
Abstract
Toll-like receptors (TLRs) are activated by pathogen-associated molecular patterns to induce innate immune responses and production of pro-inflammatory cytokines, interferons and anti-inflammatory cytokines1. TLRs activate downstream effectors through adaptors that contain Toll/interleukin-1 receptor (TIR) domains2, but the mechanisms accounting for diversification of TLR effector functions are unclear. To dissect biochemically TLR signalling, we established a system for isolating signalling complexes assembled by dimerized adaptors. Using MyD88 as a prototypical adaptor, we identified TNF receptor-associated factor 3 (TRAF3) as a new component of TIR signalling complexes that is recruited along with TRAF6. Using myeloid cells from TRAF3- and TRAF6-deficient mice, we show that TRAF3 is essential for the induction of type I interferons (IFN) and the anti-inflammatory cytokine interleukin-10 (IL-10), but is dispensable for expression of pro-inflammatory cytokines. In fact, TRAF3-deficient cells overproduce pro-inflammatory cytokines owing to defective IL-10 production. Despite their structural similarity, the functions of TRAF3 and TRAF6 are largely distinct. TRAF3 is also recruited to the adaptor TRIF (Toll/IL-1 receptor domain-containing adaptor-inducing IFN-β) and is required for marshalling the protein kinase TBK1 (also called NAK) into TIR signalling complexes, thereby explaining its unique role in activation of the IFN response.
Similar content being viewed by others

Main
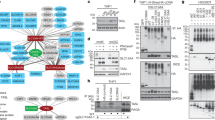
TLRs function through four TIR domain adaptors that may act in pairs: MyD88 and TIRAP/MAL, and TRIF/TICAM-1 and TRAM1. Whereas TLR4 uses all four, TLR9 signals via MyD88, and TLR3 mainly via TRIF3,4,5. Gene targeting suggests that MyD88-dependent signalling requires TRAF6 (ref. 6), but how different TIR adaptors achieve diversification of effector functions is unclear. To answer this question we used a biochemical approach. To bypass receptor-induced dimerization, we fused MyD88 or the TRAF6 effector domain to subunit B of Escherichia coli DNA gyrase (GyrB; Fig. 1a and Supplementary Fig. 1a), which dimerizes upon binding coumermycin, a bivalent antibiotic7. Coumermycin treatment of cells expressing these constructs led to the activation of p38 mitogen-activated protein kinases (MAPK) (Fig. 1b), ERK1/2, JNK1/2 and IκB kinases (IKK) (Supplementary Fig. 1 and data not shown). Activation-dependent degradation of IRAK1 was observed upon dimerization of MyD88–GyrB but not TRAF6–GyrB (Supplementary Fig. 1b), consistent with IRAK1 acting between MyD88 and TRAF6 (ref. 1). Dimerization of either MyD88–GyrB or TRAF6–GyrB induced TNF-α as efficiently as the TLR9 agonist CpG-DNA3 (Supplementary Fig. 1c). Hence, dimerization of adaptor proteins mimics TLR signalling.

a, Constructs used. DD, death domain; TIR, TIR domain; GyrB, subunit B of E. coli DNA gyrase; L, IgG hinge region linker; TAP, TAP tag; F, Flag tag. b, MyD88–GyrB-expressing RAW264.7 cells were stimulated with coumermycin (CM) or CpG-DNA, and p38 phosphorylation was analysed by immunoblotting. c, Flag–MyD88–GyrB-expressing RAW264.7 cells were stimulated with coumermycin. Lysates were prepared, immunoprecipitated with anti-Flag antibodies and analysed by immunoblotting with antibodies to TRAF6 or MyD88.
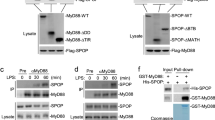
After dimerization, MyD88–GyrB efficiently recruited TRAF6 (Fig. 1c). To isolate other MyD88-interacting proteins we established RAW cells expressing MyD88–GyrB fusion proteins containing an additional TAP (tandem affinity purification) tag (Fig. 1a)8. These cells and green fluorescent protein (GFP)–TAP-expressing control cells were stimulated with coumermycin for 6 min and lysates were prepared. Protein complexes were isolated by TAP, resolved by SDS–polyacrylamide gel electrophoresis (PAGE) and stained (Fig. 2a). Bands 1 and 2 are the bait proteins. Band 4 was constitutively associated with MyD88, whereas other, barely visible bands, including band 3, were dimerization-dependent. The identity of these bands was queried by mass spectrometry (MS)9. Band 4 contained IRAK4, an essential component of all TIR signalling complexes10. In band 3, TRAF6 was identified with 12 peptides. Surprisingly, band 3 also contained four TRAF3-derived specific peptides (Supplementary Fig. 2a). TRAF3 recruitment to MyD88–GyrB and interleukin-1 receptor (IL-1R) was confirmed by immunoprecipitation and was comparable to TRAF6 recruitment (Fig. 2b and Supplementary Fig. 2b). Moreover, activation of MyD88–GyrB transfectants with CpG-DNA or Pam3Cys (TLR2)11 induced association of endogenous TRAF3 with MyD88–GyrB (Fig. 2c and Supplementary Fig. 2c). Notably, lipopolysaccharide (LPS) did not induce detectable interaction between MyD88 and TRAF3 (Fig. 2c), but both LPS and poly(I:C) (synthetic double-stranded RNA) induced association of TRAF3 with TRIF, suggesting that TRAF3 is also involved in the TRIF-dependent pathway (Fig. 2d). The kinetics of CpG-DNA-induced association of TRAF3 with MyD88 paralleled those of TBK1, but not IKK, activation (Fig. 2e). Together, the data show that TRAF3 is part of MyD88- and TRIF-dependent signalling complexes.

a, RAW264.7 cells expressing GFP–TAP or Flag–MyD88–GyrB–TAP were stimulated for 6 min with coumermycin as indicated. Cell lysates were prepared, TAP-purified, gel-separated and stained with Coomassie blue. Part of the gel is enlarged (right side). b, Flag–MyD88–GyrB stable transfectants were stimulated with coumermycin, and lysates were prepared and analysed directly or after immunoprecipitation (IP) with anti-Flag antibodies by immunoblotting (IB) as indicated. MyD88–GyrB was detected by TAP/protein A-dependent binding of antibodies used for immunoblotting. c, d, Flag–MyD88–GyrB stable transfectants were stimulated with CpG-DNA, LPS or poly(I:C). Lysates were prepared, immunoprecipitated with antibodies to TRAF3 and analysed by immunoblotting with antibodies to Flag, TRIF and TRAF3. e, RAW264.7 cells were stimulated with CpG-DNA, lysates prepared and after immunoprecipitation with indicated antibodies analysed by either immunoblotting (upper panel) or kinase assays using GST–IRF7 or GST–IκBα as substrates (lower panels).
Heretofore, TRAF3 was not implicated in TLR signalling and its biological function has been enigmatic. Traf3-/- mice die shortly after birth, exhibiting progressive hypoglycaemia and runting12. Wild-type mice reconstituted with TRAF3-deficient bone marrow are viable and we used them as a source for Traf3-/- myeloid cells. Traf3-/- bone marrow-derived macrophages (BMDMs) and wild-type counterparts were stimulated with CpG-DNA and LPS. Only small differences in JNK1/2 and p38 phosphorylation were observed, and the kinetics of IκBα degradation were comparable (Supplementary Fig. 3a). However, CpG-DNA- and LPS-induced IL-12 and IL-6 expression was strongly elevated in Traf3-/- BMDMs, whereas IL-10 production was undetectable (Fig. 3a and Supplementary Fig. 3b). Similar alterations regarding all IL-12 family members and IL-10 were observed at the messenger RNA level, whereas other genes were not affected (Supplementary Figs 3c and 4). Upregulation of the pro-inflammatory cytokines IL-12 and IL-6 was largely due to defective production of IL-10, an anti-inflammatory cytokine13, as exogenous IL-10 counteracted this effect (Supplementary Fig. 3d). Wild-type and Traf3-/- cells also differed in the regulation of IFN and IFN-dependent genes. Induction of bioactive IFN by CpG-DNA, LPS and R-848, an imidazoquinoline (TLR7)14, was defective in Traf3-/- BMDMs and in Traf3-/- Flt3 ligand (Flt3L)-induced dendritic cells, the wild-type counterparts of which are known to produce high levels of IFN (Fig. 3b, c and Supplementary Fig. 3e)15.

a, BMDMs were stimulated for 16 h with CpG-DNA or LPS, and secretion of IL-12 p40/p70 and IL-10 was determined by ELISA. b, BMDMs were stimulated for 6 h with CpG-DNA, LPS or R-848 and supernatants were bioassayed for IFN activity. c, Flt3L-induced dendritic cells were stimulated for 24 h with D19-CpG-DNA and supernatants were bioassayed for IFN activity. d, BMDMs were stimulated with CpG-DNA or poly(I:C) for 16 h, and IFN and IL-10 secretion was determined as above. e, BMDMs were exposed or not to VSV (multiplicity of infection = 1) for 2 h. After 18 h, IFN activity in culture supernatants was determined. Experiments shown were repeated at least three times and representative results are presented. co, control; ND, not determined.
Consistent with the observation that LPS activates IFN via TRIF4,5, poly(I:C), which signals solely through TRIF4,5, failed to induce IFN and IL-10 in Traf3-/- BMDMs, yet induced over-production of IL-12 (Fig. 3d and Supplementary Fig. 3f). Vesicular stomatitis virus (VSV), which can activate cells via TLR7 and MyD88 (ref. 16), failed to induce IFN in Traf3-/- BMDMs (Fig. 3e), which were also more susceptible to viral-induced killing than wild-type counterparts (data not shown).
We compared the function of TRAF3 to TRAF6. In Traf6-/- BMDMs, CpG-DNA and Pam3Cys did not induce MAPK phosphorylation or IκBα degradation, but the response to poly(I:C) proceeded normally (Fig. 4a). Thus, TRAF6 is critical for MyD88 signalling, but not TRIF signalling. Consistent with this interpretation, the LPS response was only partially reduced in the absence of TRAF6 (Fig. 4a). Accordingly, TLR9-induced gene expression was dependent on TRAF6, whereas the response to poly(I:C) was TRAF6 independent, both in BMDMs and Flt3L-induced dendritic cells (Fig. 4b, c and Supplementary Fig. 5a, b). Notably, many genes, including IL-10, IFN-β and IL-12 p40 were normally induced by LPS in Traf6-/- macrophages (Supplementary Fig. 5c). Taken together, these results strongly suggest that LPS regulates type I IFN and IL-10 via the TRIF-dependent pathway and that this pathway relies on TRAF3 rather than TRAF6.

a, BMDMs were stimulated with the indicated pathogen-associated molecular patterns and analysed for MAPK phosphorylation and IκBα degradation by immunoblotting. b, BMDMs were stimulated with CpG-DNA or poly(I:C). After 6 h the indicated mRNAs were quantified by Q-PCR. c, Flt3L-induced dendritic cells were stimulated for 6 h with D19-CpG-DNA and IFN secretion was analysed. Experiments shown (b, c) were repeated at least three times and representative results are presented. d, HoxB8-immortalized myeloid progenitors expressing Flag–MyD88–GyrB were stimulated with coumermycin, and lysates were prepared and analysed by immunoblotting with anti-TBK1 antibodies, either directly or after immunoprecipitation with antibodies to Flag. ns, non-specific. e, Model showing the different functions of TRAF3 and TRAF6 in TLR-mediated immune cell activation.
Two similar protein kinases, TBK1/NAK and IKKi/IKKɛ, are involved in type I IFN induction17,18. Notably, the TBK1 interactor TANK/i-TRAF19 is also a TRAF2/3-interacting protein20. To examine the role of TRAF3 in recruiting TBK1 to TIR complexes, we immortalized Traf3-/- fetal liver myeloid progenitors with a retrovirus expressing the HoxB8 oncogene21. These cells were transduced with the MyD88–GyrB fusion construct. Coumermycin activated typical TLR signalling pathways (Supplementary Fig. 6a) and induced efficient recruitment of TBK1 to MyD88 in wild-type cells, but not in Traf3-/- cells (Fig. 4d). Residual TBK1 recruitment in the absence of TRAF3 may be mediated by TRAF6. Immortalized Traf3-/- cells also exhibited defective IFN and IL-10 production, which was corrected by re-introduction of TRAF3 (Supplementary Fig. 6b–d). Notably, IRAK1, which is also involved in IFN expression22, was equally degraded upon stimulation of wild-type and Traf3-/- cells (data not shown), suggesting that it functions upstream of the TRAFs.
On the basis of these results we propose that TLR-induced MyD88 dimerization and IRAK4/IRAK1 activation initiates recruitment of TRAF6 and TRAF3 (Fig. 4e). The TRAF6-dependent pathway engages MAPKs and IKK, resulting in activation of transcription factors such as AP-1 and NF-κB that participate in the induction of pro-inflammatory cytokines. TRAF3, by contrast, is dispensable for MAPK and IKK activation or production of pro-inflammatory cytokines. TRAF3, however, is instrumental for recruitment of TBK1 and production of type I IFNs and IL-10. As a result of IL-10 deficiency, Traf3-/- myeloid cells overproduce the pro-inflammatory cytokines IL-12 and IL-6. TRAF3 has a much more important role than TRAF6 in TRIF-dependent signalling to the IFN and IL-10 genes, probably by marshalling TBK1 (and possibly IKKi) into the TIR signalling complex leading to activation of IRF3 and IRF7. The ratio of TRAF6 versus TRAF3 recruitment is likely to contribute to the biological specificity and consequences of TLR signalling.
Methods
Mice and cell culture
Traf6-/- mice were provided by J. Inoue6. Fetal liver transplantation has been described previously12. Bone marrow was collected from mice 6–8 weeks after reconstitution and BMDMs or Flt3L-induced dendritic cells were generated by using either L-cell conditioned medium or 50 ng ml-1 Flt3L (R&D Systems) as described23,24. Stable RAW264.7 transfectants were generated and cultured as described25. Fetal liver myeloid progenitors were immortalized using an oestrogen-responsive ER-HoxB8 oncogene in the presence of 1 µM oestrogen and GM-CSF21.
Reagents and plasmids
Antibodies are described in Supplementary Methods. Full-length mouse MyD88 was amplified by polymerase chain reaction with reverse transcription (RT–PCR) using cDNA from BMDMs, and fused to a triple Flag tag and subunit B of E. coli DNA gyrase7. Where indicated, a TAP tag was fused to the carboxy terminus of GyrB8. CpG-DNA refers to oligonucleotides (ODN) 1668 (1 µM; TIB Molbiol, TCCATGACGTTCCTGATGCT); D19-CpG refers to CpG-ODN D19 (1 µM; Invitrogen, GGTGC ATCGATGCAGGGGGG). Nucleotides with a phosphothioate backbone are in italics. Other agonists used are: LPS, 10 ng ml-1 (E. coli 0127:B8, Sigma-Aldrich); poly(I:C), 30 µg ml-1 (GE Healthcare); R-848, 300 nM (GLS); Pam3Cys (tripalmitoyl cysteinyl lipopeptide), 1 µg ml-1 (EMC Microcollections); coumermycin, 0.1 µM (Sigma-Aldrich); and IL-1β, 30 ng ml-1 (PeproTech).
Analysis of gene expression
Total cellular RNA was prepared using TRIzol (Invitrogen) and analysed by real-time quantitative polymerase chain reaction (Q-PCR)23. Primer sequences are available upon request. All values were normalized to the level of cyclophilin mRNA. To determine IFN bioactivity, a VSV-based bioassay was used4. All values represent means of duplicates. Reference murine IFN-α (NIAID/NIH) served as a standard. All ELISAs (IL-12 p40/p70, IL-6, IL-10) were from BD Biosciences.
Protein purification, analysis and mass spectrometry
TAP has been described previously8. Variations to the original protocol are described in Supplementary Methods. After TAP, samples were concentrated using Biomax-5K centrifuge filters (Millipore), separated on a 4–12% Novex Bis-Tris gel (Invitrogen) and stained with colloidal Coomassie blue (Invitrogen). Protein bands were subjected to in-gel trypsin digestion and analysed by LC-MS/MS with a QSTAR-Pulsar quadrupole time-of-flight instrument (ABI-MDS-SCIEX)9. Immunoprecipitations and kinase assays were performed as described25. Substrate proteins used in in vitro kinase assays (IκBα1–54, IRF7409–457) were expressed and purified as glutathione S transferase (GST) fusions.
References
Beutler, B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature 430, 257–263 (2004)
Yamamoto, M., Takeda, K. & Akira, S. TIR domain-containing adaptors define the specificity of TLR signalling. Mol. Immunol. 40, 861–868 (2004)
Hemmi, H. et al. A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745 (2000)
Hoebe, K. et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 424, 743–748 (2003)
Yamamoto, M. et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signalling pathway. Science 301, 640–643 (2003)
Gohda, J., Matsumura, T. & Inoue, J. Cutting edge: TNFR-associated factor (TRAF) 6 is essential for MyD88-dependent pathway but not toll/IL-1 receptor domain-containing adaptor-inducing IFN-β (TRIF)-dependent pathway in TLR signalling. J. Immunol. 173, 2913–2917 (2004)
Farrar, M. A., Alberol, I. & Perlmutter, R. M. Activation of the Raf-1 kinase cascade by coumermycin-induced dimerization. Nature 383, 178–181 (1996)
Rigaut, G. et al. A generic protein purification method for protein complex characterization and proteome exploration. Nature Biotechnol. 17, 1030–1032 (1999)
Blagoev, B., Ong, S. E., Kratchmarova, I. & Mann, M. Temporal analysis of phosphotyrosine-dependent signalling networks by quantitative proteomics. Nature Biotechnol. 22, 1139–1145 (2004)
Suzuki, N. et al. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature 416, 750–756 (2002)
Yamamoto, M. et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature 420, 324–329 (2002)
Xu, Y., Cheng, G. & Baltimore, D. Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses. Immunity 5, 407–415 (1996)
Fiorentino, D. F., Zlotnik, A., Mosmann, T. R., Howard, M. & O'Garra, A. IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 147, 3815–3822 (1991)
Hemmi, H. et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signalling pathway. Nature Immunol. 3, 196–200 (2002)
Kawai, T. et al. Interferon-α induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nature Immunol. 5, 1061–1068 (2004)
Lund, J. M. et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl Acad. Sci. USA 101, 5598–5603 (2004)
Fitzgerald, K. A. et al. IKKɛ and TBK1 are essential components of the IRF3 signalling pathway. Nature Immunol. 4, 491–496 (2003)
Hemmi, H. et al. The roles of two IκB kinase-related kinases in lipopolysaccharide and double stranded RNA signalling and viral infection. J. Exp. Med. 199, 1641–1650 (2004)
Pomerantz, J. L. & Baltimore, D. NF-κB activation by a signalling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 18, 6694–6704 (1999)
Cheng, G. & Baltimore, D. TANK, a co-inducer with TRAF2 of TNF- and CD 40L-mediated NF-κB activation. Genes Dev. 10, 963–973 (1996)
Knoepfler, P. S., Sykes, D. B., Pasillas, M. & Kamps, M. P. HoxB8 requires its Pbx-interaction motif to block differentiation of primary myeloid progenitors and of most cell line models of myeloid differentiation. Oncogene 20, 5440–5448 (2001)
Uematsu, S. et al. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-α induction. J. Exp. Med. 201, 915–923 (2005)
Park, J. M., Greten, F. R., Li, Z. W. & Karin, M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science 297, 2048–2051 (2002)
Hochrein, H. et al. Herpes simplex virus type-1 induces IFN-α production via Toll-like receptor 9-dependent and -independent pathways. Proc. Natl Acad. Sci. USA 101, 11416–11421 (2004)
Hacker, H. et al. Cell type-specific activation of mitogen-activated protein kinases by CpG-DNA controls interleukin-12 release from antigen-presenting cells. EMBO J. 18, 6973–6982 (1999)
Acknowledgements
We thank J. Inoue for Traf6-deficient mice, M. Farrar for GyrB constructs, G. Oganesyan, S. K. Saha and G. Cheng for providing cells from TRAF3-deficient mice, T. Maniatis for anti-TBK1 antibodies and L. Pfeffer for L929 cells. H.H. and V.R. were supported by fellowships from the Deutsche Forschungsgemeinschaft (DFG). Work in the laboratories of M.P.K., E.R. and M.K. was supported by grants from the National Institutes of Health. M.K. is an American Cancer Society Research Professor.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Reprints and permissions information is available at npg.nature.com/reprintsandpermissions. The authors declare no competing financial interests.
Supplementary information
Supplementary Figure 1
Characterization of coumermycin-induced cell activation through the MyD88–GyrB- and TRAF6–GyrB fusion proteins. (PDF 559 kb)
Supplementary Figure 2
TRAF3 is recruited to TIR signalling complexes. (PDF 348 kb)
Supplementary Figure 3
Role of TRAF3 in TLR-induced signal transduction and gene regulation. (PDF 1212 kb)
Supplementary Figure 4
CpG-DNA-induced gene expression in wt and TRAF3-deficient BMDM. (PDF 12 kb)
Supplementary Figure 5
CpG-DNA- and LPS-induced gene regulation in wt and TRAF6-deficient BMDM and FLT3-ligand induced DCs. (PDF 10 kb)
Supplementary Figure 6
TRAF3-reconstitution of TRAF3-deficient HoxB8-immortalized myeloid progenitor cells rescues the CpG-DNA-induced IFN and IL-10 response. (PDF 156 kb)
Supplementary Figure Legends
Contains legends to accompany Supplementary Figures 1–6. (DOC 46 kb)
Supplementary Methods
Detailed description of the tandem affinity purification (TAP) technique used to purify MyD88-GyrB-TAP and description of the antibodies used in this work. (DOC 32 kb)
Rights and permissions
About this article
Cite this article
Häcker, H., Redecke, V., Blagoev, B. et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439, 204–207 (2006). https://doi.org/10.1038/nature04369
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature04369
This article is cited by
-
Scavenger receptor B1 facilitates the endocytosis of Escherichia coli via TLR4 signaling in mammary gland infection
Cell Communication and Signaling (2023)
-
TRAF2/3 deficient B cells resist DNA damage-induced apoptosis via NF-κB2/XIAP/cIAP2 axis and IAP antagonist sensitizes mutant lymphomas to chemotherapeutic drugs
Cell Death & Disease (2023)
-
Combining single-cell RNA sequencing of peripheral blood mononuclear cells and exosomal transcriptome to reveal the cellular and genetic profiles in COPD
Respiratory Research (2022)
-
SIGIRR deficiency contributes to CD4 T cell abnormalities by facilitating the IL1/C/EBPβ/TNF-α signaling axis in rheumatoid arthritis
Molecular Medicine (2022)
-
Aging and respiratory viral infection: from acute morbidity to chronic sequelae
Cell & Bioscience (2021)
