
Abstract
Cellular energy metabolism not only promotes tumor cell growth and metastasis but also directs immune cell survival, proliferation and the ability to perform specific and functional immune responses within the tumor microenvironment. A better understanding of the molecular regulation of metabolism in different cell components in the tumor-suppressive microenvironment is critical for the development of effective strategies for human cancer treatments. Toll-like receptors (TLRs) have recently been recognized as critical factors involved in tumor pathogenesis, regulating both tumor cells and tumor-infiltrating innate and adaptive immune cells. However, little is known about the molecular crosstalk between TLR signaling and tumor or/and immune cell metabolism, although there is abundant expression of TLRs in these cells. In this review, we explore the functional role of TLR signaling in reprogramming cell metabolism in the tumor microenvironment. In particular, we discuss how malignant tumors regulate metabolism to support their growth and survival, summarize more recently identified metabolic profiles of different immune cell subsets and TLR-mediated regulation of cellular metabolism in both tumor and immune cells, and further explore potential strategies targeting cell metabolism for TLR-based cancer therapy. An improved understanding of these issues should open new avenues for the development of novel strategies via TLR-mediated metabolic reprogramming of the tumor microenvironment for cancer immunotherapy.
Similar content being viewed by others
Introduction
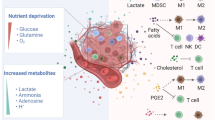
It is well established that the Warburg effect of aerobic glycolysis is a key metabolic hallmark of cancer. Malignant tumor cells selectively reprogram their metabolism to meet the rapid energy requirements for proliferation, survival and metastasis, as well as for sustaining the tumor-suppressive microenvironment.1,2,3,4,5 In addition to tumor cells, it is now well recognized that metabolic reprogramming occurs in all cell components within the tumor microenvironment that have hypoxic and acidotic conditions, including dendritic cells (DCs), macrophages and T cells.5,6 During normal activation, DCs and macrophages undergo a metabolic switch to glycolysis instead of oxidative phosphorylation (OXPHOS), which also directs the macrophage M1 or M2 phenotype switch.7,8,9 Tumor-associated macrophages (TAMs) have increased aerobic glycolysis with an inflammatory phenotype and protumorigenic effect.10,11,12 However, tumor-derived DCs with tolerogenic functions have inhibited glycolysis but increased lipid accumulation.13,14 Cellular energy metabolism also directs T-cell survival, proliferation and their specific functions.15,16,17,18,19 Aerobic glycolysis is the main metabolic pathway and is specifically required for effector function in T cells.4,15,19,20,21,22 By contrast, lipid oxidation has been found to be a primary metabolic pathway for induced Treg cells and memory CD8+ T cells.20,23,24,25 However, very limited information is known about how malignant tumors and tumor-derived microenvironmental factors affect different types of tumor-infiltrating T-cell (TIL) fate and metabolism. Recent studies have suggested that tumor cells can compete for glucose or restrict glycolysis in TILs within the tumor microenvironment, resulting in T-cell dysfunction and immune suppression.4,26,27,28 A better understanding of the molecular interactions and metabolic profiles of tumor cells and different types of immune cells within the tumor-suppressive microenvironment will facilitate the development of novel strategies for cancer therapy via metabolic reprogramming of cell fate and functions.29
Toll-like receptors (TLRs), the most well-known pattern recognition receptors, recognize molecules from invading pathogens and are important for innate immunity and inflammatory responses. It is now well recognized that TLR-mediated metabolic reprogramming is important for the activation, maturation and immunogenic functions of macrophages and DCs.7,30,31,32,33,34,35 TLRs are also widely expressed on different types of tumor cells, regulating tumor growth and functions.36,37,38 Studies from multiple preclinical animal models and clinical trials have shown that either promotion or inhibition of tumor survival and growth is mediated by different TLR stimulations in tumor cells.36,39,40,41 Recent studies suggest that TLRs directly regulate metabolism, affecting tumor behavior and function in melanoma, prostate, head and neck carcinoma and breast cancer.38,42,43,44 Furthermore, several TLR ligands, including imiquimod (TLR7) and CpG (TLR9), have shown significant promise for cancer treatment.39,45 Increasing evidence suggests that TLR signaling also affects the differentiation and function of different T-cell subsets, including Th1, Th2 and Th17 cells.46,47,48 Furthermore, human TLR8 signaling can directly reverse the suppressive function of tumor-derived CD4+, CD8+ and γδ Treg cells.49,50,51,52,53 However, whether and how TLR signaling regulates T-cell metabolism is still under investigation. Understanding functional and metabolic regulation in tumor cells and tumor-infiltrating immune cells mediated by different TLRs will be important for the development of novel TLR-based cancer immunotherapy.
Energy metabolism directs tumor growth and metastasis
In the tumor microenvironment, tumors prefer to use aerobic glycolysis instead of OXPHOS to meet their demands for growth and proliferation, especially for rapid ATP generation and biosynthesis54 (Table 1). During tumor growth with hypoxic stress in the microenvironment, hypoxia-inducible transcription factor (HIF) has a critical role in promoting the transcriptional program for this metabolic switch. HIF-1α induces expression of lactate dehydrogenase and pyruvate dehydrogenase kinase and facilitates the metabolic switch by binding to glucose transporter 1 (Glut1) and other glycolytic enzymes. This results in increased lactate production from pyruvate and limited acetyl-CoA production for the tricarboxylic acid (TCA) cycle promotion through inhibiting pyruvate dehydrogenase.55,56 The Warburg effect of cancer glycolysis occurs even in the presence of completely functional mitochondria, which significantly benefits cancer cell biosynthesis and survival. Aerobic glycolysis can also increase rapidly and maintain a higher ratio of ATP/ADP to assist the tumor to avoid apoptosis and promote proliferation than that obtained from mitochondrial respiration.2,3 In addition, glycolysis-derived metabolites, such as palmitate, NADPH and Acetyl-CoA, promote fatty-acid synthesis, cell membrane constitution and macromolecular precursor biosynthesis. Furthermore, the high rates of glycolysis in cancer cells also help their competition with TILs in the tumor microenvironment for the limited glucose, resulting in suppression of the TIL effector function and maintenance of a suppressive microenvironment.4,5 In addition to the involvement of the transcription factor HIF, many oncogenes directly regulate the metabolic rewriting of cancer cells. For example, mutated KRAS drives metabolic reprogramming of lung cancer and nutrient uptake.57 Mutated c-Myc increases the transcriptional activities of enzymes in glycolysis and glutaminolysis, contributing to tumor growth and chemoresistance.58,59 p53 expression in tumor cells also controls metabolic genes and alters glucose utilization.60 In addition to their dominant glycolysis and increased glucose metabolism, malignant tumor cells may utilize other metabolic pathways, such as fatty-acid oxidation and cholesterol synthesis, to meet their requirements for synthesizing macromolecules, such as nucleic acids, lipids and proteins, during their growth and metastasis processes.61,62 Therefore, an improved understanding of the molecular processes of tumor metabolism will facilitate the design of effective therapeutics that specifically target cancer metabolic programming.
Tumor-derived metabolites maintain a potent tumor-suppressive microenvironment
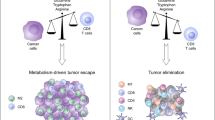
Malignant tumors display heightened glucose and glutamine consumption, resulting in the depletion of nutrients and competition with different types of tumor-infiltrating immune cells.4,5 Meanwhile, metabolic end products are also accumulated within the tumor microenvironment, including cyclic adenosine monophosphate (cAMP), indoleamine 2, 3-dioxygenase (IDO), adenosine and lactate.63 These hypoxia-derived metabolites are potent immune suppressors that can protect tumor cells from T-cell-mediated antitumor immune responses, which is one of the strategies utilized by tumor cells to create an immunosuppressive micromilieu and escape the host immune system.63,64,65 Lactate is the main metabolite of glycolysis utilized by malignant tumor cells (Warburg effect).66,67 Increased lactate production supports NAD+ regeneration in the absence of oxygen consumption and may provide other benefits to tumor cells related to altered pH, which leads to an acidified tumor microenvironment and cancer cell invasion.68 Tumor-derived lactate blocks differentiation and activation of monocytes and promotes M2 TAM polarization.69,70 Furthermore, intracellular lactate can trigger T cell and NK cell suppression and impair their tumor immunosurveillance functions.71,72 More recent studies have indicated that tumor-derived lactate promotes naive T-cell apoptosis through suppression of FAK family-interacting of 200 kDa (FIP200) and autophagy in ovarian cancer patients.28 cAMP is also a critical component of the tumor-induced hypoxic microenvironment and is a potent inhibitor of effector tumor-specific T cells.63 Furthermore, cAMP is involved in Treg-mediated suppression and is a potent inhibitor of interleukin (IL)-2 production and subsequent CD4+ T-cell proliferation.73,74 Recent studies have demonstrated that different types of tumor cells can directly induce conversion from naive/effector T cells to senescent T cells with potent suppressive activity.38,44 These studies have further identified that high concentrations of cAMP exist in tumor cells and tumor-induced senescent T cells and that tumor-derived endogenous cAMP is responsible for the induction of T-cell senescence.38,44 Adenosine is another important metabolite in tumor hypoxic microenvironments.63,75 Tumor-produced adenosine triggers immunosuppressive signaling via intracellular cyclic AMP, elevating A2A adenosine receptors on antitumor T cells. Furthermore, tumor-infiltrating Treg cells undergo apoptosis and generate adenosine to suppress T-cell-mediated tumor immunity through the A2A pathway.75 IDO expressed in tumors depletes tryptophan and inhibits T-cell proliferation.76 A better definition of the mechanistic links between tumor immunosuppression, hypoxia and metabolic dysregulation should lead to novel and alternative strategies that are capable of augmenting immune responses directly against cancer.
Metabolic regulation is crucial for immune cell development and functions
Metabolic programming also occurs in immune cell components in the tumor microenvironment. The Warburg effect is utilized by innate immune cells, including macrophages and DCs (Table 1). Activation of macrophages with different types of stimuli, such as TLR ligands, IFN-γ and certain pathogens, drives a metabolic switch from OXPHOS to glycolysis.7,30,31,32,33 Macrophages are classified into two groups, M1 and M2, based on the stimuli and their functions. M1 macrophages activated by classical stimuli, such as TLR ligands and bacteria, possess an inflammatory phenotype and affect an antitumor function. M2 microphages, polarized by IL-4 and IL-13, have an anti-inflammatory phenotype and have pro-tumoral activity.33,77 Recent studies have clearly shown that M1 and M2 macrophages have different metabolic profiles. M1 macrophages rely on glycolysis for ATP production and their increased fatty-acid synthesis, whereas M2 macrophages are supported by fatty-acid uptake and OXPHOS in the mitochondria to produce ATP.7,33 In addition, M1 macrophages have increased levels of inducible nitric oxide synthase (iNOS), HIF-1α and u-Phosphofructokinase 2, whereas M2 macrophages have increased levels of arginase 1, AMP-activated protein kinase and 6-phosphofructo-2-kinase/fructose-2,6-biSphosphatase 1.7,33 Macrophages are dominant innate immune cells in the tumor-suppressive microenvironment. TAMs have increased aerobic glycolysis with an inflammatory phenotype and pro-tumorigenic effects, which might be induced by enhanced HIF-1α.10,11,12 DCs have a very similar metabolic profile as macrophages. DCs activated by TLR agonists undergo a marked metabolic reprograming that is characterized by a switch from mitochondrial lipid oxidation and OXPHOs to increased aerobic glycolysis.7,30,34,35 Therefore, glycolysis is also important for DC activation, maturation and immunogenic functions. However, tolerogenic DCs, similar to M2 macrophages, have a distinct metabolic profile from immunogenic DCs with increased mitochondrial metabolism and OXPHOS.34,78 Tolerogenic DCs display immature and inactivated phenotypes and favor Treg induction and immune suppression.79,80 In addition, tumor-derived DCs with tolerogenic functions have inhibited glycolysis, but increased lipid accumulation, resulting in impaired APC functions and T-cell priming.13,14
Significant progress has been made in understanding T-cell metabolism over the last several years. It is now recognized that cellular energy metabolism directs T-cell survival, proliferation and their specific functions15,16,17,18,19 (Table 1). Resting T cells generate their energy using TCA to meet their limited biosynthesis. Activated T cells require more nutrients to supply them with an appropriate amount of energy from a sufficient biomass, and glucose and amino-acid metabolism are increased.81 Activated CD4+ T cells increase both glycolysis and fatty-acid metabolism, whereas CD8+ T cells dominantly shift metabolism to glycolysis to rapidly produce ATP.81,82,83 Mitochondrial fatty-acid oxidation is also required for the generation of memory T cells.24 In naive and memory T cells, stimulation of TCR and CD28 drives T-cell metabolic reprogramming, which is controlled by the transcription factor c-Myc that regulates the expression of glucose and glutamine transporters instead of HIF.84 mTOR upregulation of c-Myc and HIF is responsible for the maintenance of glycolytic activity during the process of T-cell activation.84 Notably, different T cell subsets have different metabolic profiles.16,17,18,19 Aerobic glycolysis is the main metabolic pathway and is specifically required for T cell effector function upon activation.4,15,19,20,21,22 By contrast, lipid oxidation has been found to be the primary metabolic pathway for induced Treg cells and memory CD8+ T cells.20,23,24,25 Furthermore, endogenous fatty acid synthesis and the glycolytic-lipogenic axis are important for Th17 cell development.85 In addition, both glycolysis and lipid metabolism regulate Treg differentiation, stability and homeostasis, as well as suppressive its activity.18,20,23,86,87,88,89 Several molecular signaling pathways and/or molecules, including AKT-mTOR signaling, TLR signaling, as well as the transcription factors HIF1α, c-Myc and FoxP3, have been shown to directly regulate Treg metabolic programming and development.18,23,84,86,87,88,90 Given that TILs are dysfunctional and that metabolism directs the T-cell fate and function, defining the metabolic processes of different subsets of T cells should provide alternative novel strategies for breaking the immune tolerance induced by the tumor-suppressive microenvironment.
TLR signaling involves metabolic reprogramming in tumor cells
TLRs recognize pathogen-associated molecular patterns and are critical components of the innate immune system, acting as a link between innate and adaptive immunity to orchestrate pathogen infections and inflammatory responses. In addition to immune cells, TLRs are also widely expressed on different types of tumor cells, regulating tumor growth and function, directly or indirectly, as a ‘double-edged sword’.36,37,38 Several TLR ligands, such as the TLR7 agonist imiquimod, TLR3 ligand Poly (I:C) and TLR9 ligand CpG, can directly induce TLR-positive tumor cell apoptosis or enhance tumor-infiltrating innate and tumor-specific T-cell function.39,45,91,92 By contrast, signaling through TLR2 and TLR5 can enhance the proliferation and survival of gastric cancer cells, promoting the effects of tumor metastasis.93,94,95 Lipopolysaccharide (LPS; TLR4) and Loxoribine (TLR7) favor tumor development and survival.96,97 In addition, increased expression of TLR9 seems to have a close relationship with tumor proliferation in oral squamous cell carcinoma patients, and upregulation and activation of TLR4 is associated with tumor relapse, metastasis and chemoresistance in colorectal and pancreatic cancers.41,98,99,100,101,102 Identifying the unique signaling pathways in tumor cells mediated by different TLRs will be important for exploring the molecular mechanisms involved in cancer immunopathogenesis, as well as for the development of novel TLR-based therapy for human cancer.
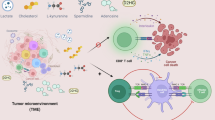
More recent studies suggest that TLRs may directly regulate cell metabolism, affecting tumor behaviors and functions (Table 2). Stimulation with the TLR3 ligand Poly (A:U) promotes metabolic reprograming of head and neck carcinoma cells, resulting in increased tumor growth.42 TLR3 induces tumor cells to switch from OXPHOS and TCA to anabolic glycolysis, showing an enhanced extracellular acid cation rate (ECAR) and lactate accumulation. The TLR3-mediated metabolic switch in cancer cells involves the transcription factor HIF-1α and hypoxic regulation, suggesting that TLR3 signaling may benefit tumors to adapt to hypoxia in the tumor microenvironment.42 Furthermore, TLR9 has been suggested to regulate lipid peroxidation and trace elements (selenium, copper, zinc, magnesium and iron) in response to oxidative stress in patients with breast diseases and breast carcinoma.43 In addition to directly affecting tumor metabolic reprograming and tumor growth, TLRs can regulate cancer cell metabolites, indirectly influencing antitumor immune responses in the tumor microenvironment. Recent studies have shown that human tumor cells can convert naive/effector T cells into senescent T cells to induce immune tolerance, which is molecularly dependent on tumor-derived endogenous metabolic cAMP.38,44 Notably, activation of TLR8 signaling in tumor cells can prevent cAMP production by tumor cells and block tumor-induced conversion of naive and tumor-specific T cells into senescent cells, resulting in enhanced antitumor immunity in vivo.38,44 In addition to the regulation of cAMP, TLR can regulate another key metabolite, IDO, during cancer immunotherapy. Administration of a TLR7 agonist, imiquimod, significantly increases IDO expression in tumor-draining lymph nodes, which inhibits tumor antigen-specific immune responses. Inhibition of IDO expression after TLR7 agonist administration could enhance the efficacy of TLR-mediated therapy.103 TLR agonists can induce enhanced expression of IDO in various cells in addition to tumor cells.104,105 These studies provide mechanistic links between innate regulation via TLR signaling, tumor immunosuppression, hypoxia and metabolic dysregulation.
TLR-mediated metabolic switch is critical for the activation and immunogenic function of macrophages and DCs
Metabolic reprograming to quickly produce ATP and energy is required for infectious pathogen-mediated activation of macrophages and DCs. It is well established that stimulation with TLR ligands from pathogens, such as LPS, can promote a metabolic switch from OXPHOS to glycolysis in macrophages, resulting in cell activation and development of M1 macrophages with inflammatory phenotypes and effector functions.7,30,31,32,33 Similarly, TLR-induced activated DCs have increased glycolysis and reduced OXPHOS.7,30,34,35 The TLR-mediated metabolic switch to glycolysis is also required for DC maturation, allowing them to acquire immunogenic functions and survive after activation.7,34,35 Several key molecules are involved in the activation of macrophages and DCs. Upon stimulation by LPS via TLR4, increased production of iNOS and nitric oxide are important for inhibition of the molecular processes toward OXPHOS.7,106 In addition, the AKT–mTOR–HIF-1α pathway controls metabolic programming and glycolysis during TLR-mediated activation of macrophages and DCs.107,108 Furthermore, recent studies have suggested that TLR-driven early glycolytic programming occurs through rapid phosphorylation and activation of the kinases AKT, TBK1 and IKKɛ, which promote the activity of glycolytic enzymes and de novo synthesis of fatty acids to support DC activation.30,109 In addition to the promotion of glycolysis for cell activation, different TLRs may have diverse biological effects on metabolic regulation in DCs and macrophages. More recent studies have shown that TLR7 and TLR9 signaling can trigger both glycolysis and lipid metabolism in plasmacytoid dendritic cells (PDCs).110,111 In mouse PDCs, TLR9 promotes late glycolysis via a type I IFN/IFNαR loop, resulting in increased fatty-acid synthesis and cholesterol metabolism.110,111 In human PDCs, TLR7 signaling triggers early glycolysis and increased ECAR, which occurs through HIF1α-mediated enhanced expression and the activity of glucose transporters and glycolytic enzymes.110 Furthermore, although stimulation with TLR2 and TLR4 ligands can promote metabolic reprogramming to glycolysis in macrophages, and LPS stimulation leads to a decrease in OXPHOS, whereas stimulation with Pam3CysSK4 can increase the oxygen consumption and mitochondrial enzyme activity involved in the upregulation of both glycolysis and OXPHOS.112,113 These studies indicate that TLR signaling is a critical trigger to control the innate immune cell fate and their functions (Table 2).
TLR-mediated metabolic switch regulates T-cell subset development and function
TLR signaling is also important for regulating the subset development, differentiation and functions of T cells.46,47 TLR2 signaling can directly enhance both proliferation and IL-17 production by Th17 cells and promote the pathogenesis of EAE.46 Activation of TLR2 can also promote and generate efficient memory T cells by producing more IFN-γ and granzyme B and enhancing the cytotoxic activity of antigen-activated CD8+ T cells.114 TLR2 signaling in DCs or Treg cells can reverse mouse Treg suppression.115,116 The TLR3 ligand Poly (I:C) and TLR9 ligand CpG can stimulate Th1 responses and mediate potent antitumor activity.47 TLR4 activation in CD4+ T cells is essential for the proliferation and survival of Th1 and Th17 cells and stimulates IL-17 production.47,117 Activation of TLR4 in CD4+CD25+ Treg cells by LPS can also directly induce Treg cell activation and promote cellular survival and proliferation.118 More recent studies have identified that TLR7 signaling activation in T cells can inhibit both Th17 and Th1 cell differentiation and function, but through different molecular mechanisms. These studies further revealed that inhibition of STAT3 signaling is responsible for TLR7-mediated suppression of Th17 cells, which is reciprocally regulated by the induction of SOCS3 and SOCS5.48 In addition, human TLR8 signaling directly reverses the suppressive functions of naturally occurring CD4+CD25+ Treg cells as well as tumor-derived CD4+, CD8+ and γδ Treg cells.49,50,51,52,53 Furthermore, various TLR signaling promotes chemoattraction and generation of Th17 cells in the tumor microenvironment through tumor cells and tumor-derived fibroblasts.37 Therefore, precisely dissecting the unique signaling pathways involved in the regulation of T-cell subsets by different TLRs will be important for the development of TLR-based tumor immunotherapy.
Although increasing and new evidence suggests that energy metabolism directs the development and functions of T cells,15,16,17,18,19 very limited information is known about the causative relationship between innate regulation through TLR signaling and T-cell metabolism and functionality (Table 2). Given that TLR signaling is also important for regulating the biology and functions of T cells, an improved understanding of the molecular basis of their crosstalk is urgent for the field of tumor immunotherapy. It has been shown that activation of TLR2 and TLR7 signaling promotes the production of IFN-γ by murine CD8+ T cells, which is mechanistically dependent on activating PI3K–AKT–mTOR signaling. This molecular action requires energy support from mitochondrial respiration rather than glycolysis mediated by TLR signaling.119 However, a more recent study suggested that TLR1 and TLR2 signaling activation in mouse Treg cells increases Treg glycolysis and proliferation, but reduces their suppressive capacity.90 TLR1/2-mediated signaling in Treg cells is also involved in the activation of PI3K–AKT–mTOR signaling and enhances expression of Glut1 and the glycolytic enzyme hexokinase 2 (HK2) in Treg cells.90 Studies from our group have demonstrated that TLR8 signaling directly reverses the suppressive functions of different types of human Treg cells, including CD4+, CD8+ and γδ Treg cells.49,50,51,52,53 TLR8-mediated reversal of Treg suppression significantly enhances antitumor immunity for tumor immunotherapy.38,49 We have also shown that tumor-derived cAMP can induce naive and effector T-cell senescence, resulting in impaired antitumor immunity.38 In addition, TLR8 signaling suppresses tumor cell metabolism, resulting in downregulated cAMP levels in both tumor cells and senescent T cells.38 Given that both glucose and lipid metabolism are required for Treg suppression89 and that cAMP is a key component of Treg cell suppression,73 we propose that TLR8 signaling in human Treg cells could also regulate Treg metabolism to inhibit Treg-suppressive activity. Future studies will facilitate our understanding of the molecular mechanisms that are involved in TLR8 signaling-based immunotherapy targeting both tumors and Treg cell metabolic reprogramming.
Rethinking TLR-based tumor immunotherapy by targeting metabolic reprogramming in the tumor microenvironment
Although several TLR agonists have been developed as immunotherapeutic drugs for use in cancer treatments, the results from clinical trials thus far have been discouraging.39,40,120 The varied effects mediated by TLR agonists on different cell types in the tumor microenvironment might be a reason for the insufficient antitumor effects that were found during patient treatment. Therefore, defining the unique molecular signaling/mechanism(s) induced by each TLR on different cell components is essential for improving TLR-mediated antitumor immunity. Recently, significant progress has been made in understanding the importance of metabolic reprogramming/switching for both tumor cells and immune cells in the tumor microenvironment, especially in different types of T cells. Furthermore, recently, many of the molecular interactions involved in TLR signaling and metabolic regulations have been identified and have been focused on by research groups. The updated information and knowledge of cellular metabolism and TLR research not only markedly facilitates our understanding of human immunity under both physiological and pathological conditions but also provides insights for us to rethink the novel and effective TLR-mediated tumor immunotherapeutic strategies that target metabolic reprogramming of both tumor and immune cells within the tumor microenvironment. The following key issues should be reconsidered when we design TLR-based immunotherapy. (a) Understand the dual effects on tumor metabolism mediated by TLRs. We first need to reconsider the fact that TLRs are a ‘double-edged sword’ with both pro-tumorigenic and antitumor effects.36,37,38 We should consider that some TLR signaling can directly promote tumor growth and metastasis by regulating their metabolic reprogramming.42,43 In addition, TLR signaling can regulate metabolites produced by tumor cells, such as cAMP and IDO, which are potent immune suppressors, maintaining the tumor-suppressive microenvironment.63,64,65 Therefore, identification of selective TLR agonists that specifically inhibit tumor metabolism and/or regulate tumor-derived metabolites is needed for TLR-based tumor vaccine/therapy. (b) Metabolically control the balance between Treg and effector T cells via suitable TLR signaling in the tumor microenvironment. It has become clear that a significant challenge in the design of novel cancer immunotherapies is to develop effective strategies for breaking the immune tolerance induced by Treg cells and regenerating the balance between tumor-specific effector T cells and Treg cells in the tumor microenvironment.121 An improved understanding of the distinct metabolic processes of Treg cells and effector T cells will provide alternative and more specific targets for controlling Treg-induced immune suppression by regulating T-cell metabolism. In addition, recent studies have shown that certain types of TLR agonists, such as TLR1/2 and TLR8, can reverse Treg suppression by regulating Treg metabolism.49,50,51,52,53,90 These studies strongly support the concept that TLR-mediated reprogramming of T-cell metabolism is a novel and feasible strategy for tumor immunotherapy. (c) Functional reconstitution of macrophages and DCs in the tumor microenvironment via TLR-mediated metabolic reprogramming. Effector functional reconstitution of macrophages and DCs in tumor sites via metabolic reprogramming through TLRs is also a potential future therapeutic approach. TAMs and tolerogenic DCs are critical cell components within the tumor-suppressive microenvironment. Both TAMs and tolerogenic DCs have distinct metabolic profiles from immunogenic macrophages and DCs.10,11,12,13,14 Developing effective strategies using the TLR-mediated metabolic switch to promote the activation and immunogenic functions of macrophage and DCs in the tumor microenvironment are also essential for tumor immunotherapy. (d) Select effective combinations of other therapeutic strategies with a TLR-based cancer treatment. It has been shown that TLR agonists that are used as a single type of antitumor drug have yielded limited success in clinical trials.40 Combined use of TLR agonists with other therapeutic strategies should be considered in the future for cancer treatments. In support of this concept, checkpoint blockage therapy with PD1/PDL1 and CTLA4 can change the metabolic balance between tumors and TILs and enhance tumor immunotherapy.29,122 Furthermore, recent studies have suggested that TLR8 signaling in tumor cells significantly downregulates tumor-derived metabolite cAMP, which induces T-cell senescence and immune suppression.38,44 Combined application of the TLR8 agonist and an adoptive transfer of T-cell therapy markedly prevents tumor-specific T-cell senescence and promotes their antitumor efficacy in vivo.38,44 Therefore, we propose that TLR-mediated metabolic reprogramming of the tumor microenvironment combined with adoptive T-cell therapy and/or checkpoint blockade therapy that targets both tumor and T cells and creates an effective condition favoring and enhancing antitumor immune responses should be an emerging concept for cancer immunotherapy.6,29
Conclusions
The tumor-suppressive microenvironments created by malignant tumors are a major obstacle for effective antitumor immunity and successful tumor immunotherapy. Cellular energy metabolism controls the fate and biological functions of both malignant tumor cells and tumor-infiltrating immune cells in the tumor microenvironment. The recently improved understanding of the metabolic profiles of different cell components, especially T-cell subsets, facilitates the development of novel strategies to reprogram cell metabolism for tumor immunotherapy. TLRs are widely expressed in both tumor cells and tumor-infiltrating immune cells and are involved in the regulation of tumor pathogenesis and antitumor immune responses. Increasing evidence strongly indicates that TLR signaling directly crosstalks with the molecular processes of cell metabolism in tumor cells and/or different subsets of immune cells. This more recent information will facilitate a rethinking of the novel role and therapeutic potential of TLR signaling in the tumor microenvironment. A better understanding of the mechanistic regulation of TLR signaling and metabolism in different cell components in the tumor-suppressive microenvironment should open new avenues for the development of novel strategies via TLR-mediated metabolic reprogramming for cancer immunotherapy.
References
Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab 2016; 23: 27–47.
Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci 2016; 41: 211–218.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324: 1029–1033.
Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015; 162: 1229–1241.
Lyssiotis CA, Kimmelman AC. Metabolic interactions in the tumor microenvironment. Trends Cell Biol 2017; 27: 863–875.
Reina-Campos M, Moscat J, Diaz-Meco M. Metabolism shapes the tumor microenvironment. Curr Opin Cell Biol 2017; 48: 47–53.
Kelly B, O'Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res 2015; 25: 771–784.
Granucci F, Lutz MB, Zanoni I. The nature of activatory and tolerogenic dendritic cell-derived signal 2. Front Immunol 2014; 5: 42.
Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015; 42: 419–430.
Arts RJ, Plantinga TS, Tuit S, Ulas T, Heinhuis B, Tesselaar M et al. Transcriptional and metabolic reprogramming induce an inflammatory phenotype in non-medullary thyroid carcinoma-induced macrophages. Oncoimmunology 2016; 5: e1229725.
Penny HL, Sieow JL, Adriani G, Yeap WH, See Chi EeP, San Luis B et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology 2016; 5: e1191731.
Wenes M, Shang M, Di Matteo M, Goveia J, Martin-Perez R, Serneels J et al. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab 2016; 24: 701–715.
Ramakrishnan R, Tyurin VA, Veglia F, Condamine T, Amoscato A, Mohammadyani D et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J Immunol 2014; 192: 2920–2931.
Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med 2010; 16: 880–886.
Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab 2014; 20: 61–72.
Pearce EL2010Metabolism in T cell activation and differentiation. Curr Opin Immunol 22: 314–320.
Zeng H, Chi H. Metabolic control of regulatory T cell development and function. Trends Immunol 2015; 36: 3–12.
Newton R, Priyadharshini B, Turka LA. Immunometabolism of regulatory T cells. Nat Immunol 2016; 17: 618–625.
MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. 2013Annu Rev Immunol 31: 259–283.
Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 2011; 186: 3299–3303.
Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O'Sullivan D et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. 2013Cell 153: 1239–1251.
Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest 2013; 123: 4479–4488.
Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 2011; 208: 1367–1376.
Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009; 460: 103–107.
Buck MD, O'Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 2016; 166: 63–76.
Sukumar M, Roychoudhuri R, Restifo NP. Nutrient competition: a new axis of tumor immunosuppression. Cell 2015; 162: 1206–1208.
Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol 2016; 17: 95–103.
Xia H, Wang W, Crespo J, Kryczek I, Li W, Wei S et al. Suppression of FIP200 and autophagy by tumor-derived lactate promotes naive T cell apoptosis and affects tumor immunity. Sci Immunol 2017; 2 doi:https://doi.org/10.1126/sciimmunol.aan4631.
O'Sullivan D, Pearce EL. Targeting T cell metabolism for therapy. Trends Immunol 2015; 36: 71–80.
Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010; 115: 4742–4749.
Pantel A, Teixeira A, Haddad E, Wood EG, Steinman RM, Longhi MP. Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol 2014; 12: e1001759.
Qualls JE, Subramanian C, Rafi W, Smith AM, Balouzian L, DeFreitas AA et al. Sustained generation of nitric oxide and control of mycobacterial infection requires argininosuccinate synthase 1. Cell Host Microbe 2012; 12: 313–323.
Geeraerts X, Bolli E, Fendt SM, Van Ginderachter JA. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front Immunol 2017; 8: 289.
Everts B, Pearce EJ. Metabolic control of dendritic cell activation and function: recent advances and clinical implications. Front Immunol 2014; 5: 203.
Dong H, Bullock TN. Metabolic influences that regulate dendritic cell function in tumors. Front Immunol 2014; 5: 24.
Huang B, Zhao J, Unkeless JC, Feng ZH, Xiong H. TLR signaling by tumor and immune cells: a double-edged sword. Oncogene 2008; 27: 218–224.
Su X, Ye J, Hsueh EC, Zhang Y, Hoft DF, Peng G. Tumor microenvironments direct the recruitment and expansion of human Th17 cells. J Immunol 2010; 184: 1630–1641.
Ye J, Ma C, Hsueh EC, Dou J, Mo W, Liu S et al. TLR8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO Mol Med 2014; 6: 1294–1311.
Smits EL, Ponsaerts P, Berneman ZN, Van Tendeloo VF. The use of TLR7 and TLR8 ligands for the enhancement of cancer immunotherapy. Oncologist 2008; 13: 859–875.
Ridnour LA, Cheng RY, Switzer CH, Heinecke JL, Ambs S, Glynn S et al. Molecular pathways: toll-like receptors in the tumor microenvironment—poor prognosis or new therapeutic opportunity. Clin Cancer Res 2013; 19: 1340–1346.
Yu L, Wang L, Chen S. Dual character of Toll-like receptor signaling: pro-tumorigenic effects and anti-tumor functions. Biochim Biophys Acta 2013; 1835: 144–154.
Veyrat M, Durand S, Classe M, Glavan TM, Oker N, Kapetanakis NI et al. Stimulation of the toll-like receptor 3 promotes metabolic reprogramming in head and neck carcinoma cells. Oncotarget 2016; 7: 82580–82593.
Karki K, Pande D, Negi R, Khanna S, Khanna RS, Khanna HD. Correlation of serum toll like receptor 9 and trace elements with lipid peroxidation in the patients of breast diseases. J Trace Elem Med Biol 2015; 30: 11–16.
Ye J, Peng G. Controlling T cell senescence in the tumor microenvironment for tumor immunotherapy. Oncoimmunology 2015; 4: e994398.
Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med 2007; 13: 552–559.
Reynolds JM, Pappu BP, Peng J, Martinez GJ, Zhang Y, Chung Y et al. Toll-like receptor 2 signaling in CD4(+) T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity 2010; 32: 692–702.
Shi G, Vistica BP, Nugent LF, Tan C, Wawrousek EF, Klinman DM et al. Differential involvement of Th1 and Th17 in pathogenic autoimmune processes triggered by different TLR ligands. J Immunol 2013; 191: 415–423.
Ye J, Wang Y, Liu X, Li L, Opejin A, Hsueh EC et al. TLR7 signaling regulates Th17 cells and autoimmunity: novel potential for autoimmune therapy. J Immunol 2017; 199: 941–954.
Peng G, Guo Z, Kiniwa Y, Voo KS, Peng W, Fu T et al. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science 2005; 309: 1380–1384.
Kiniwa Y, Miyahara Y, Wang HY, Peng W, Peng G, Wheeler TM et al. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin Cancer Res 2007; 13: 6947–6958.
Peng G, Wang HY, Peng W, Kiniwa Y, Seo KH, Wang RF. Tumor-infiltrating gammadelta T cells suppress T and dendritic cell function via mechanisms controlled by a unique toll-like receptor signaling pathway. Immunity 2007; 27: 334–348.
Ye J, Huang X, Hsueh EC, Zhang Q, Ma C, Zhang Y et al. Human regulatory T cells induce T-lymphocyte senescence. Blood 2012; 120: 2021–2031.
Ye J, Ma C, Hsueh EC, Eickhoff CS, Zhang Y, Varvares MA et al. Tumor-derived gammadelta regulatory T cells suppress innate and adaptive immunity through the induction of immunosenescence. J Immunol 2013; 190: 2403–2414.
Warburg O. On the origin of cancer cells. Science 1956; 123: 309–314.
Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006; 3: 177–185.
Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem 2001; 276: 9519–9525.
Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 2016; 531: 110–113.
Wilde BR, Ayer DE. Interactions between Myc and MondoA transcription factors in metabolism and tumourigenesis. Br J Cancer 2015; 113: 1529–1533.
Bhutia YD, Babu E, Ramachandran S, Ganapathy V. Amino acid transporters in cancer and their relevance to "glutamine addiction": novel targets for the design of a new class of anticancer drugs. Cancer Res 2015; 75: 1782–1788.
Yeung SJ, Pan J, Lee MH. Roles of p53, MYC and HIF-1 in regulating glycolysis - the seventh hallmark of cancer. Cell Mol Life Sci 2008; 65: 3981–3999.
Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016; 5: e189.
Gupta S, Roy A, Dwarakanath BS. Metabolic cooperation and competition in the tumor microenvironment: implications for therapy. Front Oncol 2017; 7: 68.
Sitkovsky MV, Kjaergaard J, Lukashev D, Ohta A. Hypoxia-adenosinergic immunosuppression: tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin Cancer Res 2008; 14: 5947–5952.
Sitkovsky M, Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat Rev Immunol 2005; 5: 712–721.
Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008; 27: 5904–5912.
Keshari KR, Sriram R, Van Criekinge M, Wilson DM, Wang ZJ, Vigneron DB et al. Metabolic reprogramming and validation of hyperpolarized 13C lactate as a prostate cancer biomarker using a human prostate tissue slice culture bioreactor. Prostate 2013; 73: 1171–1181.
Rodrigues TB, Serrao EM, Kennedy BW, Hu DE, Kettunen MI, Brindle KM. Magnetic resonance imaging of tumor glycolysis using hyperpolarized 13C-labeled glucose. Nat Med 2014; 20: 93–97.
Romero-Garcia S, Moreno-Altamirano MM, Prado-Garcia H, Sanchez-Garcia FJ. Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front Immunol 2016; 7: 52.
Dietl K, Renner K, Dettmer K, Timischl B, Eberhart K, Dorn C et al. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J Immunol 2010; 184: 1200–1209.
Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014; 513: 559–563.
Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. 2007Blood 109: 3812–3819.
Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab 2016; 24: 657–671.
Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med 2007; 204: 1303–1310.
Vang T, Torgersen KM, Sundvold V, Saxena M, Levy FO, Skalhegg BS et al. Activation of the COOH-terminal Src kinase (Csk) by cAMP-dependent protein kinase inhibits signaling through the T cell receptor. J Exp Med 2001; 193: 497–507.
Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol 2017; 18: 1332–1341.
Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol 2013; 34: 137–143.
Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014; 41: 14–20.
Ferreira GB, Kleijwegt FS, Waelkens E, Lage K, Nikolic T, Hansen DA et al. Differential protein pathways in 1,25-dihydroxyvitamin d(3) and dexamethasone modulated tolerogenic human dendritic cells. J Proteome Res 2012; 11: 941–971.
Pulendran B, Tang H, Manicassamy S. Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat Immunol 2010; 11: 647–655.
Maldonado RA, von Andrian UH. How tolerogenic dendritic cells induce regulatory T cells. Adv Immunol 2010; 108: 111–165.
Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol 2013; 14: 500–508.
Marelli-Berg FM, Fu H, Mauro C. Molecular mechanisms of metabolic reprogramming in proliferating cells: implications for T-cell-mediated immunity. Immunology 2012; 136: 363–369.
Angela M, Endo Y, Asou HK, Yamamoto T, Tumes DJ, Tokuyama H et al. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARgamma directs early activation of T cells. Nat Commun 2016; 7: 13683.
Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011; 35: 871–882.
Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med 2014; 20: 1327–1333.
Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 2013; 499: 485–490.
Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011; 146: 772–784.
De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol 2015; 16: 1174–1184.
Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M et al. The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity 2016; 44: 406–421.
Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG et al. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat Immunol 2016; 17: 1459–1466.
Glavan TM, Pavelic J. The exploitation of Toll-like receptor 3 signaling in cancer therapy. Curr Pharm Des 2014; 20: 6555–6564.
Liang X, Moseman EA, Farrar MA, Bachanova V, Weisdorf DJ, Blazar BR et al. Toll-like receptor 9 signaling by CpG-B oligodeoxynucleotides induces an apoptotic pathway in human chronic lymphocytic leukemia B cells. Blood 2010; 115: 5041–5052.
Tye H, Kennedy CL, Najdovska M, McLeod L, McCormack W, Hughes N et al. STAT3-driven upregulation of TLR2 promotes gastric tumorigenesis independent of tumor inflammation. Cancer Cell 2012; 22: 466–478.
Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009; 457: 102–106.
Song EJ, Kang MJ, Kim YS, Kim SM, Lee SE, Kim CH et al. Flagellin promotes the proliferation of gastric cancer cells via the Toll-like receptor 5. Int J Mol Med 2011; 28: 115–119.
Huang B, Zhao J, Li H, He KL, Chen Y, Chen SH et al. Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res 2005; 65: 5009–5014.
Cherfils-Vicini J, Platonova S, Gillard M, Laurans L, Validire P, Caliandro R et al. Triggering of TLR7 and TLR8 expressed by human lung cancer cells induces cell survival and chemoresistance. J Clin Invest 2010; 120: 1285–1297.
Min R, Zun Z, Siyi L, Wenjun Y, Lizheng W, Chenping Z. Increased expression of Toll-like receptor-9 has close relation with tumour cell proliferation in oral squamous cell carcinoma. Arch Oral Biol 2011; 56: 877–884.
Wang EL, Qian ZR, Nakasono M, Tanahashi T, Yoshimoto K, Bando Y et al. High expression of Toll-like receptor 4/myeloid differentiation factor 88 signals correlates with poor prognosis in colorectal cancer. Br J Cancer 2010; 102: 908–915.
Cammarota R, Bertolini V, Pennesi G, Bucci EO, Gottardi O, Garlanda C et al. The tumor microenvironment of colorectal cancer: stromal TLR-4 expression as a potential prognostic marker. J Transl Med 2010; 8: 112.
Grimmig T, Moench R, Kreckel J, Haack S, Rueckert F, Rehder R et al. Toll like receptor 2, 4, and 9 signaling promotes autoregulative tumor cell growth and VEGF/PDGF expression in human pancreatic cancer. Int J Mol Sci 2016; 17: 2060.
Yu T, Guo F, Yu Y, Sun T, Ma D, Han J et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 2017; 170: 548–563 e16.
Ito H, Ando T, Arioka Y, Saito K, Seishima M. Inhibition of indoleamine 2,3-dioxygenase activity enhances the anti-tumour effects of a Toll-like receptor 7 agonist in an established cancer model. Immunology 2015; 144: 621–630.
Von Bubnoff D, Scheler M, Wilms H, Fimmers R, Bieber T. Identification of IDO-positive and IDO-negative human dendritic cells after activation by various proinflammatory stimuli. J Immunol 2011; 186: 6701–6709.
Manches O, Munn D, Fallahi A, Lifson J, Chaperot L, Plumas J et al. HIV-activated human plasmacytoid DCs induce Tregs through an indoleamine 2,3-dioxygenase-dependent mechanism. J Clin Invest 2008; 118: 3431–3439.
Everts B, Amiel E, van der Windt GJ, Freitas TC, Chott R, Yarasheski KE et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 2012; 120: 1422–1431.
Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013; 496: 238–242.
Byles V, Covarrubias AJ, Ben-Sahra I, Lamming DW, Sabatini DM, Manning BD et al. The TSC-mTOR pathway regulates macrophage polarization. Nat Commun 2013; 4: 2834.
Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol 2014; 15: 323–332.
Wu D, Sanin DE, Everts B, Chen Q, Qiu J, Buck MD et al. Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity 2016; 44: 1325–1336.
Saas P, Varin A, Perruche S, Ceroi A. Recent insights into the implications of metabolism in plasmacytoid dendritic cell innate functions: potential ways to control these functions. F1000Res 2017; 6: 456.
Lachmandas E, Boutens L, Ratter JM, Hijmans A, Hooiveld GJ, Joosten LA et al. Microbial stimulation of different Toll-like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat Microbiol 2016; 2: 16246.
Sanin DE, Prendergast CT, Mountford AP. IL-10 production in macrophages is regulated by a TLR-driven CREB-mediated mechanism that is linked to genes involved in cell metabolism. J Immunol 2015; 195: 1218–1232.
Cottalorda A, Verschelde C, Marcais A, Tomkowiak M, Musette P, Uematsu S et al. TLR2 engagement on CD8 T cells lowers the threshold for optimal antigen-induced T cell activation. Eur J Immunol 2006; 36: 1684–1693.
Sutmuller RP, den Brok MH, Kramer M, Bennink EJ, Toonen LW, Kullberg BJ et al. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest 2006; 116: 485–494.
Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 2003; 299: 1033–1036.
Reynolds JM, Martinez GJ, Chung Y, Dong C. Toll-like receptor 4 signaling in T cells promotes autoimmune inflammation. Proc Natl Acad Sci USA 2012; 109: 13064–13069.
Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med 2003; 197: 403–411.
Salerno F, Guislain A, Cansever D, Wolkers MC. TLR-mediated innate production of IFN-gamma by CD8+ T cells is independent of glycolysis. J Immunol 2016; 196: 3695–3705.
Guha M. Anticancer TLR agonists on the ropes. Nat Rev Drug Discov 2012; 11: 503–505.
Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol 2006; 6: 295–307.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12: 252–264.
Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem 1996; 271: 32529–32537.
Acknowledgements
We apologize since we cannot cite all of the relevant references in this research area because of space limitations. This work was partially supported by grants from the American Cancer Society (RSG-10-160-01-LIB, to GP), Melanoma Research Alliance (to GP) and the NIH (AI097852, AI094478 and CA184379 to GP).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, L., Xu, H. & Peng, G. TLR-mediated metabolic reprogramming in the tumor microenvironment: potential novel strategies for cancer immunotherapy. Cell Mol Immunol 15, 428–437 (2018). https://doi.org/10.1038/cmi.2018.4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2018.4
Keywords
This article is cited by
-
Identification of a novel m6A-related lncRNAs signature and immunotherapeutic drug sensitivity in pancreatic adenocarcinoma
BMC Cancer (2024)
-
Regulatory mechanisms of PD-1/PD-L1 in cancers
Molecular Cancer (2024)
-
Pan-cancer analysis of TASL: a novel immune infiltration-related biomarker for tumor prognosis and immunotherapy response prediction
BMC Cancer (2023)
-
Applications and clinical trial landscape using Toll-like receptor agonists to reduce the toll of cancer
npj Precision Oncology (2023)
-
Molecular characteristics and therapeutic implications of Toll-like receptor signaling pathway in melanoma
Scientific Reports (2023)
