
Abstract
Selective autophagy of mitochondria, known as mitophagy, is an important mitochondrial quality control mechanism that eliminates damaged mitochondria. Mitophagy also mediates removal of mitochondria from developing erythrocytes, and contributes to maternal inheritance of mitochondrial DNA through the elimination of sperm-derived mitochondria. Recent studies have identified specific regulators of mitophagy that ensure selective sequestration of mitochondria as cargo. In yeast, the mitochondrial outer membrane protein autophagy-related gene 32 (ATG32) recruits the autophagic machinery to mitochondria, while mammalian Nix is required for degradation of erythrocyte mitochondria. The elimination of damaged mitochondria in mammals is mediated by a pathway comprised of PTEN-induced putative protein kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin. PINK1 and Parkin accumulate on damaged mitochondria, promote their segregation from the mitochondrial network, and target these organelles for autophagic degradation in a process that requires Parkin-dependent ubiquitination of mitochondrial proteins. Here we will review recent advances in our understanding of the different pathways of mitophagy. In addition, we will discuss the relevance of these pathways in neurons where defects in mitophagy have been implicated in neurodegeneration.
Similar content being viewed by others

Facts
-
Mitophagy mediates clearance of damaged mitochondria, and is also involved in the removal of mitochondria from maturing erythrocytes and eliminating sperm-derived mitochondria after fertilization.
-
In yeast, the mitochondrial outer membrane protein autophagy-related gene 32 (ATG32) recruits the core autophagic machinery to mitochondria for their selective removal.
-
A pathway containing PTEN-induced putative protein kinase 1 (PINK1) and Parkin responds to loss of mitochondrial membrane potential by targeting damaged mitochondria for clearance.
-
The PINK1/Parkin pathway is triggered when PINK1 is stabilized on the outer membrane of damaged mitochondria, where it facilitates recruitment of cytosolic Parkin.
-
Damaged mitochondria are prevented from moving and fusing with the mitochondrial reticulum in preparation for their mitophagic clearance.
Open Questions
-
How distinct are the mitophagy pathways triggered by different stimuli or conditions?
-
To what extent does Parkin activate mitophagy by causing the degradation of mitochondrial surface proteins or hyper-ubiquitinating the mitochondrial surface?
-
How do PINK1 and Parkin interact with one another and with substrates on the mitochondrial surface in causing mitophagy?
-
In contrast to the remarkable conservation of the core autophagic machinery, why are mitophagy-specific genes so little conserved between yeast and mammals?
-
How best should mitophagy be triggered by researchers probing physiologically relevant pathways?
The mitochondrion is an important actor in the life of a eukaryotic cell, but, like all good actors, a mitochondrion needs to exit the stage at the right time. Mitophagy removes mitochondria once they have played their part. This article will review the mechanism by which they are cleared and the cues that signal their removal. Alongside the critical metabolic functions of mitochondria in fatty acid oxidation, the Krebs cycle, and oxidative phosphorylation, mitochondria can also potentially damage cells.1 Reactive oxygen species (ROS), in particular superoxide anion (O2•−), hydrogen peroxide (H2O2), and hydroxyl radical (•OH) are toxic byproducts of oxidative phosphorylation.2 ROS causes oxidative damage to mitochondrial lipids, DNA, and proteins, making mitochondria further prone to ROS production. In turn, damaged mitochondria release high levels of Ca2+ and cytochrome c to the cytosol and thereby trigger apoptosis.3 Thus, like macroscopic energy sources and power plants, these microscopic power supplies are essential but prone to release hazardous materials, particularly when they have been compromised by damage or age. Accordingly, ensuring proper elimination of dysfunctional mitochondria is imperative to cellular survival, and mitochondrial damage has been implicated in aging,4 diabetes, and neurodegenerative diseases.2
To prevent cellular damage by preserving a population of healthy mitochondria, several quality control mechanisms have evolved (Figure 1). Mitochondria have their own proteolytic system, two AAA protease complexes in the inner membrane, whose function is to degrade unfolded membrane proteins.5 Cytosolic proteosomes have also been shown to mediate degradation of proteins on the inner and outer mitochondrial membrane.6 In addition to proteolytic and proteosomal degradation, recent evidence points to a lysosomal pathway in which vesicles bud from mitochondrial tubules, sequester selected mitochondrial cargos, and then deliver those mitochondrial components to the lysosome for degradation.7 This pathway is active under steady-state conditions and is further stimulated by oxidative stress. These mitochondrially derived vesicles may represent a mechanism for selective removal of oxidized mitochondrial proteins while leaving the whole organelle intact.

Three major pathways of mitochondrial quality control. Misfolded mitochondrial membrane proteins can be degraded by two AAA protease complexes with catalytic sites facing both sides of the inner membrane. Mitochondrial proteins can also be degraded by being transferred to lysosomes; vesicles budding from mitochondrial tubules sequester selected mitochondrial cargos, and deliver those mitochondrial components to the lysosome for degradation. The third pathway, known as mitophagy, involves sequestration of an entire mitochondrion within a double-membrane vesicle, the autophagosome, followed by fusion with a lysosome
The aforementioned pathways account for degradation of only a subset of mitochondrial proteins, whereas radioisotope pulse-chase assays indicate that the entire protein contents of mitochondria are turned over within a few days.8, 9 This observation implies that alternative mechanisms for bulk degradation of mitochondria exist. Accordingly, entire mitochondrial organelles were often observed in yeast vacuoles and mammalian lysosomes in electron microscopy studies.10, 11 Bulk degradation of cellular contents occurs through a regulated process called autophagy (reviewed elsewhere in this issue); however, a selective form of autophagy, termed mitophagy, is chiefly responsible for elimination of damaged or superfluous mitochondria. In this process, mitochondria are sequestered in double-membrane vesicles and delivered to lysosomes for hydrolytic degradation. In addition, although mitochondrial turnover and clearance of damaged mitochondria may be the primary functions of mitophagy, there are other specialized cases of mitophagy: the complete removal of mitochondria during erythrocyte maturation12 and the selective destruction of sperm-derived mitochondria after oocyte fertilization.13, 14 In this review, we will discuss the general features of mitophagy as a quality control pathway, and also the activation of specific types of mitophagy in response to different physiological triggers.
Overview of Autophagy
Autophagy (from Greek, meaning self-eating) is a broad term referring to different pathways for bulk degradation of cytosolic components and organelles, including mitochondria, through delivery to the lysosome. There are three distinct classes of autophagy: microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy.15 In microautophagy, invaginations of the lysosomal membrane directly engulf portions of the cytoplasm. In contrast, CMA involves the chaperone Hsc70 and its co-chaperones that recognize and unfold substrate proteins with a KFERQ amino-acid motif. The substrates bind to the lysosomal protein LAMP-2A and are translocated across the lysosomal membrane for degradation.16
Macroautophagy is the major type of autophagy and it is well conserved from yeast to mammals. Non-selective or bulk autophagy is induced by starvation to provide the cell with essential nutrients. In contrast, selective autophagy occurs to clear unwanted or damaged organelles, including mitochondria. In this process, an isolation membrane (known as phagophore) surrounds a portion of the cytoplasm or an organelle, forming a double membranous structure called the autophagosome. The autophagosome then fuses with the lysosome to form an autolysosome, in which the hydrolytic degradation of contents of the autophagosome will occur. Mitochondria are removed by a form of macroautophagy (called mitophagy), in which the core machinery of bulk macroautophagy is harnassed for the selective clearance of a mitochondrion. We will therefore focus our discussion on macroautophagy, herein referred to as autophagy.
Autophagy can be mechanistically broken down into the following five steps: initiation of the isolation membrane, elongation, closure of the isolation membrane and autophagsome formation, autophagosome–lysosome fusion, and lysosomal degradation.17 These stages are similar whether invoked for the clearance of bulk cytosol or of a mitochondrion.18 Genetic screens for defects in this pathway in the yeast Saccharomyces cerevisiae have identified nearly 30 ATGs.19 The ATG genes that form the core machinery in yeast are ATG1–10, ATG12–16, and ATG18. These core genes are highly conserved in mammals and, except for ATG13 in some circumstances, are also essential for mitochondrial autophagy.20 In the case of starvation-induced autophagy, the process begins with a complex consisting of ATG1, a kinase, and ATG13, its regulatory subunit, that exist in a complex with other autophagy proteins. Target of rapamycin complex 1 (TORC1) normally restricts ATG13 from interacting with ATG1, but starvation removes this inhibition and allows the cascade to proceed. The mammalian ortholog of these proteins, the ULK1 complex, is similarly regulated by TORC1. In the case of stress response in mammalian cells, mitophagy can similarly be signaled by the Hsp90–Cdc37 complex acting through ULK1 and ATG13.21 Other pathways for activating mitophagy are discussed below, but all seem to require the ATG1/ULK1 complex for initiation,20, 22 but do not necessarily require ATG13. The formation of the autophagosome, whether surrounding a mitochondrion or other cargo, proceeds by means of the ATG1/ULK1 complex activating a class III phosphatidylinositol 3-kinase (PI3K) complex that includes ATG14, Vps34 and Beclin1. PI3K-mediated formation of phosphatidyl inositol triphosphate (PIP3)-rich membrane domains is important for nucleation of the isolation membrane.17 In some cases, stress-induced bulk autophagy as well as mitophagy occurs independently of Beclin 1,23, 24 suggesting that alternative pathways for production of PIP3 may exist. Elongation of the isolation membrane is then mediated by two ubiquitin-like conjugation systems. In the first system, the ubiquitin fold-containing protein ATG12 is conjugated to ATG5 and an ATG5–ATG12–ATG16L1 complex then localizes at the isolation membrane. ATG12 can also be conjugated to ATG3 and this complex, although not necessary for general autophagy, promotes mitophagy when the mitochondria are depolarized.25 The mechanism by which this additional ATG12 complex figures in selective mitophagy remains unknown. The second ubiquitin-like reaction that is shared by the general autophagy pathway and mitophagy employs the ubiquitin fold-containing protein microtubule-associated protein light chain 3 (LC3) (also called MAP1LC3 or LC3B, the ortholog of yeast ATG8). It is synthesized in the pro-LC3 form, cleaved by ATG4B to form LC3-I, and conjugated in ubiquitin-like reactions to phosphatidyl ethanolamine (PE) forming LC3-II. LC3-II localizes to isolation membranes and is required for elongation and closure of this membrane to form mature autophagosomes.17
Although clearly essential for forming the defining double-membrane vesicle of the autophagosome, the origin of the membrane component remains controversial.26 De novo membrane assembly has been proposed; however, various studies have implicated the ER,26, 27 Golgi,26 plasma membrane,28 and more recently mitochondria29 as possible membrane sources for the autophagosome. Yoshi27 and co-workers observed close association of the isolation membrane with rough ER during mitophagy, implicating ER in particular as a membrane source for mitophagy. On the basis of the evidence suggesting mitochondria as a membrane source, in general autophagy, it is intriguing to speculate that, in mitophagy, the mitochondrion could supply the autophogosomal membrane for its own degradation. Clearly, further work is required to unambiguously determine the membrane sources in both general and cargo-selective autophagy.
Mitophagy as a Selective Form of Autophagy
The extensive similarity of the pathways for general autophagy and mitophagy,22, 30 as summarized above, raises an obvious question: How is an autophagosome directed selectively to mitochondria and how is that process triggered without the activation of bulk autophagy? Although mitochondria can be engulfed non-selectively along with other cytosolic contents during bulk autophagy,31 both yeast and mammalian cells can selectively degrade damaged or superfluous mitochondria by mitophagy. In yeast, mitophagy can be activated by mutations that impair mitochondrial electrochemical potential; two such mutations in Fmc1 and Mdm38 cause aggregation of the F0F1 ATPsynthase subunits and impairment of the mitochondrial H+/K+ exchanger, respectively.32, 33 Similarly, knockdown of an inhibitor of the mammalian F0F1 ATPsynthase, IF1, causes a dramatic reduction in mitochondrial mass that correlates with increased autophagic activity.34 Depolarization of mitochondria with the uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP)35 or photo-irradiation36 induces autophagic clearance of mammalian mitochondria while leaving other organelles such as peroxisomes intact. Recently, it has been reported that mitochondria carrying deleterious mutations in mtDNA can be selectively eliminated through mitophagy,37 although additional activation of macroautophagy, such as rapamycin-mediated inhibition of TORC1, may be required for efficient clearance.38 Of course, mitochondria with DNA mutations are encountered in human patients39 and so the clearance mechanism cannot be perfectly efficient; presumably, some threshold of damage must be crossed before mitophagy is likely to occur. Mitophagy also serves as a mechanism to regulate organelle number in response to developmental or physiological cues. Yeast cells that are grown on a non-fermentable carbon source require mitochondria for oxidative phosphorylation. Under such conditions, mitochondria are spared from degradation even during starvation-induced bulk autophagy, indicating differential regulation of bulk autophagy and mitophagy. In contrast, when yeast are switched from growth on non-fermentable to fermentable media, mitophagy occurs to reduce mitochondrial mass as oxidative phosphorylation is no longer needed.40 Thus, mitophagy must be mechanistically separable from other forms of autophagy so as to eliminate selectively dysfunctional or superfluous mitochondria in response to cues particular to mitophagy. Mitochondrial-specific mechanisms are beginning to emerge.
Mitophagy-Specific Effectors
To identify factors that might account for the selectivity of mitophagy, two groups independently performed screens for yeast mutants defective in mitophagy.41, 42 Taken together, they found approximately 40 genes that were distinct from known ATGs and required for mitophagy but not bulk autophagy. A previously identified gene, Uth1,43 had been shown to be required for mitochondrial removal, induced by rapamycin and starvation; conditions that would induce bulk autophagy. This gene was not isolated in the screens for selective mitophagy where mitophagy was induced by switching to fermentable media. From these screens, ATG11 emerged as an important factor in mitochondrial degradation.20, 40 ATG11 has also been implicated in other forms of selective autophagy including pexophagy (selective autophagy of peroxisomes).44 ATG11 appears to function as an adaptor between ATG8 and the proteins that function as the receptor for ATG11 on each particular organelle or cargo to be selectively removed. In the case of mitochondria, the ATG11 receptor is ATG32, another protein uncovered in the yeast screen, which localizes to mitochondria and binds ATG11 and ATG8 upon induction of mitophagy. In this manner, ATG32 recruits the canonical autophagic machinery to the mitochondria. Surprisingly, however, loss of ATG32 did not affect cellular ROS levels or growth on a non-fermentable carbon source,41 which may imply that other quality control pathways independent of ATG32-mediated mitophagy exist.
Unfortunately, ATG11 and ATG32 do not have any identifiable homologs in higher eukaryotes.20 Mammals could, however, have functional homologs of these proteins. For example, the ubiquitin-binding adaptor p62 (SQSTM1) may have an ATG11-like role. It accumulates on damaged mitochondria and has been reported to recruit mitochondria to the autophagosome by binding to ATG8/LC3 in some studies45 but not others.46 Another protein, Nix, may serve the function of both ATG32 and ATG11. Nix is a resident protein on the mitochondria and also binds directly to LC3. Nix is induced in developing reticulocytes and is required for the subsequent clearance of mitochondria during eythrocyte development.12, 47 Yet another pathway involves the ubiquitin ligase Parkin as an important player in recruiting autophagosomes to damaged mitochondria.48 We will discuss the roles of Nix and Parkin in more detail in the next sections.
Two puzzles remain. The first concerns the initiation of the autophagic pathway in selective mitophagy. Although the proteins just discussed may explain the selective recruitment of ATG8 or LC-3 to mitochondria, ATG8/LC3 represent a late stage in the autophagic pathway, downstream of several protein complexes known to be essential for mitophagy as well. It is not yet clear how conditions that trigger mitophagy, whether metabolic changes or mitochondrial damage, initiate the cascade. The second puzzle arises from a recent mammalian genomic screen that identified 96 new genes required for Parkin-mediated mitophagy.49 Lack of significant similarities in the genes identified by this screen and those in yeast may suggest extensive differences between the regulation of mitophagy in mammals and yeast. This may reflect the different physiological purposes of mitophagy in such different cell types. However, it should also be considered that the inducers of mitophagy also differed in these screens; those in yeast employed shifts in nutrient sources, while the mammalian screens used depolarization of the mitochondrial membrane potential. There may be multiple routes for the recruitment of the autophagosome to the mitochondria, all of which will converge on the core apparatus of autophagy.
Modulation of Mitochondrial Dynamics before the onset of Mitophagy
Mitochondria are dynamic organelles that move within the cell and frequently undergo fission and fusion. In many cells, a large fraction of the mitochondria at any given moment are in an interconnected network. Mitochondrial dynamics and mitophagy are closely related; a mitochondrion has to be separated from the mitochondrial network to be engulfed by the autophagosome (Figure 2). In this section, we will briefly review the molecular machinery that regulates mitochondrial dynamics and discuss the interaction of this machinery with the mitophagic pathway.

From mitochondrial network to fragmentation and mitophagy. Mitochondria typically form an interconnected network, but fission events and a block of fusion can fragment the network and thereby segregate a mitochondrion destined for mitophagy from the rest of the network. An isolation membrane, of an unknown origin, forms around this mitochondrial fragment. Closure of the isolation membrane, with the help of LC3/ATG8, gives rise to an autophagosome engulfing the mitochondrion. The autophagosome fuses with the lysosome forming an autolysosome in which the mitochondrial cargo is degraded
Fission and fusion, as well as mitochondrial trafficking on actin (in yeast) or the microtubule cytoskeleton (in higher organisms) enable the cell to adjust mitochondrial distribution to changing local needs. Mitochondrial fission and fusion are regulated by members of a family of conserved large GTPases, initially identified in yeast.50, 51 The dynamin-like GTPase, dynain-related protein 1 (Drp1) (Dnm1 in yeast), mediates fission by forming a multimeric complex that wraps around the outer membrane of mitochondrial tubules and exerts mechanical force to produce membrane scission. In contrast to fission where mitochondria are divided using only an outer membrane apparatus, two distinct machineries, Mitofusin 1 and 2 (Fzo in yeast) and optic atrophy 1 (Opa1) (Mgm1 in yeast), are required for fusion of the outer and inner membranes, respectively. Mitochondrial fusion is also regulated by mitochondrial motility, although less directly; stationary mitochondria are less likely to encounter another mitochondrion with which to fuse.52 A critical function of fusion and fission is to allow for efficient distribution of mtDNA and proteins throughout the mitochondrial network.51 Even brief fusion events can allow extensive exchanges of mitochondrial content. By following the fate of individual photo-labeled mitochondria, Twig and co-workers53 have shown that a fusion event is often followed by fission, creating daughter mitochondria with uneven mitochondrial potentials. Whereas the daughter mitochondrion with the healthier membrane potential will continue to participate in fusion and fission cycles with the mitochondrial reticulum, the depolarized daughter mitochondrion is unlikely to undergo fusion and is often degraded through mitophagy.
Depolarization causes the loss of Opa154 and Mitofusins (Mfn1 and 2),55, 56, 57 proteins necessary for the fusion of the inner and outer membranes of mitochondria, and thereby promotes mitochondrial fragmentation. In agreement with the essential role of fission in mitophagy, a yeast genetic screen revealed that Dnm1, the yeast homolog of Drp1, is essential for mitophagy.20 Similarly in mammals, genetic manipulations that cause excessive fusion, such as overexpression of Opa1 or dominant-negative Drp1, preclude autophagic degradation of mitochondria.58 This protection from autophagy by hyperfusion occurs physiologically during certain types of starvation-induced autophagy, in which a fused mitochondrial network protects mitochondria from degradation.59 In summary, a shift in the balance of mitochondrial dynamics towards increased fission and decreased fusion promotes segregation of damaged mitochondria and facilitates their clearance by mitophagy.
The PINK1/Parkin Pathway of Mitophagy
A recently identified pathway that has emerged as a paradigm for mammalian mitophagy is mediated by the PINK1 and the E3 ubiquitin ligase Parkin (Figure 3a). The genes encoding PINK160 and Parkin61 were found to be mutated in certain forms of autosomal recessive Parkinson's disease (PD). Accumulation of dysfunctional mitochondria in the brains of PD patients implied a link between PINK1 and Parkin and mitochondrial quality control. Genetic studies in Drosophila further suggested a role for PINK1 and Parkin in the regulation of mitochondrial integrity.62, 63 Loss of either protein in flies results in mitochondrial dysfunction and thereby causes degeneration of flight muscles and dopaminergic neurons. Overexpression of Parkin ameliorates the mitochondrial phenotypes in PINK1-deficient flies. This interaction and other genetic experiments demonstrate that the two genes act in the same pathway, with PINK1 upstream of Parkin. Parkin overexpression in mammalian cytoplasmic hybrid cells containing both wild-type and mutant mtDNA leads to selective elimination of mitochondria carrying mutant mtDNA.37 PINK1 and Parkin also promote removal of mitochondria damaged by chemical uncouplers, such as CCCP.35 While a role for PINK1 and Parkin in the clearance of defective mitochondria has extensive evidence, certain controversies remain. Stable knockdown of PINK1 has been reported to induce, rather than inhibit, mitophagy in neuronal cell lines.64 One explanation for this unexpected result is that PINK1 deficiency leads to higher mitochondrial oxidative stress, which in turn might trigger compensatory PINK1-independent mitophagy. In another study, it was found that while Parkin overexpression stimulates CCCP-induced mitophagy, it appears to inhibit bulk autophagy under basal conditions.65 This observation suggests that Parkin may differentially regulate distinct types of autophagy.

Pathways of mitophagy. (a) Damaged mitochondria that have lost their membrane potential (ψm) are eliminated by mitophagy through the accumulation of PINK1 and the E3 ubiquitin ligase Parkin on the mitochondrial surface. (b) In developing reticulocytes, all mitochondria are eliminated by mitophagy. The mitochondrial outer membrane protein, Nix, may serve as a receptor for targeting mitochondria to autophagosomes. (c) Sperm-derived mitochondria are selectively eliminated from fertilized oocytes through mitophagy, thereby allowing for exclusively maternal inheritance of mitochondrial DNA
As discussed in the previous section, segregation of mitochondria destined for mitophagy appears to precede their degradation. Accordingly, PINK1 and Parkin play central roles in regulating mitochondrial dynamics and the mechanistic basis of this aspect of their function is presently the best understood. A genetic interaction between the PINK1/Parkin pathway and the fission/fusion machinery was discovered in flies where overexpression of Drp1 led to suppression of mitochondrial defects in PINK1 and Parkin mutants.66 Although similar studies in mammalian systems have produced contradictory results,64, 67 the findings in the fly indicate that PINK1 and Parkin promote fission. Consistent with this conclusion, mammalian and fly studies indicate that upon mitochondrial depolarization, Parkin translocates to mitochondria and causes polyubiquitination of Mitofusin, targeting it for removal from the mitochondrial membrane by the p97 AAA-ATPase and subsequent proteosomal degradation55, 56, 57 (Figure 4bi). Degradation of Mitofusin prevents damaged mitochondria from fusing with the functional mitochondrial network and is essential for their subsequent degradation by mitophagy. However, although loss of Mitofusin is necessary for mitophagy to proceed, it is not by itself sufficient to trigger mitophagy and PINK1 and Parkin can initiate mitophagy in cells lacking Mitofusin.56, 68 Mitochondrial fragmentation in the absence of Mitofusin thus appears to be a permissive rather than a triggering event.

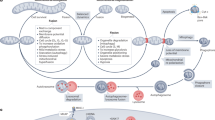
The PINK1/Parkin pathway of mitophagy. (a) In healthy mitochondria, PINK1 is imported to the inner mitochondrial membrane, presumably through the TOM/TIM complex. The TIM complex-associated protease, mitochondrial MPP, cleaves PINK1 mitochondrial targeting sequence (MTS). PINK1 is also cleaved by the inner membrane presenilin-associated rhomboid-like protease PARL and ultimately proteolytically degraded. Loss of membrane potential in damaged mitochondria prevents the import of PINK1 leading to the accumulation of unprocessed PINK1 on the outer membrane surface where it associates with the TOM complex, and recruits cytosolic Parkin, via an unknown mechanism, to damaged mitochondria. (b) Parkin promotes mitophagy of damaged mitochondria in two major ways. (i) In one mechanism, Parkin, presumably through its ubiquitin–ligase activity, causes the degradation of its substrates such as Miro and Mitofusin. In the case of Miro, its phosphorylation by PINK1 is upstream of the Parkin-dependent proteosomal degradation. Mitocondrial fragmentation and arrest of motility, through loss of Mitofusin and Miro, quarantine damaged mitochondria and promote their autophagosomal engulfment. (ii) Alternatively or in addition, Parkin-mediated hyper-ubiquitination of the mitochondrial outer membrane is recognized by ubiquitin-binding adaptors, such as p62, HDAC6, and unknown others, that may recruit damaged mitochondria to the isolation membrane through their interaction with the autophagosomal protein LC3
The PINK1 and Parkin pathway of mitophagy is also closely associated with the regulation of mitochondrial movements. Mitochondrial depolarization causes PINK1 and Parkin to associate with mitochondria Rho-GTPase (Miro), a mitochondrial adaptor protein that anchors a kinesin motor complex to the mitochondrial surface.69 This association leads to the phosphorylation of Miro by PINK1 and the subsequent proteosomal degradation of Miro in a Parkin-dependent manner (Figure 4bi). Loss of this adaptor protein releases kinesin from the mitochondrion and arrests mitochondrial motility. Because stationary mitochondria are less likely to undergo fusion,52 this arrest may serve as an additional quarantine mechanism to segregate damaged mitochondria from the rest of the mitochondrial reticulum before mitophagy. Although it remains to be seen whether degradation of Miro is a required event for mitochondrial elimination, the PINK1/Parkin-mediated degradation of both Miro and Mitofusin illustrate how regulation of mitochondrial dynamics is coupled to mitophagy and have provided insight into the mechanisms by which these enzymes act. In the following section, we will describe the molecular details of the PINK1/Parkin pathway of mitophagy.
Accumulation of PINK1 and Parkin on damaged mitochondria
PINK1 is a serine/thereonine kinase that contains a mitochondrial targeting sequence allowing for its mitochondrial localization.48 In healthy mitochondria, PINK1 is constitutively imported, probably via the TIM/TOM complex, to the inner membrane where it is cleaved by several proteases including the mitochondrial-processing protease (MPP) and the inner membrane presenilin-associated rhomboid-like protease PARL, and ultimately proteolytically degraded (Figure 4A).70, 71, 72, 73 Loss of mitochondrial membrane potential may be the event that acutely activates this pathway; loss of the potential gradient precludes import of PINK1 to the inner membrane, thereby stabilizing intact PINK1 on the mitochondrial outer membrane where it interacts with the TOM complex.74 In this manner, PINK1 serves as a sensor for mitochondrial damage. Accumulation of PINK1 on the mitochondrial surface induces translocation of Parkin from the cytosol to damaged mitochondria through a mechanism as yet unknown.71 Once recruited, Parkin promotes degradation of mitochondria through mitophagy.35 In mammalian cells, recruitment of Parkin to damaged mitochondria requires PINK1,71 although in fly studies Parkin overexpression was sufficient to rescue mitochondrial defects in PINK1-null mutants.63 Even in the absence of mitochondrial depolarization, a recombinant form of PINK1 that is stably localized on the mitochondrial outer membrane is sufficient for Parkin recruitment and induction of mitophagy.35, 71 In addition, PINK1 artificially targeted to peroxisomal membranes can recruit Parkin and mediate autophagic degradation of the peroxisome.74 Thus, PINK1 and Parkin form the minimal machinery for recruitment of the canonical autophagy players to target organelles. Although the kinase activity of PINK1 is required for it to recruit Parkin, it is unclear as to how this occurs. One hypothesis is that PINK1 binding to45, 75 and phosphorylation35, 71, 76 of Parkin serves to activate that enzyme and cause its translocation, but this remains controversial. Alternatively, it is possible that PINK1 phosphorylates an unknown substrate, and it is the primed substrate, not PINK1 per se, that binds to and recruits Parkin. This model is consistent with the sequential action of PINK1 and Parkin on the substrate Miro,69 but there is no evidence at present that Miro itself is essential for mitophagy.
Although PINK1 can clearly promote Parkin-dependent mitophagy, it is not necessarily the only means of activating the Parkin pathway. In Drosophila, Parkin overexpression rescues the mitochondrial morphology defects of a PINK1-null mutant, indicating that PINK1 is not essential for Parkin function.63, 66 Parkin overexpression can also arrest mitochondrial motility in PINK1-deficient flies.69 Furthermore, Parkin mutants had more severe phenotypes than PINK1-null flies, suggesting PINK1-independent functions of Parkin.66 On the other hand, activation of mitophagy by CCCP was not observed in mammalian cells when PINK1 was knocked down.71 This leaves open the question of whether Parkin can be activated by pathways other than mitochondrial depolarization and PINK1 activation in mammalian cells.
Parkin promotes ubiquitination of mitochondrial proteins
Following its mitochondrial translocation, the E3 ubiquitinligase activity of Parkin appears to increase.77 Parkin mediates formation of two types of polyubiquitin chains:68 lysine (K) 48 linkage associated with proteosomal degradation of the substrate; and lysine (K) 63 linkage associated with autophagic degradation.48, 78 Although Parkin-mediated polyubiquitination of mitochondrial substrates is likely to be the mechanism by which it triggers mitophagy,45, 79 (Figure 4b) the details of how this happens remain uncertain. In one model, Parkin mediates formation of K48 chains on specific mitochondrial outer membrane proteins and it is the proteosomal degradation of these proteins that triggers mitophagy. In support of this model, inhibition of proteosome activity prevents mitophagy of damaged mitochondria.56, 68 A proteomic study in HeLa cells revealed Parkin- and proteosome-dependent degradation of Miro, Mitofusin, hFis1, and Tom70, as well as others, before mitophagy. However, as mentioned above, while proteosomal elimination of Mitofusin is required, it is not sufficient for initiation of mitophagy. Moreover, proteosome inhibition in Mitofusin-null cells prevents mitophagy, indicating that Mitofusin is not the key target.68 Therefore, it is not clear whether the degradation of these candidate proteins, if any, leads to activation of mitophagy or if they are merely permissive for mitophagy by, among other things, arresting motility and inducing fragmentation (Figure 4bi). In a second model, Parkin causes formation of K63 (or 27)-linked ubiquitin chains on mitochondrial outer surface, and these ubiquitin chains, rather than inducing proteosomal degradation, initiate a signaling cascade that activates mitophagy (Figure 4bi). In support of this model, Geisler et al.45 found abundant Parkin-dependent K63 and K27 ubiquitin chains and identified ubiquitination of the mitochondrial outer membrane protein VDAC, a component of the permeability transition pore, to be essential for mitophagy. However, the requirement of VDAC ubiquitination for mitophagy could not be replicated in a different study.46 In fact, it is possible that the mere presence of high levels of ubiquitin on the mitochondrial surface, rather than ubiquitination of a particular substrate, is sufficient for induction of mitophagy. This hypothesis is consistent with the ability of Parkin ectopically targeted to peroxisomes to mediate surface ubiquitination and autophagic degradation of these organelles.74 In summary, while the ubiquitin ligase activity of Parkin is well established, further work is required to determine how Parkin-mediated ubiquitination promotes mitophagy.
Interaction of PINK1 and Parkin with the core autophagy machinery
Parkin-mediated mitophagy requires core components of the autophagic machinery. Specifically, blocking the activities of ATG3, ATG5, ATG5, or ATG 7, and class III PI3K prevents degradation of CCCP-treated mitochondria.35 How does the PINK1/Parkin pathway interact with and activate the autophagic machinery? The ubiquitin-binding adaptor protein p62/SQSTRM1 accumulates on depolarized mitochondria and it has also been proposed to facilitate recruitment of damaged mitochondria to autophagosomes by binding to LC348 (Figure 4bii). However, it is not clear whether p62 is required for Parkin-mediated mitophagy,45, 46, 80 as mitochondrial elimination has been reported to occur in the absence of p62-mediated clustering. Nor would p62-mediated recruitment of damaged mitochondria to pre-existing isolation membranes explain how Parkin might stimulate the formation of new isolation membranes.81 Recent studies suggests that both PINK1 and Parkin can directly interact with the Beclin-1 PI3K complex.82, 83 Parkin-dependent recruitment of Ambra1, an activator of the Beclin1 complex, to mitochondria upon depolarization could lead to activation of the Beclin-1 complex and nucleation of pre-autophagosomal membranes around damaged mitochondria. Therefore, both functions of Parkin, namely, ubiquitination of the mitochondrial outer membrane proteins and recruitment of Ambra1, may contribute to mitophagy of damaged mitochondria. The cytoplasmic E3 ubiquitin ligase SMURF1 has also been found to be required for Parkin-dependent mitophagy.49 The exact function of SMURF1 in mitophagy is not clear yet; however, the authors did find that its membrane targeting domain, and not its ubiquitin ligase activity, was required. On the basis of this finding, it is proposed that SMURF1 might be involved in the delivery of mitochondria to the autophagosome. In addition to its role in mitophagy of defective mitochondria, Parkin also stimulates mitochondrial biogenesis, presumably to replace damaged mitchondria with healthy and functional organelles.84 Upon depolarization, Parkin causes degradation of the trascriptional repressor PARIS. Loss of PARIS releases the mitochondrial biogenesis transcription factor PGC1α to activate its target genes.
In summary, the current body of evidence highlights the PINK1/Parkin pathway of mitophagy as an important regulator of mitochondrial homeostasis. Many questions, however, remain unanswered. How does PINK1 trigger Parkin recruitment? How does Parkin interact with various components of the core autophagic machinery and regulate mitophagy at different steps of autophagosome formation and maturation? How are the differential functions of Parkin in promoting mitophagy and mitochondrial biogenesis coordinated? Is the PINK1/Parkin pathway relevant only in cases of mitochondrial damage or does it also contribute to the steady-state turnover of mitochondria? It is noteworthy to mention that PINK1 and Parkin cannot be the only pathway for degradation of damaged mitochondria. Several observations support this conclusion. First, neither humans nor flies with mutations in the PINK1/Parkin pathway show a gross increase in their cellular mitochondrial mass. Second, PINK1 or Parkin knockout mice appear largely normal and have only subtle phenotypes,85 which would not be expected if turnover of damaged mitochondria was completely blocked. It remains to be seen what other parallel pathways account for mitochondrial turnover in the absence of PINK1 and Parkin.
What best models a physiological trigger of damage-induced mitophagy?
Because mitophagy is infrequent in healthy mammalian cells, researchers typically challenge cells to trigger the process, particularly to examine damage-induced mitophagy and the PINK1/Parkin pathway. The mitochondrial uncouplers, CCCP and FCCP, are most commonly used in this context. These drugs are nonspecific ionophores that cause a severe loss of mitochondrial membrane potential within minutes followed by recruitment of Parkin and LC3 to the mitochondria.86 For biochemical studies, the ability to depolarize all the mitochondria in a cell culture with CCCP has clear advantages.35, 69 However, how similar is this phenomenon to the process that normally removes the sporadically damaged mitochondrion within an otherwise healthy cell? Presumably, the mitophagic apparatus in most cells was not designed to cope with the immediate loss of all mitochondria. In addition, CCCP is a nonspecific ionophore that affects cellular components other than the mitochondria. In fact, CCCP causes disruption of the microtubule cytoskeleton87 and prevents lysosomal acidification.88 For gentler disruption of mitochondrial function, several other approaches have been used. These include application of valinomycin, a K+ ionophore, and antimycin A, a blocker of complex III, and these also induce recruitment of Parkin.69, 89 These compounds, although less drastic than CCCP, also affect the entire population of mitochondria. Damage to subsets of mitochondria has been achieved by photoirradiation36 and by the inducing mitochondrial DNA damage.37 These manipulations triggered selective removal of the damaged mitochondria and did so via Parkin recruitment. At present, therefore, these subtler perturbations of mitochondrial status largely confirm the conclusions from experiments with CCCP, but future work in this field may wish to focus, whenever possible, on milder techniques for induction of mitophagy that more closely resemble physiological conditions.
Mitophagy in Erythrocyte Maturation, Hypoxia, and Embryogenesis
Among the specializations that allow erythrocytes to mediate gas exchange between the lungs and peripheral tissues is the absence of internal organelles, including mitochondria. The lack of mitochondria may be important because erythrocytes experience high levels of oxidative stress during hemoglobin-mediated oxygen transport. Exposure to oxidative stress would increase mitochondrial ROS production and its consequent cellular damage. The transformation from a mitochondria-containing reticulocyte to a mature erythrocyte takes place over 2 to 3 days, during which time all their internal organelles, including mitochondria, are eliminated.90 The elimination of mitochondria in reticulocytes is mediated by a specialized form of extreme mitophagy that requires Nix (BNIP3L), a Bcl2-related mitochondria outer membrane protein with an atypical BH3 domain (Figure 3b). In fact, Nix−/− mouse retain mitochondria in their erythrocytes and develop anemia due to decreased survival of those cells.12 Nix is required specifically for elimination of reticulocyte mitochondria, but not other organelles that are lost during maturation such as ribosomes and the nucleus.12, 47 Nix-dependent clearance of mitochondria is distinct from apoptotic pathways as it does not require other Bcl2 proteins such as Bak, Bax, Bcl-xl, Bim, or Puma.47 How then does Nix participate in mitophagy? Studies on reticulocyte maturation suggest that Nix is not required for the induction stage, as LC3-rich isolation membranes form in Nix−/− mice. Instead, Nix is required for incorporation of mitochondria into autophagosomes. Nix contains a conserved LC3-binding motif known as LIR (LC3-interacting region) and therefore may act as a receptor for targeting mitochondria to autophagosomes in a manner similar to the yeast ATG32/ATG11 pair.91 Despite the involvement of Nix, however, the upstream signal that activates mitophagy in these cells remains unknown.
The function of Nix may not be restricted to erythrocyte maturation; Nix could also be involved in depolarization-induced mitophagy, as CCCP treatment enhances Nix and LC3 interaction in HeLa cells.92, 93 There is not yet consensus in these cases as to whether Nix functions upstream or downstream of mitochondrial depolarization.12, 80, 94 Unlike the clear requirement for Nix in erythrocyte maturation, treatment of reticulocytes with the uncoupling agent FCCP induces mitophagy in the absence of Nix.12 Thus, Nix can have at most a facilitating or parallel function in depolarization-induced mitophagy.
Nix, and another BH3-only protein Bnip3, are also involved in hypoxia-induced mitophagy in mammalian fibroblasts.91 Removal of mitochondria during hypoxia is important to reduce ROS production and maintain oxygen homeostasis. Activation of hypoxia-inducible factor under hypoxic conditions induces expression of Nix and Bnip3, which at high levels disrupt the interaction between Bcl2 and Beclin1. Once released, Beclin 1 is free to nucleate pre-autophagosomal membranes and induce ATG5-dependent autophagy.95 This mechanism does not explain why mitochondria are selectively degraded in hypoxia. A recent study, however, identified the mitochondrial outer membrane protein, FUNDC1, as a mitochondrial receptor for hypoxia-induced mitophagy.96 FUNDC1 binds to LC3 through a conserved LIR motif and this interaction is strengthened under hypoxic conditions, facilitating engulfment of mitochondria by autophagosomes. It is attractive to hypothesize that Nix, Bnip3, and FUNDC1 function in a coordinate manner during hypoxia with the consequence that FUNDC1 selectively targets mitochondria to autophagosomes formed through Nix- and Bnip3-mediated activation of Beclin 1.
Two groups have recently described a novel developmental role for mitophagy in the elimination of paternal mitochondria from fertilized oocytes (Figure 3c).13, 14 In most eukaryotes, only maternal mitochondrial DNA is inherited, although sperms do contain mitochondria that are present in the oocyte immediately after fertilization. These paternal mitochondria are rapidly destroyed, although the evolutionary advantage this confers is unclear. Mechanistically it had not been known whether strict maternal inheritance was due to simple dilution of paternal mtDNA or active degradation of paternal mitochondria. The two studies in Caenorhabditis elegans show that an autophagic process accounts for selective degradation of sperm-derived mitochondria in early stages of embryogenesis. When autophagy is inhibited, paternal mitochondria persist but remain fragmented and unable to fuse with maternal mitochondria. While the fusion incompetence of paternal mitochondria may predispose them to mitophagy, the signal that activates this exceptional form of mitophagy selectively on sperm-derived mitochondria remains unknown.
Mitophagy in Neurons
Neurons represent a particularly interesting cell type for mitophagy because they combine high demand for mitochondria and high mitochondrial stress with a challenging cellular architecture. Neuronal demand for mitochondria is reflected in the high local density of mitochondria at presynaptic endings, post-synaptic densities, nodes of Ranvier, and in growth cones,97, 98 where mitochondrial function is required to sustain neuronal activity. Indeed, it is estimated that the brain of a resting human is responsible for 25% of oxygen consumption, although it consists of only 2% of the volume.99 In addition to their importance for ATP production in neurons, neuronal mitochondria also serve important Ca2+-buffering roles. Neurons are post-mitotic and non-proliferating cells; therefore, their mitochondria are particularly prone to accumulation of oxidative damage over time and high levels of Ca2+ influx in neurons may be a further stressor. Not surprisingly, mitochondrial damage and dysregulation of mitophagy have been implicated in several neurodegenerative disease associated with aging, such as PD,100 Alzheimer's disease (AD), and Huntington's disease (HD).101 Significantly, it is not clear whether it is the upregulation or downregulation of mitophagy that contributes to the pathology of some of these neurodegenerative diseases. In addition, the neuronal process of mitophagy remains enigmatic as pathways of mitophagy have mainly been characterized in non-neuronal cell lines. While the role of PINK1 and Parkin in non-neuronal mitophagy is well established and mutations in these two genes are associated with neurodegeneration in hereditary PD, it remains to be seen whether our knowledge of damage-induced mitophagy can be applied to neurons. In fact, it has been reported that unlike other cell types, depolarization of neuronal mitochondria does not lead to Parkin recruitment or their autophagic clearance.102 On the other hand, recruitment of Parkin to depolarized neuronal mitochondria and their Parkin-dependent mitophagy have been observed in other studies.69, 86 Clearly, further work is required to address the relevance of the PINK1/Parkin-mediated pathway of mitophagy in neurons.
Neurons face a unique challenge for mitochondrial turnover because a large fraction of the mitochondrial mass resides in distal axonal and dendritic processes, far away from the soma where most of the biogenesis is likely to occur and most of the lysosomes are thought to be located.103 The scarcity of lysosomes in axons may suggest that mitochondrial degradation preferentially occurs in the soma. While it may take days for axonal transport to deliver an organelle to the soma for degradation, rapid elimination of dysfunctional mitochondria is probably critical for protection of neurons against oxidative damage and degeneration. Not surprisingly, perhaps, neuronal autophagy plays a critical role in the maintenance of homeostasis in distal axons. Indeed, neurons are particularly sensitive to loss of autophagic genes.104 At basal levels, neuronal autophagy is highly efficient, and as a result, autophagosomes are rarely observed in neurons.105 In fact, multiple lines of evidence suggest that regulation of autophagy in neurons is distinct from other cell types.
The life cycle of damaged axonal mitochondria is poorly understood and it is not clear whether they need to be trafficked back to the cell body at any stage during their autophagic degradation. While preferential retrograde movement of depolarized mitochondria has been controversial,106, 107 the destruction of Miro and arrest of mitochondrial movement that occurs upon mitochondrial damage will prevent the retrograde transport of mitochondria on which the PINK1/Parkin pathway has been activated.69 If retrograde transport is required for the completion of mitophagy, it is therefore not clear at which stage of the process defective mitochondria are retrieved from axons. It may be advantageous to the neuron to first sequester the damaged organelle locally within an autophagosome before transport to the soma. Retrograde transport of the autophagosome would be independent of Miro, a mitochondrion-specific adaptor.108 Indeed, autophagic markers have been detected in axons, suggesting that autophagosomes can form in neuronal processes.109 In addition, distal formation and retrograde transport of autophagosomes in axons of primary neurons have been reported and mitochondrial markers have been observed in these autophagosomes.110 Nevertheless, it is not certain that retrograde transport of an autophagosome to the soma is strictly required as lysosomal markers have also been found to colocalize with autophagosomes in distal axons, implying that lysosomal degradation can be completed outside the soma.109, 110 It bears mentioning that the above studies have looked at the total population of autophagosomes, and the fate of mitophagosomes formed in response to damage in particular have not been analyzed. Understanding the dynamics of damaged neuronal mitochondria and the molecular players of neuronal mitophagy will help elucidate the contribution of mitochondrial dysfunction to neurodegenerative diseases.
Conclusion
How diverse will the pathways to mitophagy prove to be? From one perspective, considerable evidence in both yeast and mammalian cells indicates that all modes of mitophagy are harnessing the core machinery of autophagy. The important steps of autophagy, in which autophagosomes grow and surround their cargo before fusion with a lysosome, are equally pertinent to the engulfment and destruction of the mitochondria. However, the triggers that can activate this specialized and selective form of autophagy can be quite diverse, ranging from changes in the nutrient conditions of yeast media to the developmental transition from reticulocyte to erythrocyte and to damage or drug-induced loss of mitochondrial membrane potential. Several proteins are now identified that are critical in directing autophagosomes to the mitochondria, including ATG32, Nix, p62, PINK1, and Parkin. Ubiquitination of the mitochondrion, and/or the interaction of a mitochondrial receptor with the autophagosomal protein LC3 appear to be important in targeting of the autophagosome, but in each case the details of this recruitment remain unclear. More mysterious, perhaps, is the means by which the cues for mitophagy may initiate autophagosome formation before the association with mitochondrial membranes. Finally, there is interplay between control of mitochondrial dynamics – their movements, fissions, and fusions – and the progression of mitophagy; arrest and fragmentation of mitochondria after damage is likely to facilitate the selective clearance of damaged organelles. The existence of multiple pathways for mitophagy is likely to provide distinct control mechanisms for regulating constitutive turnover of mitochondria, rapid responses to damaged mitochondria, adjustments to mitochondrial status with nutrient availability, and highly specialized developmental needs.
Abbreviations
- ROS:
-
reactive oxygen species
- LAMP2A:
-
lysosome-associated membrane protein 2A
- ATG:
-
autophagy-re;ated gene
- TORC1:
-
target of rapamycin complex 1
- PI3K:
-
phosphatidyl inositol 3-kinase
- LC3:
-
microtubule-associated protein light chain 3
- CCCP:
-
carbonyl cyanide m-chlorophenylhydrazone
- Drp1:
-
dynain-related protein 1
- Opa1:
-
optic atrophy 1
- PINK1:
-
PTEN-induced putatieve protein kinase 1
- Miro:
-
mitochondria Rho-GTPase
References
Saraste M . Oxidative phosphorylation at the fin de siecle. Science 1999; 283: 1488–1493.
Wallace DC . A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 2005; 39: 359–407.
Parsons MJ, Green DR . Mitochondria in cell death. Essays Biochem 2010; 47: 99–114.
Hagen T, Yowe D, Bartholomew J, Wehr C, Do K, Park J et al. Mitochondrial decay in hepatocytes from old rats: membrane potential declines, heterogeneity and oxidants increase. Proc Natl Acad Sci USA 1997; 94: 3064–3069.
Langer T, Kaser M, Klanner C, Leonhard K . AAA proteases of mitochondria: quality control of membrane proteins and regulatory functions during mitochondrial biogenesis. Biochem Soc Trans 2001; 29: 431–436.
Karbowski M, Youle RJ . Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol 2011; 23: 476–482.
Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA et al. A Vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 2012; 22: 135–141.
Lipsky NG, Pedersen PL . Mitochondrial turnover in animal-cells – half-lives of mitochondria and mitochondrial subfractions of rat-liver based on [bicarbonate-C-14] Incorporation. J Biol Chem 1981; 256: 8652–8657.
Miwa S, Lawless C, von Zglinicki T . Mitochondrial turnover in liver is fast in vivo and is accelerated by dietary restriction: application of a simple dynamic model. Aging Cell 2008; 7: 920–923.
Clark SL . Cellular differentiation in the kidneys of newborn mice studied with the electron microscope. J Biophys Biochem Cytol 1957; 3: 349.
Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y . Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol 1992; 119: 301–301.
Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008; 454: 232–U66.
Sato M, Sato K . Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science 2011; 334: 1141–1144.
Al Rawi S, Louvet-Vallee S, Djeddi A, Sachse M, Culetto E, Hajjar C et al. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 2011; 334: 1144–1147.
Mizushima N, Komatsu M . Autophagy: renovation of cells and tissues RID C-3635-2009. Cell 2011; 147: 728–741.
Orenstein SJ, Cuervo AM . Chaperone-mediated autophagy: molecular mechanisms and physiological relevance. Semin Cell Dev Biol 2010; 21: 719–726.
Tanida I . Autophagy basics. Microbiol Immunol 2011; 55: 1–11.
Kanki T, Klionsky D, Okamoto K . Mitochondria autophagy in yeast. CORD Conf Proc 2011; 14: 1989–2001.
Klionsky D, Cregg J, Dunn W, Emr S, Sakai Y, Sandoval I et al. A unified nomenclature for yeast autophagy-related genes RID C-6449-2009. Dev Cell 2003; 5: 539–545.
Kanki T, Wang K, Klionsky DJ . A genomic screen for yeast mutants defective in mitophagy. CORD Conf Proc 2010; 6: 278–280.
Joo JH, Dorsey FC, Joshi A, Hennessy-Walters KM, Rose KL, McCastlain K et al. Hsp90–Cdc37 chaperone complex regulates Ulk1-and Atg13-mediated mitophagy. Mol Cell 2011; 43: 572–585.
Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N . Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci 2012; 125: 1488–1499.
Chu CT, Zhu J, Dagda R . Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: implications for neurodegeneration and cell death. Autophagy 2007; 3: 663–666.
Grishchuk Y, Ginet V, Truttmann AC, Clarke PGH, Puyal J . Beclin 1-independent autophagy contributes to apoptosis in cortical neurons. Autophagy 2011; 7: 1115–1131.
Radoshevich L, Murrow L, Chen N, Fernandez E, Roy S, Fung C et al. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 2010; 142: 590–60.
Tooze SA, Yoshimori T . The origin of the autophagosomal membrane. Nat Cell Biol 2010; 12: 831–835.
Yoshii SR, Kishi C, Ishihara N, Mizushima N . Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem 2011; 286: 19630–19640.
Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC . Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 2010; 12 747-U15.
Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010; 141: 656–667.
Kanki T, Wang K, Baba M, Bartholomew CR, Lynch-Day MA, Du Z et al. A Genomic screen for yeast mutants defective in selective mitochondria autophagy. Mol Biol Cell 2009; 20: 4730–4738.
Kissova I, Salin B, Schaeffer J, Bhatia S, Manon S, Camougrand N . Selective and non-selective autophagic degradation of mitochondria in yeast. Autophagy 2007; 3: 329–336.
Priault M, Salin B, Schaeffer J, Vallette F, di Rago J, Martinou J . Impairing the bioenergetic status and the biogenesis of mitochondria triggers mitophagy in yeast. Cell Death Differ 2005; 12: 1613–1621.
Nowikovsky K, Reipert S, Devenish RJ, Schweyen RJ . Mdm38 protein depletion causes loss of mitochondrial K+/H+ exchange activity, osmotic swelling and mitophagy. Cell Death Differ 2007; 14: 1647–1656.
Campanella M . Seraphim A, Abeti R, Casswell E, Echave P, Duchen MR. IF1, the endogenous regulator of the F1Fo-ATPsynthase, defines mitochondrial volume fraction in HeLa cells by regulating autophagy. Biochim Biophys Acta 2009; 1787: 393–401.
Narendra D, Tanaka A, Suen D, Youle RJ . Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183: 795–803.
Kim I, Lemasters JJ . Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid Redox Signal 2011; 14: 1919–1928.
Suen D, Narendra DP, Tanaka A, Manfredi G, Youle RJ . Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci USA 2010; 107: 11835–11840.
Gilkerson RW, De Vries RL, Lebot P, Wikstrom JD, Torgyekes E, Shirihai OS et al. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum Mol Genet 2012; 21: 978–990.
de Vries RL, Gilkerson RW, Przedborski S, Schon EA . Mitophagy in cells with mtDNA mutations: being sick is not enough. Autophagy 2012; 8: 699–700.
Kanki T, Klionsky DJ . Mitophagy in yeast occurs through a selective mechanism. J Biol Chem 2008; 283: 32386–32393.
Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ . Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 2009; 17: 98–109.
Okamoto K, Kondo-Okamoto N, Ohsumi Y . Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 2009; 17: 87–89.
Kissova I, Deffieu M, Manon S, Camougrand N . Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem 2004; 279: 39068–39074.
He C, Song H, Yorimitsu T, Monastyrska I, Yen W, Legakis JE et al. Recruitment of Atg9 to the preautophagosomal structure by Atg11 is essential for selective autophagy in budding yeast. J Cell Biol 2006; 175: 925–933.
Geisler S, Holmstroem KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12: 119–U70.
Narendra DP, Kane LA, Hauser DN, Fearnley IM, Youle RJ . p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010; 6: 1090–1106.
Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA 2007; 104: 19500–19505.
Youle RJ, Narendra DP . Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011; 12: 9–14.
Orvedahl A, Sumpter Jr R, Xiao G, Ng A, Zou Z, Tang Y et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011; 480: 113–117.
Okamoto K, Shaw JM . Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet 2005; 39: 503–536.
Hoppins S, Lackner L, Nunnari J . The machines that divide and fuse mitochondria. Annu Rev Biochem 2007; 76: 751–780.
Twig G, Liu X, Liesa M, Wikstrom JD, Molina AJ, Las G et al. Biophysical properties of mitochondrial fusion events in pancreatic beta-cells and cardiac cells unravel potential control mechanisms of its selectivity. Am J Physiol Cell Physiol 2010; 299: C477–C478.
Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008; 27: 433–446.
Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM . Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol 2009; 187: 959–966.
Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L . The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. CORD Conf Proc 2010; 5 e10054.
Tanaka A, Cleland MM, Xu S, Narendra DP, Suen D, Karbowski M et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 2010; 191: 1367–1380.
Ziviani E, Tao RN, Whitworth AJ . Drosophila Parkin requires PINK1 for mitochondrial translocation and ubiquitinates Mitofusin. Proc Natl Acad Sci USA 2010; 107: 5018–5023.
Twig G, Shirihai OS . The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal 2011; 14: 1939–1951.
Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J . Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA 2011; 108: 10190–10195.
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004; 304: 1158–1160.
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S et al. Mutations in the Parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998; 392: 605–608.
Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ . Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA 2003; 100: 4078–4083.
Park J, Lee S, Lee S, Kim Y, Song S, Kim S et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006; 441: 1157–1161.
Dagda RK, Cherra SJ, Kulich SM, Tandon A, Park D, Chu CT . Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 2009; 284: 13843–13855.
Chen D, Gao F, Li B, Wang H, Xu Y, Zhu C et al. Parkin mono-ubiquitinates BCL-2 and regulates autophagy. J Biol Chem 2010; 285: 38214–38223.
Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ . The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USA 2008; 105: 1638–1643.
Yu W, Sun Y, Guo S, Lu B . The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum Mol Genet 2011; 20: 3227–3240.
Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RLJ et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 2011; 20: 1726–1737.
Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011; 147: 893–906.
Meissner C, Lorenz H, Weihofen A, Selkoe DJ, Lemberg MK . The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem 2011; 117: 856–867.
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. CORD Conf Proc 2010; 8 e1000298.
Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, SHY Loh et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 2011; 20: 867–879.
Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME et al. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 2012; 13: 378–385.
Lazarou M, Jin SM, Kane LA, Youle RJ . Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 2012; 22: 320–333.
Shiba K, Arai T, Sato S, Kubo S, Ohba Y, Mizuno Y et al. Parkin stabilizes PINK1 through direct interaction. Biochem Biophys Res Commun 2009; 383: 331–335.
Kim Y, Park J, Kim S, Song S, Won S, Lee S et al. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun 2008; 377: 975–980.
Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 2010; 189: 211–212.
Lim KL, Dawson VL, Dawson TM . Parkin-mediated lysine 63-linked polyubiquitination: a link to protein inclusions formation in Parkinson's and other conformational diseases? Neurobiol Aging 2006; 27: 524–529.
Lee J, Nagano Y, Taylor JP, Lim KL, Yao T . Disease-causing mutations in Parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol 2010; 189: 671–679.
Ding W, Ni H, Li M, Liao Y, Chen X, Stolz DB et al. Nix Is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin–ubiquitin–p62-mediated mitochondrial priming. J Biol Chem 2010; 285: 27879–27890.
Van Humbeeck C, Cornelissen T, Vandenberghe W, Ambra1 A . Parkin-binding protein involved in mitophagy. Autophagy 2011; 7: 1555–1556.
Van Humbeeck C, Cornelissen T, Hofkens H, Mandemakers W, Gevaert K, De Strooper B et al. Parkin interacts with Ambra1 to induce mitophagy. J Neurosci 2011; 31: 10249–10261.
Michiorri S, Gelmetti V, Giarda E, Lombardi F, Romano F, Marongiu R et al. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ 2010; 17: 962–974.
Shin J, Ko HS, Kang H, Lee Y, Lee Y, Pletinkova O et al. PARIS (ZNF746) repression of PGC-1 alpha contributes to neurodegeneration in Parkinsonss disease. Cell 2011; 144: 689–702.
Melrose HL, Lincoln SJ, Tyndall GM, Farrer MJ . Parkinson's disease: a rethink of rodent models. Exp Brain Res 2006; 173: 196–204.
Cai Q, Zakaria HM, Simone A, Sheng ZH . Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol 2012; 22: 545–552.
Maro B, Marty MC, Bornens M . In vivo and in vitro effects of the mitochondrial uncoupler FCCP on microtubules. EMBO J 1982; 1: 1347–1352.
Giusti C, Luciani MF, Klein G, Aubry L, Tresse E, Kosta A et al. Necrotic cell death: from reversible mitochondrial uncoupling to irreversible lysosomal permeabilization. Exp Cell Res 2009; 315: 26–38.
Seibler P, Graziotto J, Jeong H, Simunovic F, Klein C, Krainc D . Mitochondrial parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J Neurosci 2011; 31: 5970–5976.
Ney PA . Normal and disordered reticulocyte maturation. Curr Opin Hematol 2011; 18: 152–157.
Novak I, Dikic I . Autophagy receptors in developmental clearance of mitochondria. Autophagy 2011; 7: 301–303.
Schwarten M, Mohrlüder J, Ma P, Stoldt M, Thielmann Y, Stangler T et al. Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy 2009; 5: 690–698.
Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 2010; 11: 45–51.
Kanki T . Nix, a receptor protein for mitophagy in mammals. Autophagy 2010; 6: 433–435.
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 2008; 283: 10892–10903.
Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 2012; 14: 177–185.
Morris RL, Hollenbeck PJ . The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci 1993; 104: 917–922.
Berthold CH, Fabricius C, Rydmark M, Andersen B . Axoplasmic organelles at nodes of ranvier.1. Occurrence and distribution in large myelinated spinal root axons of the adult cat. J Neurocytol 1993; 22: 925–940.
Magistretti P . Brain energy metabolism In: Fundamental Neuroscience Zigmond MJ, Bloom FE, Landis SC, Roberts JL, Squire L (eds). Academics Publisher: San Diego, CA, 1999 p 389.
Schapira AHV . Mitochondrial pathology in Parkinson's disease. Mount Sin J Med 2011; 78: 872–881.
Batlevi Y, La Spada AR . Mitochondrial autophagy in neural function, neurodegenerative disease, neuron cell death, and aging. Neurobiol Dis 2011; 43: 46–51.
Van Laar VS, Arnold B, Cassady SJ, Chu CT, Burton EA, Berman SB . Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum Mol Genet 2011; 20: 927–940.
Holtzman E, Novikoff AB . Lysomes in the rat sciatic nerve following crush. J Cell Biol 1965; 27: 651–669.
Yue Z, Friedman L, Komatsu M, Tanaka K . The cellular pathways of neuronal autophagy and their implication in neurodegenerative diseases. Biochim Biophys Acta Mol Cell Res 2009; 1793: 1496–1507.
Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci 2008; 28: 6926–6937.
Miller KE, Sheetz MP . Axonal mitochondrial transport and potential are correlated. J Cell Sci 2004; 117: 2791–2804.
Verburg J, Hollenbeck PJ . Mitochondrial membrane potential in axons increases with local nerve growth factor or semaphorin signaling. J Neurosci 2008; 28: 8306–8315.
Glater EE, Megeath LJ, Stowers RS, Schwarz TL . Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol 2006; 173: 545–557.
Wang QJ, Ding Y, Kohtz S, Mizushima N, Cristea IM, Rout MP et al. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci 2006; 26: 8057–8068.
Maday S, Wallace KE, Holzbaur EL . Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol 2012; 196: 407–411.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by M Piacentini
Rights and permissions
About this article
Cite this article
Ashrafi, G., Schwarz, T. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20, 31–42 (2013). https://doi.org/10.1038/cdd.2012.81
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2012.81
Keywords
This article is cited by
-
DsbA-L ameliorates renal aging and renal fibrosis by maintaining mitochondrial homeostasis
Acta Pharmacologica Sinica (2024)
-
Mitochondrial transfer mediates endothelial cell engraftment through mitophagy
Nature (2024)
-
Activation of Ca2+ phosphatase Calcineurin regulates Parkin translocation to mitochondria and mitophagy in flies
Cell Death & Differentiation (2024)
-
A new perspective on mesenchymal stem cell-based therapy for liver diseases: restoring mitochondrial function
Cell Communication and Signaling (2023)
-
The worsening of skeletal muscle atrophy induced by immobilization at the early stage of remobilization correlates with BNIP3-dependent mitophagy
BMC Musculoskeletal Disorders (2023)
