
Abstract
Nuclear factor-kappa B (NF-κB) is a critical regulator of multiple biological functions including innate and adaptive immunity and cell survival. Activation of NF-κB is tightly regulated to preclude chronic signaling that may lead to persistent inflammation and cancer. Ubiquitination of key signaling molecules by E3 ubiquitin ligases has emerged as an important regulatory mechanism for NF-κB signaling. Deubiquitinases (DUBs) counteract E3 ligases and therefore play a prominent role in the downregulation of NF-κB signaling and homeostasis. Understanding the mechanisms of NF-κB downregulation by specific DUBs such as A20 and CYLD may provide therapeutic opportunities for the treatment of chronic inflammatory diseases and cancer.
Similar content being viewed by others


Introduction
Nuclear factor-kappa B (NF-κB)/Rel represents a eukaryotic transcription factor family that is a master regulator of genes that control innate and adaptive immune responses 1. NF-κB proteins all share an approximately 300-amino-acid Rel homology domain (RHD) that regulates DNA binding, nuclear localization and dimerization 2. NF-κB family members form homo- and heterodimers, including p65/RelA, c-Rel, RelB, p105/p50 and p100/p52 3. NF-κB proteins are sequestered in the cytoplasm as latent complexes by inhibitory proteins, or IκBs, that prevent NF-κB nuclear translocation and DNA binding 4. Members of the IκB family, consisting of IκBα, IκBβ, IκBɛ, NF-κB1, NF-κB2, IκBζ (also known as MAIL) and Bcl-3, all contain a series of ankyrin repeat domains that mediate binding with NF-κB subunits 3. Whereas the majority of IκBs serve as inhibitors of NF-κB, the IκBζ and Bcl-3 isoforms instead potentiate NF-κB transactivation in the nucleus 5, 6. The NF-κB1 and NF-κB2 proteins, also known as p105 and p100, respectively, are precursor forms of the p50 and p52 NF-κB subunits 7.
There are two unique NF-κB signaling pathways, termed canonical and noncanonical, that have distinct biological roles 8. In the canonical NF-κB pathway, proinflammatory cytokines such as TNF-α or IL-1β, or pathogen-derived components such as bacterial lipopolysaccharide (LPS), trigger the rapid nuclear translocation of NF-κB subunits 1. The canonical NF-κB pathway plays an important role in many physiological processes such as innate and adaptive immunity and cell survival. A wide variety of stimuli activate canonical NF-κB signaling, all of which converge at the IκB kinase (IKK), consisting of catalytic subunits IKKα and IKKβ and the regulatory subunit IKKγ (also known as NEMO) 9, 10, 11. The IKKβ and IKKγ subunits serve essential and non-redundant roles in the activation of NF-κB in the canonical pathway 12, 13. IKK phosphorylates IκB proteins at two amino (N)-terminal regulatory serine residues, triggering their ubiquitination and degradation by the proteasome, thus allowing NF-κB to enter the nucleus and activate stimulus-specific gene programs 14. In response to specific stimuli, such as proinflammatory cytokines, NF-κB is activated transiently due to a series of tightly regulated negative feedback loops including the induction of inhibitory proteins such as IκBα that downregulate NF-κB signaling 15. Terminating an NF-κB response is essential to prevent persistent NF-κB activation that may lead to chronic inflammation and/or tumorigenesis.
In the noncanonical pathway, NF-κB2 is processed by the proteasome to generate p52, which together with RelB regulates a distinct subset of target genes controlling B lymphocyte survival and lymphoid organogenesis 8. The noncanonical pathway is triggered by a subset of TNF superfamily ligands, such as BAFF, LT-β and CD40L 16, 17, 18, 19. The MAP 3 kinase MAP3K14, also known as NF-κB inducing kinase or NIK, is a central player in the noncanonical pathway 20. NIK is predominantly regulated post-translationally with extremely low levels of NIK in most cell types 21. Low levels of NIK are maintained by persistent degradation of NIK by a multi-subunit E3 ubiquitin ligase complex consisting of TNF-receptor-associated factor (TRAF) 2, TRAF3, cIAP1 and cIAP2 22, 23, 24. BAFF-R, CD40 and LT-βR ligation all trigger the degradation of TRAF3, resulting in the inactivation of the TRAF/cIAP E3 ubiquitin ligase complex and the concomitant stabilization of NIK 24. NIK, in turn, phosphorylates IKKα, which phosphorylates NF-κB2 to trigger its processing by the proteasome 25. A recent study indicates post-translational regulation of NIK function via its K63-linked polyubiquitination by the zinc finger protein 91 (ZFP-91) 26. NIK ubiquitination promotes its stabilization through an unknown mechanism.
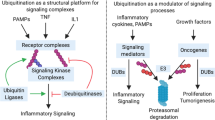
Ubiquitination is a reversible post-translational modification involving the covalent attachment of one or more ubiquitin monomers to a protein substrate, best known as a targeting signal for proteasome-mediated degradation. However, recent studies have demonstrated numerous nonproteolytic functions of ubiquitination including protein trafficking and activation of kinases and phosphatases 27. Therefore, ubiquitination plays a central role in the regulation of many signaling pathways. Ubiquitination consists of a three-step enzymatic cascade initiating with the activation of ubiquitin by a ubiquitin-activating enzyme (E1) followed by transfer of the activated ubiquitin to the active-site cysteine of a ubiquitin-conjugating enzyme (E2) and finally, with the assistance of an E3 or ubiquitin ligase, transfer of the ubiquitin to a lysine on a substrate protein to form an isopeptide bond 28. There are two E1s, approximately 50 E2s and 600 E3s encoded in the human genome 29. The majority of E2 enzymes have been linked to K48-linked protein ubiquitination and degradation. However, the dimeric E2 complex Ubc13/Uev1a is specific for the synthesis of K63-linked polyubiquitin chains 30, 31. The E3s provide specificity for ubiquitination of substrates and consist of three groups: RING, HECT and F-box 32. RING domain and the closely related F-Box E3 ligases catalyze the transfer of ubiquitin from E2s to substrates, whereas HECT domain E3 ligases directly transfer ubiquitin to substrates 32.
Ubiquitination of a substrate may proceed with a single ubiquitin (monoubiquitination) or a chain of covalently linked polyubiquitin molecules (polyubiquitination) 28. Ubiquitin contains seven internal lysine residues (K6, K11, K27, K29, K33, K48 and K63) that can each support polyubiquitination chains 28. Indeed, isopeptide linkages involving every lysine residue in ubiquitin have been detected by mass spectrometry of total protein lysates 33. K48-linked polyubiquitin chains are recognized by the 26S proteasome, leading to protein degradation. K63-linked polyubiquitin chains do not trigger protein degradation but instead have nonproteolytic functions such as protein trafficking or kinase and phosphatase activation 29, 34. In addition, ubiquitin chains can also be linked in a linear fashion, whereby the C-terminal glycine is attached to an N-terminal methionine resulting in head-to-tail polyubiquitination 35. Two RING-type E3 ligases, HOIL1 and HOIP, specifically assemble linear polyubiquitin chains that play an important role in NF-κB regulation 36. Other types of ubiquitin modifications have also been observed, including K11-mediated polyubiquitination. A novel K11-linkage-specific antibody was used to demonstrate that K11-linked ubiquitin chains are highly upregulated during mitosis and are linked to protein degradation 37. There have also been reports indicating that K27-linked polyubiquitin chains regulate the targeting of Jun to lysosomes 38. In addition, AIP4/Itch catalyzes the formation of K29-linked polyubiquitin chains on Notch receptor to induce its lysosomal-mediated degradation 39. However, the biological roles of these noncanonical ubiquitin linkages remain poorly understood.
Ubiquitination is reversible and is counter-regulated by a family of deubiquitinases (DUBs) of which there are nearly 100 encoded in the human genome 40. There are five families of DUBs characterized by specific structural domains: ubiquitin C-terminal hydrolases (UCHs), ubiquitin-specific proteases (USPs), ovarian tumor proteases (OTUs), Josephins and JAB1/MPN/MOV34 metalloenzymes (JAMMs). UCHs, USPs, OTUs and Josephins function as cysteine proteases, whereas JAMMs are zinc-dependent metalloproteases 41. The USPs compose the largest subfamily of DUBs with over 50 members 41. Many DUBs harbor ubiquitin-binding domains (UBDs) that coordinate recognition and recruitment of ubiquitinated substrates. Conversely, as will be discussed later, a number of DUBs rely on ubiquitin-binding adaptor molecules to confer specificity. In this review, we will describe the roles of DUBs in the regulation of NF-κB signaling.
Ubiquitin-dependent activation of NF-κB signaling
Engagement of the TNFR or IL-1R/TLR4 pathways leads to recruitment and activation of specific TRAFs. TRAFs 2-7 all have N-terminal RING domains that confer E3 ligase activity and TRAF domains that mediate homo- and heterotypic protein interactions 42. TRAF1 lacks a RING domain and functions as a negative regulator of TNF signaling and NF-κB activation 43. TRAF6 activates IKK in a K63-ubiquitin chain-dependent manner that requires the dimeric E2 enzyme complex Ubc13/Uev1a as well as the transforming growth factor receptor-β-activated kinase 1 (TAK1) and its regulatory subunits TAB1 and TAB2 31. Genetic studies in mice support essential roles for TRAF6 and TAK1 in the activation of NF-κB by multiple stimuli 44, 45, 46. Surprisingly, genetic ablation of Ubc13 does not affect activation of NF-κB by multiple stimuli in B cells, macrophages or MEFs 47. Rather, Ubc13 appears to play an important role in MAP kinase activation, partly through NEMO ubiquitination 47. Contrary to these results, haploinsufficient Ubc13+/− mice, generated by an independent group, are resistant to LPS-induced lethality 48. Ubc13-deficient macrophages and splenocytes derived from these mice are impaired in the induction of proinflammatory cytokines by LPS 48. Furthermore, Ubc13+/− macrophages display defects in NF-κB, p38 and JNK activation as a result of impaired LPS-induced TRAF6 ubiquitination 48. It is unclear why the two different models of Ubc13 deficiency result in distinct phenotypes and conclusions, possibly due to the different strategies used to generate the knockout mice.
Binding of IL-1 to IL-1R triggers the recruitment of Myd88, IRAK1, IRAK4 and TRAF6 to form a receptor-associated complex 49. TRAF6 undergoes oligomerization, activation and K63-linked polyubiquitination that leads to the recruitment of the TAB1-TAB2-TAK1 complex and subsequent activation of TAK1, which, in turn phosphorylates IKKβ 50. However, the role of TRAF6 autoubiquitination is unclear since reconstitution of TRAF6-deficient MEFs with a TRAF6 lysine-deficient mutant rescues IL-1- and RANK-induced NF-κB activation similar to wild-type TRAF6 51. The RING domain of TRAF6 is important for downstream NF-κB signaling, indicating that TRAF6-catalyzed ubiquitination of other substrates is more important than TRAF6 autoubiquitination. Indeed, IRAK1 undergoes TRAF6-dependent K63-linked polyubiquitination upon IL-1 stimulation that regulates binding to NEMO 52, 53. A recent study also suggests that unanchored polyubiquitin chains synthesized by TRAF6 and Ubc13/Uev1a are key regulators of NF-κB signaling by directly activating TAK1 via binding to TAB2 54. TRAF6 also functions together with UbcH5c to synthesize unanchored polyubiquitin chains that activate IKK 54. Unanchored polyubiquitin chains also regulate antiviral signaling 55, and may indeed play a pervasive role in regulating signaling pathways.
TNF-α binding to TNFR promotes the rapid formation of a TRADD, TRAF2, cIAP1, cIAP2 and RIP1 receptor-associated complex 56. RIP1 undergoes K63-linked polyubiquitination on lysine 377 (K377) in response to TNF-α stimulation 57, 58. Although TRAF2 was originally thought to ubiquitinate RIP1, TRAF2-deficient MEFs exhibit only a mild defect in TNF-α-mediated NF-κB signaling 59. This may be due to the functional redundancy with TRAF5, since combined deletion of TRAF2 and TRAF5 completely impairs TNF-mediated NF-κB activation 60. More recent studies suggest that cIAP1 and cIAP2 serve as direct E3 ubiquitin ligases for RIP1, suggesting that TRAFs may instead serve as adaptors for RIP1 61. RIP1 ubiquitination plays a critical role in the recruitment of the TAK1 complex, which is responsible for the phosphorylation and activation of IKK 62, 63. RIP1 is therefore a key target of DUBs that downregulate TNF-α-induced NF-κB signaling.
A20 function and the mechanisms of NF-κB inhibition
A20 (also known as TNFAIP3) was originally identified as a TNF-inducible gene in human umbilical vein endothelial cells (HUVEC) 64. In most cell types A20 is expressed at very low levels but can be rapidly induced by proinflammatory cytokines or mitogens 64. Conversely, thymocytes and peripheral T lymphocytes express high levels of A20 that are subsequently downregulated upon stimulation with T cell receptor agonists 65. A20 contains an N-terminal OTU domain and seven zinc finger domains (C2H2) in the C-terminus (Figure 1). A20 harbors DUB activity mediated by the OTU domain 66, 67. However, A20 does not exert a global effect on ubiquitinated proteins, suggesting specificity for its substrates. Recently, great progress has been made in understanding how A20 regulates inflammatory signaling pathways, as well as its novel role as a tumor suppressor that will be discussed later.

Structural domains in A20 and CYLD. (A) A20 contains an N-terminal OTU domain responsible for the DUB activity of A20. The catalytic cysteine residue Cys103 is also important for binding to the E2 enzymes Ubc13 and UbcH5c. There is an IKKβ phosphorylation site at Ser381 downstream of the OTU domain. A20 contains seven zinc finger domains (C2H2) in its C-terminus. ZnF4 confers A20 E3 ligase activity and is also involved in TAX1BP1 interactions. ZnF6 and ZnF7 are important for targeting A20 to lysosomes. In addition, A20 zinc fingers have been implicated in A20 oligomerization and A20 binding to ABINs and NEMO. (B) CYLD contains a C-terminal USP domain responsible for its DUB activity. The N-terminus of CYLD contains three CAP-Gly (CAP) domains and two proline-rich (PR) motifs. The first two CAP domains mediate binding to microtubules whereas the third CAP domain regulates NEMO interactions. CYLD also contains a TRAF2 binding site (PVQES) and a phosphorylation site cluster between CAP domains 2 and 3.
A20 was first described as a zinc finger protein that protects cells from TNF cytotoxicity 68. Subsequent studies, based mainly on overexpression, implicated A20 as a negative regulator of NF-κB signaling in multiple pathways 69, 70, 71. Generation of A20-deficient mice (Tnfaip3−/−) confirmed the anti-apoptotic and NF-κB inhibitory functions of A20. A20-deficient mice die prematurely due to multi-organ inflammation and cachexia as a result of dysregulated NF-κB signaling 72. Tnfaip3−/− mice are also hyper-responsive to proinflammatory stimuli in vivo and succumb to sub-lethal doses of TNF-α or LPS 72. Consistently, A20-deficient MEFs exhibit persistent TNF-α and IL-1-induced NF-κB signaling as observed by prolonged IKK activation and IκBα degradation 72. MEFs deficient in A20 are also sensitized to TNF-mediated cell death, illustrating the anti-apoptotic function of A20 72.
The uncontrolled inflammation in Tnfaip3−/− mice is independent of the adaptive immune response, since Tnfaip3−/− mice on a Rag1−/− background lacking T and B lymphocytes are not rescued from premature lethality 72. Although A20 was initially described as a negative regulator of TNFR signaling, the spontaneous inflammation and cachexia in Tnfaip3−/− mice still occurs in both Tnfaip3−/− Tnfα−/− and Tnfaip3−/− Tnfr1−/− double-mutant mice, indicating that A20 also restricts TNF-independent signaling pathways in vivo 73. All Toll-like receptor pathways use the common adaptor Myd88 to activate NF-κB and MAP kinase pathways to induce expression of proinflammatory cytokines 74. Tnfaip3−/− Myd88−/− double-mutant mice are rescued from lethal inflammation, suggesting that TLR signals drive the spontaneous inflammation in the absence of A20 75. Consistent with these observations, treatment of A20-deficient mice with broad-spectrum antibiotics rescues the severe inflammation and lethality, indicating that a dysregulated response to commensal intestinal flora is responsible for the constitutive TLR signaling in Tnfaip3−/− mice 75. Therefore, A20 plays an essential role in restricting TLR signaling and maintaining immune homeostasis in the intestine.
The spontaneous inflammation and premature lethality of Tnfaip3−/− mice has precluded the use of these mice in experimentally induced inflammatory disease models. Therefore, conditional gene targeting was used to specifically delete A20 in intestinal epithelial cells (IECs) by crossing with Cre transgenic mice driven by the IEC-specific promoter villin 76. The A20 IEC conditional knockout (A20IEC-KO) mice develop normally, appear healthy and do not develop spontaneous colitis 76. However, these mice are more susceptible to experimentally induced colitis after injection with dextran sulphate sodium (DSS), and experience pronounced colon shortening, crypt loss and immune cell infiltration 76. The increased DSS-induced colitis susceptibility of A20IEC-KO mice is triggered by enhanced TNF-mediated apoptosis of IECs, leading to a breakdown of the intestinal barrier and systemic inflammation 76. Crossing A20IEC-KO mice to TNFR1-deficient mice ameliorates colitis symptoms, indicating that uncontrolled TNFR1 signaling contributes to the intestinal pathogenesis 76. These findings also underscore the anti-apoptotic function of A20 in the intestinal epithelium in a pathophysiological setting.
In addition to IECs, A20 has also been selectively deleted in B lymphocytes by crossing with CD19-Cre transgenic mice 77. Although A20 is constitutively expressed in B cells, stimulation with agonistic CD40 antibodies further upregulates A20 expression via NF-κB 78. B cell-specific A20 knockout (A20CD19-KO) mice develop normally, but have increased numbers of immature (CD19+IgMhi) and germinal center (GC) B cells 77. Furthermore, splenic B cells from A20CD19-KO mice exhibit enhanced proliferation and upregulation of costimulatory molecules upon stimulation with α-CD40, LPS or CpG 77. CD40-induced IκBα phosphorylation and degradation as well as p100 phosphorylation are all enhanced in splenic B cells from A20CD19-KO mice 77, indicating the importance of A20 in inhibiting canonical and noncanonical NF-κB signaling downstream of CD40 ligation in B cells. As a result of accumulated immature and GC B cells, A20CD19-KO mice develop symptoms of autoimmunity, including the presence of autoantibodies 77. In this regard, it is interesting that polymorphisms in the vicinity of the human A20 gene have been linked to development of systemic lupus erythematosus (SLE) 79. Furthermore, splenic B cells from A20CD19-KO mice are resistant to Fas-mediated programmed cell death due to enhanced expression of the antiapoptotic molecule Bcl-x 77. This result is somewhat surprising given the known anti-apoptotic effects of A20 in TNFR signaling. Nevertheless, the collective results firmly establish A20 as an essential regulator of B cell homeostasis.
A20 functions as a negative feedback inhibitor of NF-κB in multiple innate immune signaling pathways including TNFR and Toll-like receptor pathways (Figure 2). In the TNFR pathway, A20 expression is rapidly upregulated in an NF-κB-dependent manner and targets ubiquitinated RIP1 for inactivation in a two-step sequential process 64, 67. A20 first removes K63-linked polyubiquitin chains from RIP1 in an OTU-dependent manner, then catalyzes formation of K48-linked polyubiquitin chains onto RIP1 to trigger proteasome-mediated degradation 67. Remarkably, recombinant A20 functions as an E3 ligase despite the lack of a RING or HECT domain. The E3 ligase activity of A20 is dependent on the fourth zinc finger domain (ZNF4), suggesting a novel type of E3 ligase domain 67. Thus, A20 functions as a unique dual function ubiquitin-editing enzyme that inactivates RIP1 via sequential DUB and E3 ligase activities. In addition, A20 may also target substrates for degradation via the lysosomal pathway. A fraction of cellular A20 localizes to lysosomes in a zinc finger-dependent manner and promotes the degradation of TRAF2 in lysosomes 80, 81.

NF-κB signaling pathways downstream of TNFR1, TLR4 and TCR/CD28. TNFR1, TLR4 and TCR/CD28 engagement trigger the K63-linked ubiquitination of RIP1, TRAF6 and MALT1, respectively. Each of these ubiquitinated signaling molecules are targeted for deubiquitination by either A20, CYLD, Cezanne or USP21, as indicated. MALT1 cleaves A20 to inactivate its function. CYLD also removes polyubiquitin chains from TAK1 in the TCR pathway.
A20-deficient mice exhibit spontaneous multi-organ inflammation due to persistent activation of Toll-like receptor signaling. A20 expression is rapidly induced by LPS in bone marrow-derived macrophages (BMDMs) 73. A20-deficient macrophages produce more proinflammatory cytokines in response to LPS stimulation, suggesting that A20 is an important negative regulator of TLR4 signaling in macrophages. Indeed, LPS-induced NF-κB activation is elevated and persistent in Tnfaip3−/− macrophages. A20 downregulates NF-κB signaling upstream of IKK at the level of TRAF6. Overexpression of A20, but not a catalytically inactive DUB mutant C103A, inhibits TRAF6 ubiquitination, and A20-deficient MEFs also exhibit persistent LPS-induced TRAF6 ubiquitination 73. Although A20 clearly antagonizes TRAF6 ubiquitination, it appears that the mechanisms of TRAF6 downregulation are distinct from those of RIP1 in TNFR signaling since TRAF6 is not degraded upon IL-1 or LPS stimulation 82.
Recently, a new mechanism of A20 inhibition of NF-κB signaling has emerged. At early times after LPS stimulation, A20 disrupts the binding of TRAF6 with the E2 enzymes Ubc13 and UbcH5c 82. Similarly, A20 also disrupts the binding of TRAF2 and cIAP1 with Ubc13. At later times (4-6 h) of LPS stimulation, A20 also triggers the proteasome-mediated degradation of the E2 enzymes Ubc13 and UbcH5c 82. Ubc13 is also ubiquitinated and degraded in response to TNF-α stimulation (Figure 3). A20 disruption of E2-E3 enzymes is dependent on ZnF4 and Cys103 in the OTU domain consistent with previous studies 67, 82. Therefore, it appears that A20 uses multiple complementary mechanisms to downregulate NF-κB signaling.

Temporal regulation of NF-κB signaling downstream of TNFR1. During early stages of TNFR1 signaling (signal initiation—15-30 min after receptor engagement), RIP1 undergoes K63-linked ubiquitination, which activates IKK and NF-κB. CYLD is inactivated by IKK-mediated phosphorylation. At later times (0.5-6 h), A20 and IκBα expression is induced by NF-κB as part of a negative feedback loop. A20 interacts with TAX1BP1, Itch, RNF11 and possibly ABIN-1 and YMER to form the A20 ubiquitin-editing complex. A20 removes K63-linked ubiquitin chains from RIP1 and catalyzes the formation of K48-linked chains to trigger RIP1 degradation. A20 also disrupts cIAP1 binding to Ubc13 and promotes Ubc13 ubiquitination and degradation.
A20 also negatively regulates signaling by nucleotide-binding oligomerization domain containing 2 (NOD2), a cytoplasmic pathogen recognition receptor that recognizes bacterial cell wall components, specifically muramyl dipeptide (MDP), and activates NF-κB. RIP2 (also known as RICK) is a key downstream regulator of NOD2 that facilitates IKK activation upon K63-linked polyubiquitination and oligomerization 83. A20-deficient macrophages display enhanced NOD2-mediated RIP2 ubiquitination, NF-κB signaling and generation of proinflammatory cytokines 84. Tnfaip3−/− mice exhibit pronounced in vivo responses in response to MDP challenge as shown by greater amounts of serum IL-6 84. Mechanistically, A20 inhibits NOD2 signaling by deubiquitinating K63-linked polyubiquitin chains from RIP2. The E3 ligase Itch also negatively regulates MDP-induced NF-κB activation, although Itch was shown to function as an E3 ligase for RIP2 85.
In addition to regulating innate immunity, A20 also is important for adaptive immune responses. A20 is an important negative regulator of NF-κB signaling in both T and B lymphocytes. Its regulation is unique in T lymphocytes because of the high basal levels compared to other cell types 65. Nevertheless, overexpression of wild-type A20, but not an A20 DUB mutant, attenuates NF-κB activation triggered by agonistic antibodies to CD3 and CD28 86. T-cell receptor ligation promotes the recruitment of A20 to the paracaspase MALT1 and the adaptor protein Bcl-10, where MALT1 cleaves A20 to impair its NF-κB inhibitory function and fine-tune NF-κB signaling 87. A20 also removes MALT1 polyubiquitin chains to inhibit interactions between ubiquitinated MALT1 and the downstream IKK complex (Figure 2) 88. Therefore, the balance between A20 cleavage by MALT1 and A20 deubiquitination of MALT1 determines NF-κB activation in T lymphocytes. A similar cross-regulation between A20 and MALT1 is thought to exist in B lymphocytes, and is dysregulated in B-cell lymphomas which are characterized by uncontrolled NF-κB activation. MALT1 paracaspase activity is constitutive in B-cell lymphomas, and inhibition of MALT1 catalytic activity preserves full-length A20, which then inhibits NF-κB and promotes toxicity towards B-cell lymphoma cells 89.
A20 is also a potent negative regulator of dendritic cell (DC) maturation and antigen presentation. Silencing of A20 expression using siRNA in mouse myeloid DCs triggers spontaneous and enhanced expression of proinflammatory cytokines and costimulatory molecules 90. A20-silenced DCs are particularly effective in anti-tumor responses by both inhibiting T regulatory cells and hyperactivating tumor-infiltrating cytotoxic T cells 90. Due to the elevated expression of proinflammatory cytokines IL-6 and TNF-α in A20-silenced DCs, it is likely that constitutive NF-κB activation is largely responsible for the marked activation and antigen presentation.
Regulation of A20 by ubiquitin-binding proteins and adaptors
Given that A20 does not exhibit a strong preference for deubiquitinating K63-linked polyubiquitin chains in vitro 91, it is not surprising that A20 cooperates with several other proteins to inhibit NF-κB signaling. The NF-κB inhibitory function of A20 is dependent on several other proteins including Tax1 binding protein 1 (TAX1BP1), Itch, Ring finger protein (RNF) 11 and possibly others, including ABIN-1 and YMER, that together form a complex of proteins that we have termed the “A20 ubiquitin-editing complex” (Figure 3). TAX1BP1 was first identified in a yeast two-hybrid screen using the HTLV-I Tax oncoprotein as bait 92. Shortly after, TAX1BP1 was also identified as a novel A20 binding protein that functions as an anti-apoptotic protein regulating the function of A20 93.
Disruption of the Tax1bp1 locus by gene trapping results in embryonic lethality of homozygous mutant mice due to systemic hemorrhaging and cardiac defects at approximately E13.5 94. On the other hand, Tax1bp1−/− mice, generated by conventional gene targeting methods, succumb to age-dependent inflammatory cardiac valvulitis 95. Similar to Tnfaip3−/− mice, Tax1bp1−/− mice are hypersensitive to proinflammatory cytokines and succumb to sub-lethal doses of TNF-α and IL-1β 95. Tnfaip3−/− and Tax1bp1−/− MEFs display enhanced NF-κB activation and a defect in the termination of NF-κB signaling in response to TNF-α or IL-1β stimulation 94, 95. TAX1BP1 interacts with RIP1 downstream of TNFR and counteracts its ubiquitination since RIP1 ubiquitination is persistent upon TNF-α stimulation in Tax1bp1−/− MEFs 94. Together, these studies indicate that TAX1BP1 is a critical negative regulator of NF-κB signaling.
TAX1BP1 contains an N-terminal SKIP (skeletal muscle and kidney enriched inositol phosphatase) carboxyl homology (SKICH) domain, originally described as a membrane targeting domain in the SKIP and PIPP phosphatases 96. TAX1BP1 also contains three coiled-coil domains and two C-terminal zinc finger domains. Due to the striking functional similarities between Tnfaip3−/− and Tax1bp1−/− MEFs, and the fact that TAX1BP1 lacks a DUB domain, it was hypothesized that TAX1BP1 inhibited NF-κB through A20. Indeed, TAX1BP1 is required for A20 to interact with RIP1 upon TNF stimulation and also mediates the NF-κB inhibitory function of A20 94, 95. Interestingly, one of the TAX1BP1 zinc finger domains functions as a novel UBD 95. Therefore, TAX1BP1 likely serves as an adaptor by recruiting A20 to ubiquitinated substrates that are first recognized by the TAX1BP1 UBD.
The zinc finger domains of TAX1BP1 also harbor highly conserved 'PPXY' (where P=proline, X=any amino acid and Y=tyrosine) motifs that are precisely positioned in the center of both zinc finger domains 97. PPXY motifs are well-established binding motifs for modular 'WW' (where W=tryptophan) domain-containing proteins 98. Reconstitution of TAX1BP1-deficient MEFs with TAX1BP1 PPXY mutants fails to restore transient NF-κB signaling, indicating the importance of these sites for TAX1BP1 function 97. The HECT domain E3 ligase Itch contains multiple WW domains and interacts with TAX1BP1 via the PPXY motifs 99. The TAX1BP1/Itch binding is inducible and occurs rapidly following TNF-α stimulation 97. Itch also is essential for A20 recruitment to RIP1 upon TNF-α stimulation and the concomitant downregulation of RIP1 ubiquitination 97. Consistent with these findings, Itch−/− MEFs exhibit enhanced and prolonged NF-κB signaling as observed in Tnfaip3−/− and Tax1bp1−/− MEFs. Despite the functional similarities observed between Itch−/− and Tnfaip3−/− MEFs, there are clear differences in the phenotypes of the knockout mice. Whereas A20-deficient mice exhibit spontaneous inflammation in multiple organs, the inflammation in Itch-deficient mice is mainly confined to the lungs and skin 100. Furthermore, in Tnfaip3−/− mice, the inflammation is independent of adaptive immunity, unlike Itch−/− mice which are rescued from lethal inflammation by crossing with Rag1−/− mice 97. These in vivo differences may indicate a cell-type-specific functional interaction between A20 and Itch. Also, Itch clearly has functions in signaling and cell death that are independent of A20.
RNF11 is a RING-type E3 ligase overexpressed in breast, prostate and pancreatic cancers 101, 102. RNF11 interacts with several regulators of the transforming growth factor beta (TGF-β) pathway such as Smurf2 and SMAD7 to potentiate TGF-β signaling 103, 104. A high-throughput yeast two-hybrid screen identified over 70 putative binding proteins of RNF11 including A20, Itch, TAX1BP1, NEMO and ABIN-1 105, suggesting a possible link between RNF11 and NF-κB signaling. Indeed, RNF11 inducibly interacts with TAX1BP1 and Itch in response to TNF-α or IL-1 stimulation 106. Furthermore, siRNA knockdown of RNF11 enhances NF-κB signaling, whereas overexpression of RNF11 downregulates NF-κB, suggesting that RNF11 is a negative regulator of NF-κB 106. Knockdown of RNF11 is also associated with elevated levels of RIP1 and TRAF6 ubiquitination upon TNF-α or LPS stimulation, respectively 106. A20 is also dependent on RNF11 to downregulate NF-κB signaling, suggesting that RNF11 is a component of the multi-subunit A20 ubiquitin-editing protein complex together with A20, TAX1BP1 and Itch. Because the PPXY motif in RNF11 is essential for its NF-κB regulatory function and is known to mediate binding to Itch 102, RNF11 may function as an adaptor for Itch or, alternatively, may regulate its catalytic activity. Determining the precise function of RNF11 in vivo will require the generation of gene-targeted mice.
ABIN-1 was initially identified in a yeast two-hybrid screen as an A20-interacting protein 70. Overexpression of ABIN-1 inhibits NF-κB signaling by multiple stimuli and mimics the NF-κB inhibitory function of A20 70, 107. ABIN-1 is required for A20 to interact with and deubiquitinate NEMO, suggesting that ABIN-1 serves as an adaptor molecule that bridges A20 and NEMO 108. Consistent with these findings, a recent study demonstrates that the Shigella effector protein IpaH9.8, which functions as an E3 ligase, hijacks ABIN-1 to interact with and ubiquitinate NEMO 109. Furthermore, the ubiquitin-binding domain of ABIN-1 plays an important role in the adaptor function of ABIN-1 by promoting the interaction between IpaH9.8 and NEMO 109. However, the exact role of NEMO ubiquitination in inflammatory signaling pathways remains unclear and ABIN-1-deficient MEFs do not exhibit major defects in NF-κB regulation 110.
Another protein termed YMER (also known as CCDC50) was shown to interact with A20 in a yeast two-hybrid screen 111. Overexpression of YMER downregulates NF-κB signaling, whereas knockdown of YMER by siRNA enhances NF-κB activation 111. YMER contains a K63-linked polyubiquitin-binding motif that is essential for inhibiting NF-κB 111. Consistent with these findings, YMER was also identified in a screen for polyubiquitin-binding proteins 112. Because YMER interacts with RIP1 and promotes an interaction of A20 with K63-linked polyubiquitin chains 111, YMER may also function as a ubiquitin-binding adaptor for A20. However, gene targeting studies of YMER are necessary to firmly establish its role as a regulator of A20 and NF-κB.
Regulation of A20 deubiquitinase activity
The crystal structure of the OTU domain of A20 was determined by two groups, both suggesting a unique mechanism of catalytic function distinct from papain-like cysteine proteases 91, 113. Although the overall structure of A20 differs from other cysteine proteases, the arrangements of catalytic histidine and cysteine residues are similar and conserved 113. The catalytic cysteine residue (Cys103) in A20 resides within an alpha helix domain that is predicted to form a platform for ubiquitin binding 91. Accordingly, Cys103 plays an important role in A20-mediated NF-κB downregulation, suggesting a requirement for A20 DUB activity 67, 73, 94. Conversely, others have reported that Cys103, and hence DUB activity, is dispensable for A20 to inhibit NF-κB 66, 80. Although the reasons for these discrepancies are unclear, they may be attributed to experimental factors such as varying amounts of transfected A20 and/or different genetic backgrounds of the cells (i.e. wild-type or A20-deficient). Nevertheless, it is clear that A20 inhibits antiviral signaling in a DUB-independent manner 114, 115, 116. A20, together with TAX1BP1, inhibits antiviral signaling by disrupting a TRAF3-TBK1-IKKi protein complex via the C-terminal zinc finger domains 115.
Given that A20 opposes K63-linked polyubiquitination of RIP1 and TRAF6, it is surprising that A20 preferentially cleaves K48-linked polyubiquitin chains in vitro 91, 117. It is likely that additional factors, possibly TAX1BP1, Itch and/or RNF11, determine A20 specificity in vivo. Another surprise from the A20 structural studies is that A20 cleaves polyubiquitin chains at the TRAF6/ubiquitin interface instead of disassembling ubiquitin chains in a step-wise manner 91. Therefore, the specificity and catalytic activity of A20 are distinct from other OTU family cysteine proteases.
Although A20 is mainly regulated at the transcriptional level in a negative feedback loop, A20 is also subject to regulation by post-translational mechanisms. A20 is phosphorylated by IKKβ on Ser381, leading to enhanced NF-κB inhibition by A20 118, although it is not clear if the DUB function of A20 is regulated by phosphorylation. A20 phosphorylation provides yet another layer of control by the NF-κB pathway to terminate its activation in a negative feedback loop.
The role of A20 in autoimmune diseases and cancer
Dysregulation of A20 expression has been implicated in a number of autoimmune diseases and cancers. Polymorphisms within the A20 genomic locus increase susceptibility to a number of human autoimmune diseases, including type 1 diabetes, psoriasis, rheumatoid arthritis, inflammatory bowel disease, celiac disease, coronary artery disease and SLE 119, 120, 121, 122, 123, 124. Presumably, the identified polymorphisms influence either the expression or function of A20. A recent study examined the functional effects of the African-derived A20 polymorphism A125V, which lies in the DUB domain 125. The A20 A125V mutation impairs A20-mediated degradation, as well as deubiquitination, of TRAF2 125. The functional defects due to the mutation may result from a conformational change in A20 that alters protein-protein interactions.
A20 has also been implicated as a tumor suppressor in several subsets of B-cell lymphomas. Previous studies have demonstrated that chromosome 6q is frequently deleted in several types of non-Hodgkin lymphomas 126. Inactivating point mutations and deletions are frequently found in the A20 gene, encoded on chromosome 6q, in activated B-cell-like (ABC) diffuse large B-cell lymphoma (DLBCL), marginal zone lymphoma, MALT lymphoma, and Hodgkin lymphoma 127, 128, 129, 130. The A20 promoter may also become methylated, leading to downregulation of A20 in MALT lymphoma 131. Reintroduction of A20 into lymphoma cell lines with biallelic loss of A20 induces apoptosis, suggesting that A20 is indeed a tumor suppressor in B-cell lymphoma 128, 129.
Loss of A20 expression has also been demonstrated during oncogenic transformation of MEFs. Transformation of primary MEFs by E1A and Ras results in impaired TNF-α-induced expression of A20, despite upregulation of other NF-κB-dependent genes 132. Loss of A20 sensitizes transformed MEFs to TNF-α-mediated cell death despite enhanced NF-κB activation 132. The low A20 expression in transformed MEFs is attributed to impaired Bcl-3 binding to the A20 promoter, suggesting yet another distinct mechanism of A20 downregulation in tumor cells.
Because A20 also functions as a potent prosurvival gene, A20 may even function as an oncogene in certain tumor types such as breast cancer and glioma. In breast cancer, A20 is an estrogen-regulated gene which confers resistance to the proapoptotic effects of tamoxifen 133. A20 is overexpressed in estrogen receptor negative (ER-), progesterone receptor negative (PR-) and high-grade breast tumors suggesting that A20 may be a useful prognostic marker for aggressive breast carcinomas 133. A20 is also overexpressed in glioma and is required for survival of glioma stem cells suggesting that the tumor stem cells may be “addicted” to A20 overexpression for survival 134. Although A20 is commonly lost or mutated in B-cell lymphomas, recent large scale sequencing efforts of several tumor types (Catalogue of Somatic Mutations in Cancer (COSMIC): http://www.sanger.ac.uk/genetics/CGP/cosmic/) suggest that A20 is not commonly mutated in other cancers. However, two novel A20 mutations V377M and P656L have been identified in lung carcinomas, although it is unclear if these mutations impact the function or stability of A20.
CYLD function and the regulation of NF-κB
The cylindromatosis (CYLD) gene encodes a deubiquitinating enzyme of the USP family with a C-terminal catalytic domain, three N-terminal Cap-Gly domains and two proline-rich motifs (Figure 1) 135. CYLD was originally identified as a tumor suppressor gene since mutations in the Cyld gene predispose individuals to tumors of skin appendages 136. Three simultaneous publications demonstrated that CYLD negatively regulates NF-κB signaling via DUB activity. Using an shRNA approach, Brummelkamp et al. showed that CYLD inhibits NF-κB signaling by counteracting TRAF2 ubiquitination 137. Two other groups used yeast two-hybrid screens to identify CYLD as a novel NEMO-interacting protein and negative regulator of NF-κB activation 138, 139. CYLD DUB activity is abolished by mutation of the conserved catalytic residue Cys601 139, and CYLD DUB mutants are impaired in inhibiting NF-κB signaling 139. In addition to regulating NF-κB, CYLD regulates a number of other pathways including c-Jun amino terminal kinase (JNK), p38 and retinoic acid-inducible gene I (RIG-I)-mediated innate antiviral signaling 140, 141, 142, 143.
CYLD-deficient mice have been generated by multiple groups, revealing important roles for CYLD in immune cell development and homeostasis, as well as unexpected roles in osteoclastogenesis and spermatogenesis. Cyld−/− mice exhibit reduced numbers of single positive CD4+ and CD8+ T cells in the thymus and periphery indicating an important role of CYLD in T-cell development 144. Mechanistically, in thymocytes CYLD removes polyubiquitin chains from the Src family kinase Lck and promotes an interaction with its substrate Zap70 144. Therefore, CYLD is required for T-cell receptor (TCR) signaling in thymocytes. Conversely, peripheral T cells from Cyld−/− mice are hyper-responsive to TCR stimulation and mediate spontaneous inflammation in the colon 145. Thus, CYLD has developmentally-specific roles in regulating TCR signaling. In agreement with these results, thymocyte-restricted DUB-defective CYLD mutant mice lacking exon 9 exhibit severe defects in thymocyte development 146, underscoring the importance of CYLD DUB activity in thymocyte development. CYLD also plays essential roles in the development of natural killer T (NKT) cells by providing survival signals for immature NKT cells 147.
In mature T cells, CYLD targets TAK1 for inactivation to downregulate IKK and NF-κB activation 145. Specifically, CYLD removes K63-linked polyubiquitin chains from TAK1 that leads to its inactivation (Figure 2) 145. CYLD also plays important roles in the development and activation of a variety of other immune cells. For example, B cells from Cyld−/− mice display markers indicative of activation and are hyper-responsive to activation via the B-cell receptor (BCR) 148. Although CYLD does not play a major role in B-cell development, there is a marked increase in marginal zone B cells in CYLD-deficient mice 148. CYLD prevents the spontaneous activation of NF-κB in B lymphocytes since Cyld−/− B cells exhibit constitutive IKK and NF-κB activation 148. Taken together, these studies indicate that CYLD is essential to prevent spontaneous activation of NF-κB in peripheral T and B lymphocytes and is important for the maintenance of immune homeostasis.
In addition to its crucial immune regulatory functions, CYLD also plays other important physiological roles including that in osteoclastogenesis and spermatogenesis. Cyld−/− mice develop osteoporosis as a result of enhanced osteoclast differentiation 149. CYLD-deficient osteoclast precursors are hypersensitive to RANKL signaling and produce more osteoclasts upon stimulation with RANKL 149. CYLD inhibits RANK signaling by deubiquitinating TRAF6, and CYLD requires the adaptor molecule p62 to interact with TRAF6 149.
CYLD is also an important regulator of spermatogenesis 150. In the absence of CYLD, NF-κB is turned on constitutively in testicular cells leading to the upregulation of antiapoptotic genes such as Bcl-2 and Bcl-xL and a block in the early wave of germ cell apoptosis 150. Cyld−/− testicular cells exhibit marked upregulation of RIP1 ubiquitination together with enhanced RIP1-IKK interactions 150. Therefore, CYLD-mediated deubiquitination of RIP1 is an essential step in spermatogenesis.
A number of other Cyld−/− or mutant mice have been generated further confirming the role of CYLD as an important negative regulator of NF-κB and inflammation. B and T lymphocytes and macrophages from Cyld−/− mice are hypersensitive to activating stimuli, resulting in enhanced NF-κB and JNK signaling possibly due to increased TRAF2 and NEMO ubiquitination 151. The Cyld−/− mice have increased inflammation and tumor formation in a colitis-associated cancer model 151. Another mouse strain was engineered to express a naturally occurring Cyld splice variant lacking exons 7 and 8 containing the TRAF2 and NEMO binding domains 152. Mice expressing the Cyld splice variant have enlarged spleens and secondary lymphoid organs due to a dramatic accumulation of B lymphocytes as a result of enhanced survival and expression of NF-κB and TRAF proteins 152.
Because CYLD is an important tumor suppressor in the skin, CYLD-deficient mice are more prone to chemically induced skin tumors 153. Cyld−/− keratinocytes hyperproliferate and express higher levels of cyclin D1 in response to the mitogen 12-O-tetradecanoylphorbol-13 acetate (TPA) or ultraviolet (UV) light 153. CYLD inducibly translocates to a perinuclear region where it binds to the IκB family member Bcl-3 and removes K63-linked polyubiquitin chains, thus preventing nuclear translocation of Bcl-3-p50 and Bcl-3-p52 complexes 153. Therefore, CYLD regulates Bcl-3 and cell proliferation in keratinocytes in a noncanonical pathway.
Since CYLD and A20 target many of the same substrates for deubiquitination, it is interesting to consider why no functional redundancy exists between these two proteins. It is likely that CYLD and A20 inhibit NF-κB at different times during an inflammatory response. Whereas CYLD prevents spontaneous activation of NF-κB, A20 is essential to terminate NF-κB signaling in a negative feedback loop 135. A20 is expressed at low levels in most cell types and is induced by NF-κB activating stimuli. Therefore, A20 and CYLD serve precise and temporally distinct roles in inhibiting NF-κB. It is also essential for NF-κB to overcome repression by CYLD in response to activating stimuli like TNF-α or LPS. Indeed, CYLD is phosphorylated on multiple serine residues in a stimulus-dependent manner by IKK 154. The phosphorylation is transient and is dependent on the NEMO subunit of IKK 154. Furthermore, CYLD phosphorylation is required for TRAF2 ubiquitination and downstream activation of NF-κB and JNK 154. Therefore, IKK phosphorylates CYLD to inactivate its DUB function thus providing a window of NF-κB activation prior to signal-induced termination by A20. This mechanism of CYLD inactivation has been subverted by the breast cancer oncogene IKKɛ, which phosphorylates CYLD and inactivates its DUB function 155. Presumably, inactivation of CYLD predisposes to enhanced NF-κB activation and tumorigenesis.
Another key difference between A20 and CYLD is the specificity of CYLD for K63-linked polyubiquitin chains. In vitro, A20 preferentially cleaves K48-linked polyubiquitin chains whereas CYLD cleaves both K63-linked polyubiquitin chains as well as linear ubiquitin chains 91, 117, 156. Therefore, A20 likely depends on ubiquitin-binding adaptor proteins such as TAX1BP1 and others to provide specificity. CYLD requires p62 as an adaptor molecule to interact with and deubiquitinate TRAF6 in RANK signaling in osteoclasts 149. Whether CYLD uses distinct adaptors for other substrates such as RIP1 is unknown.
The tumor suppressor role of CYLD
Cyld was originally identified as a gene mutated in familial cylindromatosis, an autosomal dominant condition characterized by the growth of benign tumors or cylindromas on skin appendages 136. Familial cylindromatosis patients usually have germline mutations in Cyld and exhibit loss of heterozygosity of the remaining allele in the skin lesions 136. The majority of the cylindromatosis mutations result in truncated CYLD proteins that lack DUB activity underscoring the importance of CYLD catalytic activity in its tumor suppressor function 157. Emerging studies suggest that CYLD may play a broader role as a tumor suppressor for multiple cancers. For example, inactivating mutations have been identified in a number of NF-κB regulatory proteins, including CYLD in multiple myeloma, a plasma cell malignancy exhibiting persistent canonical and noncanonical NF-κB signaling 158, 159. In T-cell acute lymphoblastic leukemia (T-ALL), CYLD expression is repressed by the Notch/Hes1 pathway to promote persistent IKK activation and cell survival 160. CYLD expression is also downregulated in malignant melanoma by the transcription factor Snail 161. As a result of CYLD downregulation, Bcl-3 displays persistent nuclear expression and upregulates cyclin D1 and N-cadherin, leading to enhanced proliferation and invasion of melanoma cells 161. CYLD is also downregulated in other malignancies including colon and hepatocellular carcinomas 162.
It is likely that the tumor suppressor function of CYLD may not be entirely due to NF-κB inhibition, since CYLD possesses a number of NF-κB-independent functions. An shRNA screen for mitotic regulatory proteins identified CYLD; accordingly, knockdown of CYLD by siRNA delays entry of cells into mitosis 163. This function of CYLD is independent of NF-κB although it requires its catalytic activity 163. A putative substrate of CYLD, Plk1, was also identified by mass spectrometry as a binding partner of CYLD 163. Since loss of Plk1 or CYLD have a similar cell cycle phenotype, CYLD may regulate Plk1 activity by deubiquitinating K63-linked ubiquitin chains 163. However, since CYLD regulation of the cell cycle may be considered pro-tumorigenic, it is not clear how this particular function of CYLD relates to cancer. Paradoxically, another study indicated that CYLD inhibits the mitotic kinase Aurora B by promoting its dephosphorylation via the PP2A phosphatase 164. Additional studies are necessary to determine the complex roles of CYLD in cell cycle regulation.
CYLD contains three CAP-Gly domains commonly found in microtubule-interacting proteins. Indeed, CYLD has been shown to interact with microtubules, predominantly via the first and second CAP-Gly domains 165. CYLD promotes microtubule assembly by enhancing tubulin polymerization into microtubules 165. CYLD also induces the acetylation of α-tubulin by inhibiting HDAC6 and thus counteracts the depolymerization of microtubules leading to an overall stabilization of the microtubule network 166. As a result of its microtubule assembly function, CYLD also regulates angiogenesis by mediating the spread and migration of vascular endothelial cells 167. The cell cycle regulatory function of CYLD may also be partly dependent on the modulation of microtubule stability by CYLD.
The Wnt signaling pathway is a key developmental pathway that is also linked to cancer. An siRNA screen identified CYLD as a negative regulator of Wnt signaling and β-catenin activation 168. CYLD functions upstream of β-catenin by removing K63-linked polyubiquitin chains from the adaptor molecule Dishevelled (Dvl) 168. Therefore, Wnt signaling is hyperactive in cylindromatosis skin tumors, suggesting a potential tumorigenic role in cylindromatosis. It will be interesting to examine Wnt/β-catenin signaling in other tumor types exhibiting loss of CYLD.
Other DUBs targeting NF-κB
Although A20 and CYLD clearly play vital and distinct roles in inhibiting NF-κB, other DUBs may also regulate NF-κB. For example, Cezanne is a member of the A20 family of cysteine protease DUBs 169. Similar to A20, Cezanne is rapidly upregulated upon TNF-α stimulation and recruited to the TNFR, where it inhibits RIP1 ubiquitination and downstream IKK and NF-κB signaling (Figure 2) 170. Cezanne requires its DUB domain since a catalytically inactive mutant is impaired in inhibiting NF-κB and RIP1 ubiquitination 170. A recent study indicates that Cezanne preferentially hydrolyzes K11-linked polyubiquitin chains 171. How this activity of Cezanne regulates NF-κB signaling is unknown.
Ubiquitin-specific peptidase 21 (USP21) inhibits TNF-induced NF-κB signaling by also promoting the deubiquitination of RIP1 (Figure 2) 172. However, it is unclear why several DUBs including A20, Cezanne and USP21 are necessary to remove polyubiquitin chains from RIP1. One possibility is that these proteins may be working cooperatively in a large protein complex to inhibit RIP1 polyubiquitination. Regardless, gene targeting studies are essential to confirm the in vivo roles of Cezanne and USP21 in the regulation of NF-κB signaling.
DUBs may also function downstream of RIP1 and IKK in the NF-κB signaling cascade. For example, the COP9 signalsome (CSN) inducibly interacts with IκBα and regulates IκBα stability and NF-κB activation 173. USP15 is a CSN-associated DUB that removes K48-linked polyubiquitin chains from IκBα to enhance its stability and fine-tune NF-κB activation 173.
Pathogens modulating NF-κB activity via DUBs
Given the importance of A20 and CYLD in the negative regulation of NF-κB signaling, it is not surprising that oncogenic viruses target these proteins for inactivation to promote persistent NF-κB signaling. The human T-cell leukemia virus type I (HTLV-I) is etiologically linked to the development of a CD4+CD25+ malignancy termed adult T-cell leukemia (ATL). The HTLV-I genome encodes an oncogenic regulatory protein Tax that serves as a potent activator of NF-κB 174. Tax upregulates the expression of A20 via an NF-κB binding site in the A20 promoter 175. Tax also interacts with TAX1BP1, the ubiquitin-binding adaptor molecule for A20 92, 176. Indeed, it appears that Tax interacts with TAX1BP1 as a mechanism to disrupt its functions as an NF-κB inhibitor and a coactivator for nuclear receptors 94, 176. In the presence of Tax, TAX1BP1 binding to A20 and the E3 ligase Itch is impaired 94. Furthermore, both A20 and Itch are impaired in the inducible binding to RIP1 when Tax is expressed in T cells 94. Similarly, Tax also prevents the recruitment of A20 to TRAF6 in response to IL-1 stimulation 82. Finally, Tax also prevents TNF-α-induced interaction of A20 with Ubc13 and the subsequent degradation of Ubc13 82. Taken together, it appears that Tax targets TAX1BP1, in part, to inhibit the function of A20 and promote persistent NF-κB activation.
Other oncogenic viruses may also target A20 and its adaptor molecules. TAX1BP1 interacts with the human papillomavirus virus (HPV) E2 protein and enhances E2-dependent transcription and prevents its proteasomal degradation 177. However, it is unclear if TAX1BP1 plays any role in NF-κB activation in the context of HPV infection. LMP1, the Epstein Barr Virus (EBV)-encoded oncogenic protein, induces NF-κB-dependent expression of A20 and also interacts with A20 178, 179. Although A20 displaces TRAF1 from the LMP1 complex 179, it is unclear if A20 retains NF-κB inhibitory activity in the presence of LMP1 which promotes constitutive NF-κB signaling. It is possible that LMP1 either inactivates A20 or turns on NF-κB signaling in a manner that is insensitive to the inhibition by A20. A recent study has reported that A20 deubiquitinates IRF7 in EBV-infected Raji cells, thus providing evidence that A20 is indeed functional in LMP1-expressing cells 180.
The HPV-encoded oncogenic protein E6 promotes hypoxia-induced NF-κB activation by triggering the ubiquitination and proteasome-mediated degradation of CYLD 181. Inactivation of CYLD leads to TRAF6 ubiquitination and IKK activation, thus promoting anchorage-independent growth of tumor cells 181. Therefore, E6 targeting of CYLD appears to be an essential step in HPV-mediated tumorigenesis.
Conversely, pathogens such as viruses or bacteria may encode DUBs as a mechanism to inhibit NF-κB and evade host immune responses. The Crimean Congo hemorrhagic fever virus (CCHFV) is a human nairovirus that causes hemorrhagic fever and lethality in 30% of infections 182. The CCHFV-encoded L protein contains an OTU domain in its N-terminal region 183. The OTU domain is also found in related nairoviruses Dugbe virus (DUGV) and Nairobi sheep disease virus. The L protein of CCHFV reduces levels of ubiquitinated and ISGylated proteins and downregulates TNF-α-mediated NF-κB activation, presumably as a mechanism of immune evasion 183. The bacterial pathogen Yersinia, the causative agent of plague, encodes a virulence factor YopJ that functions as a DUB for TRAF2, TRAF6 and IκBα to inhibit NF-κB and host proinflammatory responses 184. YopJ does not appear to have specificity for K63-linked polyubiquitin chains since it also cleaves K48-linked polyubiquitin chains 184. Collectively, certain pathogenic viruses and bacteria encode DUBs as a unique strategy of immune evasion by inhibiting NF-κB.
Concluding remarks
Recent years have brought dramatic advances in our understanding of the mechanisms of NF-κB regulation. DUBs such as A20 and CYLD have been shown to play prominent roles in inhibiting NF-κB. However, many outstanding questions remain regarding the specific roles of DUBs in downregulating NF-κB. For example, although it is clear that A20 relies on a number of adaptor and effector molecules to inhibit NF-κB, what is the precise function of each of the molecules in the A20 ubiquitin-editing complex? Do these molecules play tissue-specific roles in NF-κB regulation? Why are there several DUBs (i.e. A20, CYLD, Cezanne and USP21) that inhibit RIP1 ubiquitination downstream of TNFR signaling? What is the precise role of noncanonical polyubiquitin chains (i.e. K11-, K27-, K29-linked ubiquitin chains) in NF-κB signaling and are these polyubiquitin chains counteracted by specific DUBs? Does CYLD require ubiquitin-binding adaptors to inhibit RIP1 ubiquitination? A more thorough understanding of how DUBs regulate NF-κB signaling may contribute to the design of novel therapies for inflammatory disorders and cancer.
References
Vallabhapurapu S, Karin M . Regulation and function of NF-κB transcription factors in the immune system. Annu Rev Immunol 2009; 27:693–733.
Chen LF, Greene WC . Shaping the nuclear action of NF-κB. Nat Rev Mol Cell Biol 2004; 5:392–401.
Ghosh S, Hayden MS . New regulators of NF-κB in inflammation. Nat Rev Immunol 2008; 8:837–848.
Scheidereit C . IκB kinase complexes: gateways to NF-κB activation and transcription. Oncogene 2006; 25:6685–6705.
Bours V, Franzoso G, Azarenko V, et al. The oncoprotein Bcl-3 directly transactivates through κB motifs via association with DNA-binding p50 homodimers. Cell 1993; 72:729–739.
Kitamura H, Kanehira K, Okita K, Morimatsu M, Saito M . MAIL, a novel nuclear IκB protein that potentiates LPS-induced IL-6 production. FEBS Lett 2000; 485:53–56.
Beinke S, Ley SC . Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem J 2004; 382 (Pt 2):393–409.
Sun SC, Ley SC . New insights into NF-κB regulation and function. Trends Immunol 2008; 29:469–478.
Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M . The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell 1997; 91:243–252.
Yamaoka S, Courtois G, Bessia C, et al. Complementation cloning of NEMO, a component of the IκB kinase complex essential for NF-κB activation. Cell 1998; 93:1231–1240.
Rothwarf DM, Zandi E, Natoli G, Karin M . IKKγ is an essential regulatory subunit of the IκB kinase complex. Nature 1998; 395:297–300.
Li ZW, Chu W, Hu Y, et al. The IKKβ subunit of IκB kinase (IKK) is essential for NF-κB activation and prevention of apoptosis. J Exp Med 1999; 189:1839–1845.
Rudolph D, Yeh WC, Wakeham A, et al. Severe liver degeneration and lack of NF-κB activation in NEMO/IKKγ-deficient mice. Genes Dev 2000; 14:854–862.
Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U . Control of IκBα proteolysis by site-specific, signal-induced phosphorylation. Science 1995; 267:1485–1488.
Sun SC, Ganchi PA, Ballard DW, Greene WC . NF-κB controls expression of inhibitor IκBα: evidence for an inducible autoregulatory pathway. Science 1993; 259:1912–1915.
Claudio E, Brown K, Park S, Wang H, Siebenlist U . BAFF-induced NEMO-independent processing of NF-κB2 in maturing B cells. Nat Immunol 2002; 3:958–965.
Kayagaki N, Yan M, Seshasayee D, et al. BAFF/BLyS receptor 3 binds the B cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-κB2. Immunity 2002; 17:515–524.
Dejardin E, Droin NM, Delhase M, et al. The lymphotoxin-β receptor induces different patterns of gene expression via two NF-κB pathways. Immunity 2002; 17:525–535.
Coope HJ, Atkinson PG, Huhse B, et al. CD40 regulates the processing of NF-κB2 p100 to p52. EMBO J 2002; 21:5375–5385.
Xiao G, Harhaj EW, Sun SC . NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol Cell 2001; 7:401–409.
Qing G, Qu Z, Xiao G . Stabilization of basally translated NF-κB-inducing kinase (NIK) protein functions as a molecular switch of processing of NF-κB2 p100. J Biol Chem 2005; 280:40578–40582.
Liao G, Zhang M, Harhaj EW, Sun SC . Regulation of the NF-κB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem 2004; 279:26243–26250.
Zarnegar BJ, Wang Y, Mahoney DJ, et al. Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol 2008; 9:1371–1378.
Vallabhapurapu S, Matsuzawa A, Zhang W, et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat Immunol 2008; 9:1364–1370.
Senftleben U, Cao Y, Xiao G, et al. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science 2001; 293:1495–1499.
Jin X, Jin HR, Jung HS, et al. An atypical E3 ligase zinc finger protein 91 stabilizes and activates NF-κB-inducing kinase via K63-linked ubiquitination. J Biol Chem 2010; 285:30539–30547.
Yang WL, Zhang X, Lin HK . Emerging role of Lys-63 ubiquitination in protein kinase and phosphatase activation and cancer development. Oncogene 2010; 29:4493–4503.
Kerscher O, Felberbaum R, Hochstrasser M . Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol 2006; 22:159–180.
Bhoj VG, Chen ZJ . Ubiquitylation in innate and adaptive immunity. Nature 2009; 458:430–437.
Hofmann RM, Pickart CM . Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell 1999; 96:645–653.
Deng L, Wang C, Spencer E, et al. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000; 103:351–361.
Pickart CM . Mechanisms underlying ubiquitination. Annu Rev Biochem 2001; 70:503–533.
Peng J, Schwartz D, Elias JE, et al. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol 2003; 21:921–926.
d'Azzo A, Bongiovanni A, Nastasi T . E3 ubiquitin ligases as regulators of membrane protein trafficking and degradation. Traffic 2005; 6:429–441.
Kirisako T, Kamei K, Murata S, et al. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J 2006; 25:4877–4887.
Tokunaga F, Sakata S, Saeki Y, et al. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat Cell Biol 2009; 11:123–132.
Matsumoto ML, Wickliffe KE, Dong KC, et al. K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol Cell 2010; 39:477–484.
Ikeda H, Kerppola TK . Lysosomal localization of ubiquitinated Jun requires multiple determinants in a lysine-27-linked polyubiquitin conjugate. Mol Biol Cell 2008; 19:4588–4601.
Chastagner P, Israel A, Brou C . AIP4/Itch regulates Notch receptor degradation in the absence of ligand. PLoS ONE 2008; 3:e2735.
Skaug B, Jiang X, Chen ZJ . The role of ubiquitin in NF-κB regulatory pathways. Annu Rev Biochem 2009; 78:769–796.
Reyes-Turcu FE, Ventii KH, Wilkinson KD . Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem 2009; 78:363–397.
Chung JY, Park YC, Ye H, Wu H . All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J Cell Sci 2002; 115:679–688.
Tsitsikov EN, Laouini D, Dunn IF, et al. TRAF1 is a negative regulator of TNF signaling. enhanced TNF signaling in TRAF1-deficient mice. Immunity 2001; 15:647–657.
Lomaga MA, Yeh WC, Sarosi I, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev 1999; 13:1015–1024.
Wan YY, Chi H, Xie M, Schneider MD, Flavell RA . The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat Immunol 2006; 7:851–858.
Sato S, Sanjo H, Takeda K, et al. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol 2005; 6:1087–1095.
Yamamoto M, Okamoto T, Takeda K, et al. Key function for the Ubc13 E2 ubiquitin-conjugating enzyme in immune receptor signaling. Nat Immunol 2006; 7:962–970.
Fukushima T, Matsuzawa S, Kress CL, et al. Ubiquitin-conjugating enzyme Ubc13 is a critical component of TNF receptor-associated factor (TRAF)-mediated inflammatory responses. Proc Natl Acad Sci USA 2007; 104:6371–6376.
Verstrepen L, Bekaert T, Chau TL, et al. TLR-4, IL-1R and TNF-R signaling to NF-κB: variations on a common theme. Cell Mol Life Sci 2008; 65:2964–2978.
Wang C, Deng L, Hong M, et al. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001; 412:346–351.
Walsh MC, Kim GK, Maurizio PL, Molnar EE, Choi Y . TRAF6 autoubiquitination-independent activation of the NF-κB and MAPK pathways in response to IL-1 and RANKL. PLoS ONE 2008; 3:e4064.
Conze DB, Wu CJ, Thomas JA, Landstrom A, Ashwell JD . Lys63-linked polyubiquitination of IRAK-1 is required for IL-1R- and TLR-mediated NF-κB activation. Mol Cell Biol 2008; 28:3538–3547.
Windheim M, Stafford M, Peggie M, Cohen P . Interleukin-1 (IL-1) induces the Lys63-linked polyubiquitination of IL-1 receptor-associated kinase 1 to facilitate NEMO binding and the activation of IKK. Mol Cell Biol 2008; 28:1783–1791.
Xia ZP, Sun L, Chen X, et al. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature 2009; 461:114–119.
Zeng W, Sun L, Jiang X, et al. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 2010; 141:315–330.
Micheau O, Tschopp J . Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003; 114:181–190.
Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ . Activation of IKK by TNFα requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell 2006; 22:245–257.
Li H, Kobayashi M, Blonska M, You Y, Lin X . Ubiquitination of RIP is required for TNFα-induced NF-κB activation. J Biol Chem 2006; 281:13636–13643.
Yeh WC, Shahinian A, Speiser D, et al. Early lethality, functional NF-κB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity 1997; 7:715–725.
Tada K, Okazaki T, Sakon S, et al. Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-κB activation and protection from cell death. J Biol Chem 2001; 276:36530–36534.
Bertrand MJ, Milutinovic S, Dickson KM, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 2008; 30:689–700.
Takaesu G, Surabhi RM, Park KJ, et al. TAK1 is critical for IκB kinase-mediated activation of the NF-κB pathway. J Mol Biol 2003; 326:105–115.
Kanayama A, Seth RB, Sun L, et al. TAB2 and TAB3 activate the NF-κB pathway through binding to polyubiquitin chains. Mol Cell 2004; 15:535–548.
Opipari AW Jr, Boguski MS, Dixit VM . The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J Biol Chem 1990; 265:14705–14708.
Tewari M, Wolf FW, Seldin MF, et al. Lymphoid expression and regulation of A20, an inhibitor of programmed cell death. J Immunol 1995; 154:1699–1706.
Evans PC, Ovaa H, Hamon M, et al. Zinc-finger protein A20, a regulator of inflammation and cell survival, has de-ubiquitinating activity. Biochem J 2004; 378:727–734.
Wertz IE, O'Rourke KM, Zhou H, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 2004; 430:694–699.
Opipari AW Jr, Hu HM, Yabkowitz R, Dixit VM . The A20 zinc finger protein protects cells from tumor necrosis factor cytotoxicity. J Biol Chem 1992; 267:12424–12427.
Song HY, Rothe M, Goeddel DV . The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-κB activation. Proc Natl Acad Sci USA 1996; 93:6721–6725.
Heyninck K, De Valck D, Vanden Berghe W, et al. The zinc finger protein A20 inhibits TNF-induced NF-κB-dependent gene expression by interfering with a RIP- or TRAF2-mediated transactivation signal and directly binds to a novel NF-κB-inhibiting protein ABIN. J Cell Biol 1999; 145:1471–1482.
Jaattela M, Mouritzen H, Elling F, Bastholm L . A20 zinc finger protein inhibits TNF and IL-1 signaling. J Immunol 1996; 156:1166–1173.
Lee EG, Boone DL, Chai S, et al. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science 2000; 289:2350–2354.
Boone DL, Turer EE, Lee EG, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol 2004; 5:1052–1060.
Kawai T, Akira S . Signaling to NF-κB by Toll-like receptors. Trends Mol Med 2007; 13:460–469.
Turer EE, Tavares RM, Mortier E, et al. Homeostatic MyD88-dependent signals cause lethal inflammation in the absence of A20. J Exp Med 2008; 205:451–464.
Vereecke L, Sze M, Guire CM, et al. Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. J Exp Med 2010; 207:1513–1523.
Tavares RM, Turer EE, Liu CL, et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity 2010; 33:181–191.
Sarma V, Lin Z, Clark L, et al. Activation of the B-cell surface receptor CD40 induces A20, a novel zinc finger protein that inhibits apoptosis. J Biol Chem 1995; 270:12343–12346.
Graham RR, Cotsapas C, Davies L, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet 2008; 40:1059–1061.
Li L, Hailey DW, Soetandyo N, et al. Localization of A20 to a lysosome-associated compartment and its role in NF-κB signaling. Biochim Biophys Acta 2008; 1783:1140–1149.
Li L, Soetandyo N, Wang Q, Ye Y . The zinc finger protein A20 targets TRAF2 to the lysosomes for degradation. Biochim Biophys Acta 2009; 1793:346–353.
Shembade N, Ma A, Harhaj EW . Inhibition of NF-κB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 2010; 327:1135–1139.
Hasegawa M, Fujimoto Y, Lucas PC, et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-κB activation. EMBO J 2008; 27:373–383.
Hitotsumatsu O, Ahmad RC, Tavares R, et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity 2008; 28:381–390.
Tao M, Scacheri PC, Marinis JM, et al. ITCH K63-ubiquitinates the NOD2 binding protein, RIP2, to influence inflammatory signaling pathways. Curr Biol 2009; 19:1255–1263.
Stilo R, Varricchio E, Liguoro D, Leonardi A, Vito P . A20 is a negative regulator of BCL10- and CARMA3-mediated activation of NF-κB. J Cell Sci 2008; 121:1165–1171.
Coornaert B, Baens M, Heyninck K, et al. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-κB inhibitor A20. Nat Immunol 2008; 9:263–271.
Duwel M, Welteke V, Oeckinghaus A, et al. A20 negatively regulates T cell receptor signaling to NF-κB by cleaving Malt1 ubiquitin chains. J Immunol 2009; 182:7718–7728.
Ferch U, Kloo B, Gewies A, et al. Inhibition of MALT1 protease activity is selectively toxic for activated B cell-like diffuse large B cell lymphoma cells. J Exp Med 2009; 206:2313–2320.
Song XT, Evel-Kabler K, Shen L, et al. A20 is an antigen presentation attenuator, and its inhibition overcomes regulatory T cell-mediated suppression. Nat Med 2008; 14:258–265.
Lin SC, Chung JY, Lamothe B, et al. Molecular basis for the unique deubiquitinating activity of the NF-κB inhibitor A20. J Mol Biol 2008; 376:526–540.
Gachon F, Peleraux A, Thebault S, et al. CREB-2, a cellular CRE-dependent transcription repressor, functions in association with Tax as an activator of the human T-cell leukemia virus type 1 promoter. J Virol 1998; 72:8332–8337.
De Valck D, Jin DY, Heyninck K, et al. The zinc finger protein A20 interacts with a novel anti-apoptotic protein which is cleaved by specific caspases. Oncogene 1999; 18:4182–4190.
Shembade N, Harhaj NS, Liebl DJ, Harhaj EW . Essential role for TAX1BP1 in the termination of TNF-α-, IL-1- and LPS-mediated NF-κB and JNK signaling. EMBO J 2007; 26:3910–3922.
Iha H, Peloponese JM, Verstrepen L, et al. Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective NF-κB activation. EMBO J 2008; 27:629–641.
Gurung R, Tan A, Ooms LM, et al. Identification of a novel domain in two mammalian inositol-polyphosphate 5-phosphatases that mediates membrane ruffle localization. The inositol 5-phosphatase skip localizes to the endoplasmic reticulum and translocates to membrane ruffles following epidermal growth factor stimulation. J Biol Chem 2003; 278:11376–11385.
Shembade N, Harhaj NS, Parvatiyar K, et al. The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat Immunol 2008; 9:254–262.
Sudol M, Chen HI, Bougeret C, Einbond A, Bork P . Characterization of a novel protein-binding module--the WW domain. FEBS Lett 1995; 369:67–71.
Matesic LE, Copeland NG, Jenkins NA . Itchy mice: the identification of a new pathway for the development of autoimmunity. Curr Top Microbiol Immunol 2008; 321:185–200.
Perry WL, Hustad CM, Swing DA, et al. The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18H mice. Nat Genet 1998; 18:143–146.
Subramaniam V, Li H, Wong M, et al. The RING-H2 protein RNF11 is overexpressed in breast cancer and is a target of Smurf2 E3 ligase. Br J Cancer 2003; 89:1538–1544.
Kitching R, Wong MJ, Koehler D, et al. The RING-H2 protein RNF11 is differentially expressed in breast tumours and interacts with HECT-type E3 ligases. Biochim Biophys Acta 2003; 1639:104–112.
Azmi P, Seth A . RNF11 is a multifunctional modulator of growth factor receptor signalling and transcriptional regulation. Eur J Cancer 2005; 41:2549–2560.
Li H, Seth A . An RNF11: Smurf2 complex mediates ubiquitination of the AMSH protein. Oncogene 2004; 23:1801–1808.
Colland F, Jacq X, Trouplin V, et al. Functional proteomics mapping of a human signaling pathway. Genome Res 2004; 14:1324–1332.
Shembade N, Parvatiyar K, Harhaj NS, Harhaj EW . The ubiquitin-editing enzyme A20 requires RNF11 to downregulate NF-κB signalling. EMBO J 2009; 28:513–522.
Heyninck K, Kreike MM, Beyaert R . Structure-function analysis of the A20-binding inhibitor of NF-κB activation, ABIN-1. FEBS Lett 2003; 536:135–140.
Mauro C, Pacifico F, Lavorgna A, et al. ABIN-1 binds to NEMO/IKKγ and co-operates with A20 in inhibiting NF-κB. J Biol Chem 2006; 281:18482–18488.
Ashida H, Kim M, Schmidt-Supprian M, et al. A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKγ to dampen the host NF-κB-mediated inflammatory response. Nat Cell Biol 2010; 12:66–73; sup pp 61–69.
Oshima S, Turer EE, Callahan JA, et al. ABIN-1 is a ubiquitin sensor that restricts cell death and sustains embryonic development. Nature 2009; 457:906–909.
Bohgaki M, Tsukiyama T, Nakajima A, et al. Involvement of Ymer in suppression of NF-κB activation by regulated interaction with lysine-63-linked polyubiquitin chain. Biochim Biophys Acta 2008; 1783:826–837.
Fenner BJ, Scannell M, Prehn JH . Identification of polyubiquitin binding proteins involved in NF-κB signaling using protein arrays. Biochim Biophys Acta 2009; 1794:1010–1016.
Komander D, Barford D . Structure of the A20 OTU domain and mechanistic insights into deubiquitination. Biochem J 2008; 409:77–85.
Wang YY, Li L, Han KJ, Zhai Z, Shu HB . A20 is a potent inhibitor of TLR3- and Sendai virus-induced activation of NF-κB and ISRE and IFN-β promoter. FEBS Lett 2004; 576:86–90.
Parvatiyar K, Barber GN, Harhaj EW . TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J Biol Chem 2010; 285:14999–15009.
Lin R, Yang L, Nakhaei P, et al. Negative regulation of the retinoic acid-inducible gene I-induced antiviral state by the ubiquitin-editing protein A20. J Biol Chem 2006; 281:2095–2103.
Komander D, Reyes-Turcu F, Licchesi JD, et al. Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep 2009; 10:466–473.
Hutti JE, Turk BE, Asara JM, et al. IKKβ phosphorylates the K63 deubiquitinase A20 to cause feedback inhibition of the NF-κB pathway. Mol Cell Biol 2007; 27:7451–7461.
Musone SL, Taylor KE, Lu TT, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet 2008; 40:1062–1064.
Fung EY, Smyth DJ, Howson JM, et al. Analysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/TNFAIP3 as a susceptibility locus. Genes Immun 2009; 10:188–191.
Trynka G, Zhernakova A, Romanos J, et al. Coeliac disease-associated risk variants in TNFAIP3 and REL implicate altered NF-κB signalling. Gut 2009; 58:1078–1083.
Wang K, Baldassano R, Zhang H, et al. Comparative genetic analysis of inflammatory bowel disease and type 1 diabetes implicates multiple loci with opposite effects. Hum Mol Genet 2010; 19:2059–2067.
Boonyasrisawat W, Eberle D, Bacci S, et al. Tag polymorphisms at the A20 (TNFAIP3) locus are associated with lower gene expression and increased risk of coronary artery disease in type 2 diabetes. Diabetes 2007; 56:499–505.
Nair RP, Duffin KC, Helms C, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-κB pathways. Nat Genet 2009; 41:199–204.
Lodolce JP, Kolodziej LE, Rhee L, et al. African-derived genetic polymorphisms in TNFAIP3 mediate risk for autoimmunity. J Immunol 2010; 184:7001–7009.
Thelander EF, Ichimura K, Corcoran M, et al. Characterization of 6q deletions in mature B cell lymphomas and childhood acute lymphoblastic leukemia. Leuk Lymphoma 2008; 49:477–487.
Honma K, Tsuzuki S, Nakagawa M, et al. TNFAIP3/A20 functions as a novel tumor suppressor gene in several subtypes of non-Hodgkin lymphomas. Blood 2009; 114:2467–2475.
Kato M, Sanada M, Kato I, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature 2009; 459:712–716.
Compagno M, Lim WK, Grunn A, et al. Mutations of multiple genes cause deregulation of NF-κB in diffuse large B-cell lymphoma. Nature 2009; 459:717–721.
Schmitz R, Hansmann ML, Bohle V, et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med 2009; 206:981–989.
Chanudet E, Huang Y, Ichimura K, et al. A20 is targeted by promoter methylation, deletion and inactivating mutation in MALT lymphoma. Leukemia 2010; 24:483–487.
Huang HL, Yeh WC, Lai MZ, et al. Impaired TNFα-induced A20 expression in E1A/Ras-transformed cells. Br J Cancer 2009; 101:1555–1564.
Vendrell JA, Ghayad S, Ben-Larbi S, et al. A20/TNFAIP3, a new estrogen-regulated gene that confers tamoxifen resistance in breast cancer cells. Oncogene 2007; 26:4656–4667.
Hjelmeland AB, Wu Q, Wickman S, et al. Targeting A20 decreases glioma stem cell survival and tumor growth. PLoS Biol 2010; 8:e1000319.
Sun SC . Deubiquitylation and regulation of the immune response. Nat Rev Immunol 2008; 8:501–511.
Bignell GR, Warren W, Seal S, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet 2000; 25:160–165.
Brummelkamp TR, Nijman SM, Dirac AM, Bernards R . Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-κB. Nature 2003; 424:797–801.
Kovalenko A, Chable-Bessia C, Cantarella G, et al. The tumour suppressor CYLD negatively regulates NF-κB signaling by deubiquitination. Nature 2003; 424:801–805.
Trompouki E, Hatzivassiliou E, Tsichritzis T, et al. CYLD is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature 2003; 424:793–796.
Reiley W, Zhang M, Sun SC . Negative regulation of JNK signaling by the tumor suppressor CYLD. J Biol Chem 2004; 279:55161–55167.
Lim JH, Stirling B, Derry J, et al. Tumor suppressor CYLD regulates acute lung injury in lethal Streptococcus pneumoniae infections. Immunity 2007; 27:349–360.
Friedman CS, O'Donnell MA, Legarda-Addison D, et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep 2008; 9:930–936.
Zhang M, Wu X, Lee AJ, et al. Regulation of IκB kinase-related kinases and antiviral responses by tumor suppressor CYLD. J Biol Chem 2008; 283:18621–18626.
Reiley WW, Zhang M, Jin W, et al. Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat Immunol 2006; 7:411–417.
Reiley WW, Jin W, Lee AJ, et al. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J Exp Med 2007; 204:1475–1485.
Tsagaratou A, Trompouki E, Grammenoudi S, Kontoyiannis DL, Mosialos G . Thymocyte-specific truncation of the deubiquitinating domain of CYLD impairs positive selection in a NF-κB essential modulator-dependent manner. J Immunol 2010; 185:2032–2043.
Lee AJ, Zhou X, Chang M, et al. Regulation of natural killer T-cell development by deubiquitinase CYLD. EMBO J 2010; 29:1600–1612.
Jin W, Reiley WR, Lee AJ, et al. Deubiquitinating enzyme CYLD regulates the peripheral development and naive phenotype maintenance of B cells. J Biol Chem 2007; 282:15884–15893.
Jin W, Chang M, Paul EM, et al. Deubiquitinating enzyme CYLD negatively regulates RANK signaling and osteoclastogenesis in mice. J Clin Invest 2008; 118:1858–1866.
Wright A, Reiley WW, Chang M, et al. Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev Cell 2007; 13:705–716.
Zhang J, Stirling B, Temmerman ST, et al. Impaired regulation of NF-κB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J Clin Invest 2006; 116:3042–3049.
Hovelmeyer N, Wunderlich FT, Massoumi R, et al. Regulation of B cell homeostasis and activation by the tumor suppressor gene CYLD. J Exp Med 2007; 204:2615–2627.
Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R . CYLD inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-κB signaling. Cell 2006; 125:665–677.
Reiley W, Zhang M, Wu X, Granger E, Sun SC . Regulation of the deubiquitinating enzyme CYLD by IKKγ-dependent phosphorylation. Mol Cell Biol 2005; 25:3886–3895.
Hutti JE, Shen RR, Abbott DW, et al. Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKɛ promotes cell transformation. Mol Cell 2009; 34:461–472.
Komander D, Lord CJ, Scheel H, et al. The structure of the CYLD USP domain explains its specificity for Lys63-linked polyubiquitin and reveals a B box module. Mol Cell 2008; 29:451–464.
Saggar S, Chernoff KA, Lodha S, et al. CYLD mutations in familial skin appendage tumours. J Med Genet 2008; 45:298–302.
Annunziata CM, Davis RE, Demchenko Y, et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007; 12:115–130.
Keats JJ, Fonseca R, Chesi M, et al. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell 2007; 12:131–144.
Espinosa L, Cathelin S, D'Altri T, et al. The Notch/Hes1 pathway sustains NF-κB activation through CYLD repression in T cell leukemia. Cancer Cell 2010; 18:268–281.
Massoumi R, Kuphal S, Hellerbrand C, et al. Down-regulation of CYLD expression by Snail promotes tumor progression in malignant melanoma. J Exp Med 2009; 206:221–232.
Hellerbrand C, Bumes E, Bataille F, et al. Reduced expression of CYLD in human colon and hepatocellular carcinomas. Carcinogenesis 2007; 28:21–27.
Stegmeier F, Sowa ME, Nalepa G, et al. The tumor suppressor CYLD regulates entry into mitosis. Proc Natl Acad Sci USA 2007; 104:8869–8874.
Sun L, Gao J, Huo L, et al. Tumour suppressor CYLD is a negative regulator of the mitotic kinase Aurora-B. J Pathol 2010; 221:425–432.
Gao J, Huo L, Sun X, et al. The tumor suppressor CYLD regulates microtubule dynamics and plays a role in cell migration. J Biol Chem 2008; 283:8802–8809.
Wickstrom SA, Masoumi KC, Khochbin S, Fassler R, Massoumi R . CYLD negatively regulates cell-cycle progression by inactivating HDAC6 and increasing the levels of acetylated tubulin. EMBO J 2010; 29:131–144.
Gao J, Sun L, Huo L, et al. CYLD regulates angiogenesis by mediating vascular endothelial cell migration. Blood 2010; 115:4130–4137.
Tauriello DV, Haegebarth A, Kuper I, et al. Loss of the tumor suppressor CYLD enhances Wnt/β-catenin signaling through K63-linked ubiquitination of Dvl. Mol Cell 2010; 37:607–619.
Evans PC, Taylor ER, Coadwell J, et al. Isolation and characterization of two novel A20-like proteins. Biochem J 2001; 357:617–623.
Enesa K, Zakkar M, Chaudhury H, et al. NF-κB suppression by the deubiquitinating enzyme Cezanne: a novel negative feedback loop in pro-inflammatory signaling. J Biol Chem 2008; 283:7036–7045.
Bremm A, Freund SM, Komander D . Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat Struct Mol Biol 2010; 17:939–947.
Xu G, Tan X, Wang H, et al. Ubiquitin-specific peptidase 21 inhibits TNF-α-induced NF-κB activation via binding to and deubiquitinating receptor-interacting protein 1. J Biol Chem 2010; 285:969–978.
Schweitzer K, Bozko PM, Dubiel W, Naumann M . CSN controls NF-κB by deubiquitinylation of IκBα. EMBO J 2007; 26:1532–1541.
Harhaj EW, Harhaj NS . Mechanisms of persistent NF-κB activation by HTLV-I tax. IUBMB Life 2005; 57:83–91.
Laherty CD, Perkins ND, Dixit VM . Human T cell leukemia virus type I Tax and phorbol 12-myristate 13-acetate induce expression of the A20 zinc finger protein by distinct mechanisms involving nuclear factor κB. J Biol Chem 1993; 268:5032–5039.
Chin KT, Chun AC, Ching YP, Jeang KT, Jin DY . Human T-cell leukemia virus oncoprotein Tax represses nuclear receptor-dependent transcription by targeting coactivator TAX1BP1. Cancer Res 2007; 67:1072–1081.
Wang X, Naidu SR, Sverdrup F, Androphy EJ . TAX1BP1 interacts with papillomavirus E2 and regulates E2-dependent transcription and stability. J Virol 2009; 83:2274–2284.
Laherty CD, Hu HM, Opipari AW, Wang F, Dixit VM . The Epstein-Barr virus LMP1 gene product induces A20 zinc finger protein expression by activating nuclear factor κB. J Biol Chem 1992; 267:24157–24160.
Fries KL, Miller WE, Raab-Traub N . The A20 protein interacts with the Epstein-Barr virus latent membrane protein 1 (LMP1) and alters the LMP1/TRAF1/TRADD complex. Virology 1999; 264:159–166.
Ning S, Pagano JS . The A20 deubiquitinase activity negatively regulates LMP1 activation of IRF7. J Virol 2010; 84:6130–6138.
An J, Mo D, Liu H, et al. Inactivation of the CYLD deubiquitinase by HPV E6 mediates hypoxia-induced NF-κB activation. Cancer Cell 2008; 14:394–407.
Whitehouse CA . Crimean-Congo hemorrhagic fever. Antiviral Res 2004; 64:145–160.
Frias-Staheli N, Giannakopoulos NV, Kikkert M, et al. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe 2007; 2:404–416.
Zhou H, Monack DM, Kayagaki N, et al. Yersinia virulence factor YopJ acts as a deubiquitinase to inhibit NF-κB activation. J Exp Med 2005; 202:1327–1332.
Acknowledgements
We apologize for omitting any relevant references due to space limitations. The laboratory of EWH is supported by NIH grants PO1CA128115, RO1CA135362 and RO1GM083143.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Harhaj, E., Dixit, V. Deubiquitinases in the regulation of NF-κB signaling. Cell Res 21, 22–39 (2011). https://doi.org/10.1038/cr.2010.166
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2010.166
