,700. The same analysis based on the population mutation parameters of the YH, Watson, Venter, and NCBI36 genomes gives an estimate of 3,300 for the effective human population size, which is closer to the estimation based on HapMap data17, but lower than the estimated 10,000–15,000 ancestral population size.
Known phenotypic or disease risk variant screen
The primary goal of personal genome sequencing is to allow identification of disease risk genotypes. We surveyed 1,495 alleles of 116 genes in the YH genome in the Online Mendelian Inheritance in Man (OMIM)18 database and found one mutation in the GJB2 gene, which is associated with a recessive deafness disorder. This allele was heterozygous, thus there was no expectation of, or evidence for, deafness in this individual, but it does raise the possibility of offspring having this disorder.
A preliminary search of genes and variants associated with common, complex phenotypes or disorders using OMIM data (Table 3) identified several genotypes that confer risk for tobacco addiction and Alzheimer’s disease. This donor is a heavy smoker, as is consistent with individuals of similar genotypes in tobacco addiction studies. The donor contains 9 (56.3%) of the 16 identified Alzheimer’s disease risk alleles3, including two APOE alleles19 and 7 SORL1 alleles20. These findings indicate an increased risk for Alzheimer’s disease, but there are no available data from any family members to assess whether there is a family history of Alzheimer’s disease.
Discussion
Here we present the first genome sequence of an Asian individual. This sequence, which was accomplished using next-generation short-read sequencing technology, is one of the first genome sequences from a single individual (the genome sequences of J. D. Watson and J. C. Venter were accomplished using 454 and Sanger sequencing technology, respectively).
Our analysis of the YH genome, including consensus assembly, assessment of genome coverage, variation detection and validation, demonstrated the ability of this technology for sequencing large eukaryotic genomes given the availability of a reference genome. This sequencing method also resulted in sequence redundancy reaching an average 36-fold; significantly deeper than the ∼7-fold coverage of the Watson and Venter genomes. Thus, the YH consensus sequence accuracy is higher and is especially suitable for calling heterozygous alleles.
Next-generation sequencing technologies have a very high throughput, as a hundred million DNA fragments can be sequenced in parallel on the chip. The Illumina GA sequencing used in this study can provide up to 4–8 Gb high-quality data per week. In this regard, the time needed to decipher a human genome (1–2 months using five next-generation sequencers), as well as the cost of sequencing (less than half a million US dollars), are substantially reduced.
The use of paired-end sequencing for structural variation detection allowed the identification of small but accurate insert sizes, making the attainable resolution excellent for deletion and small insertion identification, but limited for detection of insertions longer than the paired-end insert sizes. Using a combination of both short and long insert sizes in the future will enable the identification of a larger variety of structural variations.
We were also able to phase a large number of heterozygous SNPs that overlapped with sites of inferred haplotypes of the Asian population from the HapMap data. However, to phase all the heterozygous SNPs of the assembled diploid genome with two sites covered by two reads belonging to a pair, we require different sized, long paired-end sequences. Improvement in haplotype prediction and heterozygote phasing will require genome sequences from many individuals in a population.
Adding to such advances, a recently formed international collaborative project, called the 1,000 Genome Project, aims to catalogue a detailed set of human genetic variations, which will serve as a multiple-genome-sequence blueprint for building genetic maps and extend our knowledge on genetic difference between individuals and between different ethnic populations. Ultimately, we predict an increase in the number of people who will be able to afford having their own genomes sequenced. Personal genome sequencing may eventually become an essential tool for diagnosis, prevention and therapy of human diseases.
Methods Summary
Library preparation followed the manufacturer’s instructions (Illumina). Cluster generation was performed using the Illumina cluster station and the workflow was as follows: template hybridization, isothermal amplification, linearization, blocking, denaturation and sequencing primer hybridization. The fluorescent images were processed to sequences using the Illumina base-calling pipeline (SolexaPipeline-0.2.2.6). The human reference genome, together with the annotation of genes and repeats, were downloaded from the UCSC database (http://genome.ucsc.edu/), in line with NCBI build 36.1. dbSNP v128 and HapMap release 23 were used. The SNP set of the Venter genome was downloaded from the public FTP of JCVI, and the SNP set of the Watson genome was provided by Baylor College of Medicine.
We used SOAP to align all short reads onto the human reference genome (NCBI 36), and we used a statistical model based on Bayesian theory and the Illumina quality system to calculate the probability of each possible genotype at every position from the alignment of short reads on the NCBI reference genome. The genotype of each position was assigned as the allele types that had the highest probability. The final consensus probabilities were transformed to quality scores in Phred scale. We grouped abnormally mapped paired-end reads with coordinate distances smaller than the maximum insert size on both ends into diagnostic paired-end (PE) clusters. To avoid misalignment, PE clusters with <4 pairs were discarded. Common structural variations such as deletions, translocations, duplications, inversions and so on were examined and summarized into alignment models. The reads were assembled locally to verify the specific coordinate of structural variation elements.
Online Methods
DNA library construction and sequencing
Genomic DNA was extracted from peripheral venous blood, and the blood sample was collected using the guidelines dictated by the institutional review board of the Beijing Genomics Institute (BGI).
Library preparation followed the manufacturer’s instructions (Illumina). Briefly, 2–5 μg of genomic DNA in 50 μl TE buffer were fragmented by nebulization with compressed nitrogen gas at 32 p.s.i. for 9 min. Nebulization generated double-stranded DNA fragments with blunt ends or with 3′ or 5′ overhangs. The overhangs were converted to blunt ends using T4 DNA polymerase and Klenow polymerase, after which an ‘A’ base was added to the ends of double-stranded DNA using Klenow exo- (3′ to 5′ exo minus). Next, DNA adaptors (Illumina) with a single ‘T’ base overhang at the 3′ end were ligated to the above products. These products were then separated on a 2% agarose gel, excised from the gel at a position between 150 and 250 bp, and purified (Qiagen Gel Extraction Kit). The adaptor-modified DNA fragments were enriched by PCR with PCR primers 1.1 and 2.1 (Illumina). Separate 8-, 10-, 12-, 15- and 18-cycle reactions were used for sequencing. The concentration of the libraries was measured by absorbance at 260 nm.
The template DNA fragments of the constructed libraries were hybridized to the surface of flow cells and amplified to form clusters. After double-stranded DNA was denatured to single-stranded DNA and nonspecific sites were blocked, genomic DNA sequencing primers were hybridized for DNA sequencing initiation. In brief, cluster generation was performed on the Illumina cluster station, and the basic workflow (based on the standard Illumina protocol) was as follows: template hybridization, isothermal amplification, linearization, blocking and denaturisation, and hybridization of the sequencing primers. The fluorescent images were converted to sequence using the Illumina base-calling pipeline (SolexaPipeline-0.2.2.6).
Public data used
The human reference genome, together with genes and repeats annotation, was downloaded from the UCSC database (http://genome.ucsc.edu/), which has the same sequence as the NCBI build 36.1. The NCBI reference genes with prefix ‘NM’ were mapped to the reference genome using BLAT by UCSC. Hits with >90% identity were retained for further analysis, and only one transcript was retained for each gene. dbSNP v128 and HapMap release 23 were used. The SNP set from the Venter genome was downloaded from the public FTP site of JCVI (ftp://ftp.jcvi.org/pub/data/huref/), and the SNP set of the Watson genome was provided by Baylor College of Medicine.
Short reads alignment
We used SOAP to align each read or read-pair to a position on a chromosome of the NCBI36 human reference genome that had least number of nucleotide differences between the read and the reference genome, and called this a ‘best hit’. If a read had only a single best hit, it was considered uniquely aligned. Reads that had more than one ‘best hit’ (meaning they could be aligned to multiple positions that each had the same number of mismatches) were considered repeatedly aligned. For repeatedly aligned reads a random position was chosen from all of its best hits for placement on the reference genome for sequencing depth calculation.
In the specific alignment process, at most two mismatches were allowed between a read and the reference, and best hits were selected. Because errors can accumulate during sequencing, the quality of the last several base pairs at the end of reads can be relatively low. We thus set option –c 52 during our alignment procedure. Thus, if a read could not be aligned, we discarded the first base, and iteratively trimmed 2 bp at the 3′ end until the read could be aligned or the remaining sequence was shorter than 27 bp. For paired-end reads, two reads belonging to a pair were aligned with both being in the correct orientation and proper span size on the reference genome. If a pair could not be aligned without gaps but allowing at most two mismatches on each read, a gapped alignment was then performed with a maximum gap size of 3 bp. If the two reads could not be aligned as a pair, they were aligned independently.
Consensus assembly
We used a statistical model based on Bayesian theory and the Illumina quality system to calculate the probability of each possible genotype at every position from the alignment of short reads on the NCBI reference genome. A calibration matrix was built based on all uniquely mapped reads to estimate the probability for a given genotype T to have an observed base X located at a position k of its original read with quality score S. For a variety of reasons, similar sequencing errors are often repeated, thus, the ith occurrence of base X covering a particular position would contribute less to denote an X in consensus by an adjustment formula. In brief, likelihood P(X|T) is a function of (k, S, i, X, T), not simply of F(S). The total likelihood of all observed bases (O) covering a site P(O|T) is the product of each one.
From observed prior probability, the SNP rate is expected to be about 0.1%, and the most common SNPs should already be present in dbSNP. Therefore, for positions without known polymorphisms, on one haploid, the reference bases will dominate the prior probability as 0.999; others will share the remaining 0.1% mutation rate. Because sequencing errors would look like heterozygous (HET) SNPs, a penalty factor of 0.001 is multiplied to the HET prior probability. At dbSNP sites, bases already observed dominate the prior probability equally and the HET penalty factor is 0.01. As a result, the prior probabilities were as follows: (1) 0.45 for a homozygote and 0.1 for a heterozygote at a SNP site that has been validated in dbSNP; (2) 0.495 for a homozygote and 0.01 for a heterozygote at a SNP site that has not been validated in dbSNP; and (3) 1 × 10-6 for a homozygote and 2 × 10-6 for a heterozygote at a potentially novel SNP site (one that is absent in dbSNP).
Using the information above, we calculated the posterior probability of each genotype using a Bayesian formula. The genotype of each position was assigned as the allele type that had the highest probability. A rank sum test was applied to adjust for the probability of heterozygotes. The final consensus probabilities were transformed to quality scores in Phred scale.
Calling SNPs
We used six steps to filter out unreliable portions of the consensus sequence: (1) we used a Q20 quality cutoff; (2) we required at least four reads; (3) the overall depth, including randomly placed repetitive hits, had to be less than 100; (4) the approximate copy number of flanking sequences had to be less than 2 (this was done to avoid misreading SNPs as heterozygotes caused by the alignment of similar reads from repeat units or by copy number variations (CNVs)); (5) there had to be at least one paired-end read; and (6) the SNPs had to be at least 5 bp away from each other. For chromosome X and Y, condition (2) was altered by requiring only two unique reads with at least 1 paired-end (PE) read. In the SOAP algorithm, a gap-free alignment is done first and then a gapped alignment. Thus, we required condition (6) because most of the discrepancies between the YH genome and the NCBI reference genome that are too close to each other are due to mismatches across indels. After filtering, we were confident in the calculated YH consensus sequence, and discrepancies between the YH genome and NCBI reference genome were called as SNPs.
Identification of short indels
As the number of SNPs is roughly one order of magnitude larger than that of indels, in the first stage of alignment we did not allow any gaps. Thus, some read pairs containing real indels could not be mapped when PE requirements were satisfied. After the first alignment stage, we mapped the unmapped read pairs by allowing up to 3-bp indels to enable them to meet PE requirements. This limited the indels that could be detected in our study to gaps of 1–3 bp in length. If different read pairs provided the same outer coordinates in mapping, they are likely to be duplicated products of a single fragment during PCR. We merged these redundant pairs before looking for indels. Gaps that were supported by at least three non-redundant paired-end reads were extracted. If the number of ungapped reads that crossed a possible indel was no more than twice that of gapped reads, then an indel was called. In chromosome X and Y, we required all indel sites to be covered by only gapped reads because valid indels on sex chromosomes are expected to be homozygous.
Accession codes
Primary accessions
GenBank/EMBL/DDBJ
Data deposits
The data have been deposited in the EBI/NCBI short read archive (accession number ERA000005). These data, together with all the associated analyses, are freely availably at http://yh.genomics.org.cn. SNPs and indels have been submitted to NCBI dbSNP and will be available in dbSNP version 130.
References
International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001)
Venter, J. C. et al. The sequence of the human genome. Science 291, 1304–1351 (2001)
Levy, S. et al. The diploid genome sequence of an individual human. PLoS Biol. 5, e254 (2007)
Wheeler, D. A. et al. The complete genome of an individual by massively parallel DNA sequencing. Nature 452, 872–876 (2008)
Church, G. M. The personal genome project. Mol. Syst. Biol. 1 doi: 10.1038/msb4100040 (2005)
Li, R., Li, Y., Kristiansen, K. & Wang, J. SOAP: short oligonucleotide alignment program. Bioinformatics 24, 713–714 (2008)
Sherry, S. T. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 (2001)
Iafrate, A. J. et al. Detection of large-scale variation in the human genome. Nature Genet. 36, 949–951 (2004)
Korbel, J. O. et al. Paired-end mapping reveals extensive structural variation in the human genome. Science 318, 420–426 (2007)
Kidd, J. M. et al. Mapping and sequencing of structural variation from eight human genomes. Nature 453, 56–64 (2008)
Bovee, D. et al. Closing gaps in the human genome with fosmid resources generated from multiple individuals. Nature Genet. 40, 96–101 (2008)
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008)
Stephens, M., Smith, N. J. & Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 68, 978–989 (2001)
Tang, H., Peng, J., Wang, P. & Risch, N. J. Estimation of individual admixture: analytical and study design considerations. Genet. Epidemiol. 28, 289–301 (2005)
Wright, S. Evolution in Mendelian populations. Genetics 16, 97–159 (1931)
Noonan, J. P. et al. Sequencing and analysis of Neanderthal genomic DNA. Science 314, 1113–1118 (2006)
Tenesa, A. et al. Recent human effective population size estimated from linkage disequilibrium. Genome Res. 17, 520–526 (2007)
McKusick, V. A. Mendelian Inheritance in Man and its online version, OMIM. Am. J. Hum. Genet. 80, 588–604 (2007)
Coon, K. D. et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J. Clin. Psychiatry 68, 613–618 (2007)
Rogaeva, E. et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nature Genet. 39, 168–177 (2007)
Acknowledgements
We are indebted to the faculty and staff of Beijing Genomics Institute at Shenzhen, whose names were not included in the author list, but who contributed to this work. This project is funded by the Shenzhen Municipal Government and the Yantian District local government of Shenzhen, and Shenzhen University assisted in this work. This study is also supported by the National Natural Science Foundation of China (30890032, 30725008, 90403130, 90608010, 30221004, 90612019, 30392130), the Ministry of Science and Technology of China (973 program: 2007CB815701, 2007CB815703, 2007CB815705; 863 program: 2006AA02Z334, 2006AA10A121, 2006AA02Z177), the Chinese Academy of Sciences (GJHZ0701-6, KSCX2-YWN-023), the Beijing Municipal Science and Technology Commission (D07030200740000), the Danish Platform for Integrative Biology, the Ole Rømer grant from the Danish Natural Science Research Council, a pig bioinformatics grant from Danish Research Council and the Solexa project (272-07-0196), and the Lundbeck Foundation Centre of Applied Medical Genomics for Personalized Disease Prediction, Prevention and Care (LUCAMP). We thank Illumina Inc. for their assistance in setting up the Illumina Genome Analyzer platform and providing technology support. We appreciate the help of R. Gibbs and D. Wheeler with the SNP data set of the genome of J. D. Watson, and S. Levy’s help for providing HuRef novel sequences. LUDAOPEI Hospital provided karyotyping of the DNA sample.
Author Contributions Ju.W., W.W., R.L. and Yi.L. contributed equally to this work. Ju.W., H.Y. and Ji.W. managed the project. W.W., G.T., Jun.Z., Jua.Z., Ya.L., Hu.L., Yi.Z., Zhenz.Y., X.Y., B.Y., W.L., Da.L., Q.H., Yu.L., S.Y., F.C., L.L., K.Z., Y.R., L.Y., Y.G., G.Y., Zhu.L., Xiaol.F., K.K., L.B. and X.Z. performed sequencing. Ju.W., R.L. and Yi.L. designed the analyses. R.L., Yi.L., W.F., J.L., Y.G., B.F., He.L., Xiaod.F., Z.D., Dong Li, Y.H., H.Z., I.H., M.I., J.P., Jin.Z., J.D., Ya.Z., J.Q., L.M., G.L., Zhent.Y., G.Z., C.Y., F.L, S.L, P.N., J.R., Q.L., Hongm.Z., Dongy.L, Zhi.L., N.L., G.G., Jia.Z., J.Y., L.F., Q.C., Y.S., A S., C.P., Hongk.Z., G.W., R.N., R.D. and S.L. performed the data analyses. Ju.W., R.L., Yi.L. and L.G. wrote the paper.
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Information
This file contains Supplementary Figures S1-S8, with Legends, Supplementary Tables S1, S2, S5 and S7 (See separate files for Supplementary Tables S3, S4, S6 and S8-S11, Supplementary Notes and Consent Forms to participate and donate to The Yanhuang Project-Phase 1. (PDF 725 kb)
Supplementary Table S3
Supplementary Table S3 shows a full list of the PCR validation of the SNP sites which are inconsistent between the assembled consensus and genotyping. (XLS 27 kb)
Supplementary Table S4
Supplementary Table S4 shows a full list of the PCR validation of the indel sites. (XLS 25 kb)
Supplementary Table S6
Supplementary Table S6 shows a list of genes overlapped with SVs. (XLS 87 kb)
Supplementary Table S8
Supplementary Table S8 shows a full list of genes containing non-synonymous SNPs. (XLS 681 kb)
Supplementary Table S9
Supplementary Table S9 shows a full list of genes containing frameshift indels. (XLS 41 kb)
Supplementary Table S10
Supplementary Table S10 shows a full list of genes involved in predicted selective sweeps. (XLS 145 kb)
Supplementary Table S11
Supplementary Table S11 shows a list of HGMD alleles which are positive in YH genome. (XLS 74 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Wang, J., Wang, W., Li, R. et al. The diploid genome sequence of an Asian individual. Nature 456, 60–65 (2008). https://doi.org/10.1038/nature07484
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature07484
This article is cited by
-
A review of the pangenome: how it affects our understanding of genomic variation, selection and breeding in domestic animals?
Journal of Animal Science and Biotechnology (2023)
-
The chemistry of next-generation sequencing
Nature Biotechnology (2023)
-
A biallelic multiple nucleotide length polymorphism explains functional causality at 5p15.33 prostate cancer risk locus
Nature Communications (2023)
-
SparkGC: Spark based genome compression for large collections of genomes
BMC Bioinformatics (2022)
-
Characterizing sensitivity and coverage of clinical WGS as a diagnostic test for genetic disorders
BMC Medical Genomics (2021)
