
Abstract
Autophagy is a lysosomal pathway for cellular homeostasis control. Both non-selective bulk autophagy and selective autophagy of specific proteins or organelles have been found. Selective autophagy prevents cells from pathogen invasion and stress damage, but its role in regulating transcriptional factors is not clear. Using a macrophage cell differentiation model, the role of autophagy in nuclear factor-κB (NF-κB) regulation is investigated. The bone marrow-derived macrophages (BMDMs) will differentiate into a M2-like phenotype in the presence of hepatoma tumor cell condition medium (CM). The TLR2 signaling drives this M2 polarization and causes NF-κB p65 degradation via lysosome-dependent pathway. The CM-induced ubiquitinated- NF-κB p65 forms aggresome-like structures (ALS) in the cytoplasm of cultured and hepatoma-associated M2 macrophages. This NF-κB p65-contained ALS is recognized by p62/SQSTM1 and degraded by selective autophagy. Treatment with the lysosomal inhibitor bafilomycin A1 or the knockdown of Atg5 can prevent CM-induced NK-κB p65 degradation and induce M2 macrophages to produce a high level of pro-inflammatory cytokines. Furthermore, TLR2 signal induces sustained phosphorylation of extracellular signal-regulated kinase 1/2 to facilitate this autophagy-dependent NF-κB regulation. Our finding provides a novel pathway of NF-κB regulation by p62/SQSTM1-mediated selective autophagy.
Similar content being viewed by others


Main
Autophagy is an evolutionarily conserved lysosome-dependent system in eukaryotes to regulate the turnover of cellular proteins and organelles. This self-eating system has been shown to control various cellular functions, including immune activation and cancer generation.1, 2 During autophagy, target proteins or organelles are delivered into double-membrane autophagosomes for lysosomal degradation. Although different kinds of cargos are found in autophagosomes, emerging studies have indicated that specialized target protein aggregates will selectively be recognized by autophagy, particularly ubiquitylated protein aggregates or pathogens.3, 4 The ubiquitin-binding proteins, such as p62/SQSTM1 (sequestosome 1), histone deacetylase 6 (HDAC6), neighbor of BRCA1 gene 1 (NBR1), nuclear dot protein 52 kDa (NDP52) and optineurin are responsible for this selective autophagy degradation.5, 6, 7, 8, 9
The transcriptional factor nuclear factor-κB (NF-κB) plays a central role in the regulation of inflammatory responses and macrophage differentiation. Both classical M1 and alternative M2 macrophage differentiation require NF-κB activation.10 Most tumor infiltrating macrophages are characterized by an anti-inflammatory M2 phenotype with a high production of IL-10, but low IL-12. Although NF-κB activation is essential for M2 macrophage induction, isolated tumor-associated macrophages (TAM) from various tumors represent less NF-κB activity.10, 11 It is still not clear how these NF-κBs are downregulated. NF-κB can be activated by toll-like receptor (TLR) signaling in myeloid cells to induce degradation of the NF-κB inhibitor, IkBα, and cause its nuclear translocation.12 After activation, NF-κB will be terminated by two established mechanisms, described previously. One is to export the activated-NF-κB from the nucleus with newly synthesized IkBα by NF-κB itself, and the other is through proteasome-dependent nuclear degradation of ubiquitinated-NF-κB by two E3 ligases, copper metabolism gene MURR1 domain (COMMD1) and protein-containing nuclear LIM domain (PDLIM2).13, 14, 15 Since autophagy is also induced by TLR signaling in myeloid cells,16 we hypothesized that ubiquitinated-NF-κB by TLR stimulation might act as a target for selective autophagy to terminate NF-κB activation.
In this study, we investigated the role of autophagy on NF-κB regulation in hepatoma condition medium (CM)-induced M2 macrophage differentiation. We found that a TLR2-dependent signal from CM is required for M2 macrophage differentiation. CM induced NF-κB p65 activation, ubiquitination and the formation of aggresome-like structures (ALS) in the cytoplasm of bone marrow-derived macrophages (BMDMs). The NF-κB p65-containing ALS is recognized by p62/SQSTM1 and degraded via selective autophagy. A sustained phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2) by a TLR2-related signal is required to facilitate this selective autophagy. Our finding provides a novel pathway of NF-κB regulation by p62/SQSTM1-mediated selective autophagy.
Results
Tumor cell CM induced bone marrow-derived macrophage to undergo M2 differentiation by TLR2-dependent signal
Tumor-derived factors from different origins have been shown to polarize macrophages to a M2-like phenotype. Recently, a study reported that lung cancer cells can activate macrophages by TLR2 signaling to promote tumor metastasis.17 Here we examined the role of TLR2 in hepatoma cell-induced macrophage differentiation from bone marrow-derived macrophages (BMDM). We found that mouse hepatoma cell line ML-14a CM (MCM) stimulated BMDM to express arginase-1 and SOCS-3 (Figure 1a). Furthermore, the reduced production of TNF-α and IL-12p40, but the increased production of IL-10 in MCM-treated BMDM after lipopolysaccharides (LPS) stimulation, indicated that a M2-phenotype macrophage was induced by hepatoma-derived factors (Supplementary Figure 1). In contrast, no arginase-1 and SCOS3 were induced in MCM-treated TLR2−/− BMDMs, whereas STAT-1 phosphorylation, high expression of IL-12p40 and MHC-II were detected in MCM-treated TLR2−/− BMDMs, suggesting that MCM can induce a TLR2-dependent M2 polarization (Figures 1b and c).

Tumor cell CM induced bone marrow-derived macrophages to undergo M2 differentiation by TLR2-dependent signal. (a) MCM induces TLR2-dependent M2 macrophage differentiation. Cell lysates of MCM-stimulated BMDMs from WT or TLR2−/− mice were collected at indicated time. M2-related protein expressions were detected by Western blotting. (b) Cytokine profiles of MCM-stimulated wild type (WT) or TLR2−/− BMDMs. BMDMs from B6 WT or TLR2−/− mice were treated with MCM for 24 h, and the supernatants were collected for further detection of IL-10, IL-12p40 and TNF-α by ELISA. (c) The surface molecule expression on MCM-treated BMDMs. BMDMs from WT or TLR2−/− mice were cocultured with MCM for 24 h to analyze the surface molecules by flow cytometry. *P<0.05; **P<0.001; ***P<0.0001
NF-κB p65 degradation by autophagy in MCM-treated BMDMs
NF-κB activities are usually suppressed in TAMs.18 On proceeding to check the activation status of NF-κB in hepatoma-polarized M2-like macrophages, it was found that while MCM would stimulate NF-κB p65 nuclear translocation from 3 to 12 h post treatment in WT BMDMs, this was only observed at 12 h in TLR2−/− BMDMs (Supplementary Figure 2). However, the protein level of NF-κB p65 in the cytoplasm was dramatically decreased after 24 h post MCM stimulation in WT BMDMs. This decline in NF-κB p65 expression was attenuated in MCM-stimulated TLR2−/− BMDMs (Figure 2a). To verify the role of lysosomes or proteasomes in MCM-induced NF-κB p65 degradation, the lysosomal inhibitor, bafilomycin A1, or proteasome inhibitor, MG132, was used. Pretreatment with bafilomycin A1, but not MG132, recovered the protein level of NF-κB p65 and increased both NF-κB transcriptional activity and suppressor of cytokine signaling 3 (SOCS3) expression in MCM-treated BMDMs (Figures 2b and c). Furthermore, pretreatment of bafilomycin A1 drove MCM-treated BMDMs to produce pro-inflammatory cytokines, such as IL-6, IL-12 p40 and TNF-α, but not IL-10 (Figure 2d). This lysosome-dependent NF-κB degradation by MCM was limited to p65, and did not include p50 (Figure 2e). These results indicate that MCM induces NF-κB p65 degradation by lysosomes to regulate its activity. Autophagy is a lysosomal degradation process for cytoplasmic substances and organelles, and so when proteins are targeted by autophagy, this leads to lysosome-dependent degradation. Next we investigated the role of autophagy in MCM-induced NF-κB p65 degradation in macrophages. According to the results shown in Figures 3a–c, MCM induced a double-layer membrane of autophagosome formation, light chain 3-II (LC3-II) accumulation and conversion in BMDMs. Pretreatment of autophagy inhibitor, 3-methyladenine, inhibited the MCM-induced NF-κB p65 degradation at 24 h. To further examine the role of autophagy in MCM-induced NF-κB p65 degradation, the Atg5 was silenced by a lentiviral vector containing Atg5 shRNA in RAW 264.7 cells. The knockdown of Atg5 by lentiviral vector-introduced shRNA can prevent MCM-induced NF-κB p65 degradation in a MCM dose-dependent manner, suggesting that NF-κB p65 was degraded by autophagy (Figures 3d and e). These results indicate that MCM induced autophagy-dependent NF-κB degradation in M2 macrophage differentiation.

Lysosomal degradation of NF-κB p65 limits M1 polarization by MCM. (a) TLR2-related NF-κB p65 degradation induced by MCM. WT or TLR2−/− BMDMs were treated with MCM and the cell lysates were collected at indicated time. The expression of NF-κB p65, arginase-1 (arg-1), and SOCS3 was determined by western blotting. (b and c) Lysosome-dependent NF-κB p65 degradation in MCM-treated BMDMs. BMDMs were pretreated with DMSO, bafilomycin A1 (Baf-A1, 25 nM) or MG132 (1 μM) for 30 min prior to adding the MCM. After 24 h post treatment, the cell lysates were collected to detect NF-κB p65, LC3-I/II and SOCS3 expression by western blotting. To determine the NF-κB activities, RAW-Blue cells were pretreated with DMSO, bafilomycin A1 or MG132 for 30 min and incubated with DMDM or MCM. All cell supernatants were collected to analyze NF-κB related activity as described in materials and methods. (d) Lysosome-dependent production of pro-inflammatory cytokines in MCM-treated BMDMs. BMDMs were pretreated with Baf-A1 (25 nM) for 30 min before adding MCM. After 24 h post treatment, cell supernatants were collected to detect the level of several cytokines by ELISA. (e) Lysosome-related NF-κB p65 degradation, but not p50, in MCM-treated BMDMs. BMDMs were pretreated with DMSO or Baf-A1 for 30 min, and MCM was then added for further 24, 48 and 72 h. The cell lysates were collected at indicated times to determine NF-κB p65, p50, arginase-1 (Arg-1), SOCS3 and LC3-I/II expression by western blotting. **P<0.001; ***P<0.0001

NF-κB p65 degradation is primarily mediated by autophagy (a) Electron micrographs of MCM-treated BMDMs. The autophagic vesicles with a double-layer (arrow head) are shown. (b) MCM induces LC3 aggregation. BMDMs were treated with MCM for 6 h and stained with anti-LC3 antibody. The LC3 aggregation was observed by confocal microscopy. (c) MCM induces LC3-II conversion. BMDMs were treated with various dilutions of MCM for 24 h. The LC3-II conversion was determined by western blotting. (d) Inhibition of autophagy suppresses MCM-induced NF-κB p65 degradation. BMDMs were pretreated with DMSO, Baf-A1 (25 nM) or 3-methyladenine (3-MA, 2 mM) for 30 min prior to adding MCM. The cell lysates were collected at indicated time to determine NF-κB p65 and LC3-I/II expression by western blotting. (e) Knockdown of Atg 5 by shRNA attenuates MCM-induced NF-κB p65 degradation. RAW264.7 cells which stably expressed luciferase or Atg5 shRNA were treated with various dilutions of MCM for 24 h. The expression level of NF-κB p65, Atg5, and LC3-I/II were determined by western blotting
p62/SQSTM1-mediated selective autophagy degrades ubiquitinated-NF-κB formed ALS
We next investigated how NF-κB is targeted by autophagy. By immunostaining observation, NF-κB p65 mainly translocated to the nucleus at 3 h, then was exported to the cytoplasm with IκBα and formed ALS in BMDMs from 6 h post treatment of MCM. However, some NF-κB p65-containing ALS did not incorporate with IκBα after 12 h post treatment (indicated by an arrow, Figure 4a). In addition to IκBα, NF-κB p65 was colocalized with LC3, p62/SQSTM1 or ubiquitin in ALS at 12 h post treatment of MCM in BMDMs (Figure 4b). This suggests that MCM might induce NF-κB p65 ubiqutination, which is then recognized by an ubiquitin-binding adapter protein p62/SQSTM1. The immunoprecipitation assay was performed to examine the interaction of ubiquitin, p62/SQSTM1 and NF-κB p65. It was found that ubiquitinated-NF-κB p65 was formed, and persisted from 3 to 12 h post MCM treatment in BMDMs (Figure 4c). The induction of ubiquitinated-NF-κB p65 by MCM, as well as LTA-SA or LPS, was mainly in the cytoplasm of BMDMs and associated with p62/SQSTM1 (Figures 4d and e). The nuclear ubiquitinated-NF-κB p65 only presented in LPS-treated BMDMs. To examine the role of p62/SQSTM1 in autophagy-dependent NF-κB p65 degradation, the p62/SQSTM1 was silenced by a lentiviral vector containing p62/SQSTM1 shRNA in RAW 264.7 cells. According to the results shown in Figure 4f, compared with the control shRNA-expressed RAW 264.7 cells, MCM-induced NF-κB p65 degradation and SOCS3 induction were both suppressed when p62/SQSTM1 was silenced in RAW 264.7 cells. These results demonstrate that p62/SQSTM1-mediated selective autophagy degrades NF-κB p65 in MCM-treated BMDMs.

p62/SQSTM1 mediates selective autophagy to degrade NF-κB p65. (a) NF-κB p65, coupled with IκBα, forms ALS in MCM-treated BMDMs. BMDMs were treated with MCM for 3 to 12 h and stained with anti-NF-κB p65 and anti-IκBα antibodies at indicated time. The distributions of these proteins were analyzed by confocal microscopy. Arrows indicate NF-κB p65-ALS without incorporation of IκBα. (b) Ubiquitinated NF-κB p65 was associated with p62/SQSTM1 and LC3 to form ALS. BMDMs were treated with MCM for 12 h to fix and stain with anti-NF-κB p65, ubiquitin, p62/SQSTM1 and LC3. The distributions of these proteins were analyzed by confocal microscopy. (c) MCM induces ubiquitination of NF-κB p65. After treatment of MCM at indicated time, the NF-κB p65 of BMDMs was immunoprecipitated with anti-NF-κB p65 antibody. The bound ubiquitin on NF-κB p65 was analyzed by western blotting. (d) Ubiquitination of cytoplasmic and nuclear NF-κB p65 by MCM, LTA-SA and LPS. The cytoplasmic and nuclear extracts from MCM, LTA-SA or LPS-treated BMDMs were immunoprecipitated with anti-NF-κB p65 antibody and immunoblotted for ubiquitin. (e) Immunoprecipitation with NF-κB p65 and p62/SQSTM1. Cell extracts from MCM, LTA-SA or LPS-treated BMDMs were immunoprecipitated with anti-NF-κB p65 antibody and immunoblotted for p62/SQSTM1. (f) p62/SQSTM1 is required for MCM-induced NF-κB p65 degradation. RAW264.7 cells which stably expressed control or p62/SQSTM1 shRNA were treated with various dilutions of MCM for 24 h. The expression levels of NF-κB p65, SOCS3 and p62/SQSTM1 were determined by western blotting
Hepatoma-associated M2 macrophages, but not M1-polarized macrophages, carry NF-κB p65-containing ALS
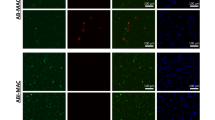
We showed that MCM induces BMDMs to differentiate into M2 macrophages with NF-κB p65-containng ALS formation, which is then degraded by autophagy. Next, we examined this ALS-formed NF-κB p65 induction and its turnover in M1-polarized macrophages. TNF-α, IFN-γ, IFN-γ/TNF-α and LPS were used to induce BMDMs to M1 polarization.19 As shown in Supplementary Figures 3a and b, IFN-γ, IFN-γ/TNF-α and LPS stimulated a high expression of MHC-II or inducible NO synthase in BMDMs, which suggests that M1 macrophages were induced. However, compared to MCM- or LTA-treated BMDMs, no declined NF-κB p65 or NF-κB p65-LC3-containing ALS was detected in these polarized M1 macrophages (Supplementary Figures 3b and c). This indicates that the activity of NF-κB p65 is not inhibited by autophagy in M1 macrophage polarization. Since hepatoma cell CM can force NF-κB p65 to form ALS and limit its activity in cultured BMDMs, we next proceeded to check NF-κB p65-containing ALS in hepatoma-associated macrophages. Mice hepatoma cells ML-14a were implanted in mice to establish in situ hepatoma in mice liver as previously described.20 After two weeks post inoculation, the liver tissue sections were stained for CD68, a macrophage marker, and NF-κB p65 by immunostaining. According to the results shown in Figure 5, compared with the non-tumor area, a lot of infiltrating-CD68+ macrophages were found in the tumor area. Many of these hepatoma-associated macrophages carried NF-κB p65-containing ALS (indicated by arrowheads), while no such NF-κB p65 aggregates were found in non-tumor area macrophages. These results indicate that hepatoma-derived factors induce NF-κB p65 to form ALS in cultured BMDMs and hepatoma-associated macrophages, which might restrict NF-κB p65 activities in these M2 polarized macrophages.

Hepatoma-associated macrophages carry NF-κB p65-containing ALS. Intrasplenic-injection of ML-14a cells established tumor nodule formation in the liver of BABL/c mice. After two weeks of ML-14a inoculation, the liver sections of hepatoma-bearing mice were collected to stain with CD68 (indicated by green), NF-κB p65 (indicated by purple), and Hoechst (indicated by blue). The hepatoma-associated macrophages which carry NF-κB p65-containing ALS are indicated by arrowheads
Sustained phosphorylation of ERK1/2 by TLR2 stimulation is crucial for autophagy-dependent NF-κB p65 degradation
MCM-induced NF-κB p65 degradation was partially reversed in TLR2−/− BMDMs, suggesting that the TLR2 signal can trigger this lysosome-mediated degradation pathway. To further determine the role of TLR2 or TLR4 signaling in autophagy-dependent NF-κB p65 degradation, BMDMs were stimulated with TLR2- or TLR4-specific ligands, LTA-SA or LPS, respectively, in the following experiments. It was found that LTA-SA and MCM, but not LPS, induced TLR2-dependent NF-κB p65 degradation and LC3-II conversion in BMDMs (Figure 6a). The LTA-SA-caused NF-κB p65 degradation was inhibited by lysosomal inhibitors, such as bafilomycin A1, chloroquine (CQ) and NH4Cl (Figure 6b). Although NF-κB p65 was associated with p62/SQSTM1 under LTA-SA and LPS stimulation, the subsequent LC3-II recruitment to NF-κB p65 was only observed in LTA-SA-treated BMDMs (Figures 4e and 6c). These results suggest that distinct downstream signals from TLR2 and TLR4 might control this lysosomal regulation of NF-κB p65. A previous study has indicated that sustained phosphorylation of ERK1/2 triggered by TLR2 will instruct dendritic cells to induce T helper cell responses which are different from those of TLR4.21 Here, we found that LTA-SA induced sustained phosphorylation of ERK1/2 from 3 to 12 h post treatment in BMDMs, which represents an early conversion to LC3-II and NF-κB p65 degradation (24 h). However, only transient phosphorylation of ERK1/2 was observed (at 0.5 and 3 h) in LPS-treated BMDMs (Figure 7a). To further examine the role of this sustained phosphorylation of ERK1/2 in TLR2-caused NF-κB p65 degradation, an inhibitor, U0126, was used to interfere with the ERK1/2 phosphorylation at various time points. As shown in Figure 7b, inhibition of ERK1/2 phosphorylation by U0126 at 0, 3, 6 and 12 h, but not 18 h post treatment was able to restore the NF-κB p65 protein level in LTA-SA-treated BMDMs, indicating that sustained ERK1/2 phosphorylation before 12 h is required for LTA-SA-caused NF-κB p65 degradation. Although this sustained-phosphorylated ERK1/2 was not responsible for NF-κB p65 targeting by p62/SQSTM1 or autophagosomes, we further found that it is crucial for triggering NF-κB p65-containing autophagosomes to fuse with lysosomes (Supplementary Figure 4 and Figure 7c). Thus, our results demonstrate that TLR2-triggered sustained phosphorylation of ERK1/2 regulates the autophagy process to degrade NF-κB p65.

TLR2-dependent lysosomal degradation of NF-κB p65. (a) TLR2-depedent NF-κB p65 degradation. WT or TLR2−/− BMDMs were treated with MCM, LTA-SA and LPS for 24 h. The cell lysates were collected to detect NF-κB p65 and LC3-I/II expression by western blotting. Densitometric analysis of NF-κB p65 band intensity was normalized to each β-actin level. (b) LTA-SA induces lysosomal degradation of NF-κB p65. BMDMs were pretreated with Baf-A1 (25 nM), CQ (20 μM) or NH4Cl (20 mM) for 30 min prior to adding LTA-SA or LPS. After 24 h post treatment, the cell lysates were collected to detect NF-κB p65 and LC3-I/II expression by western blotting. (c) NF-κB p65 cellular distribution in LTA-SA or LPS-treated BMDMs. BMDMs were treated with LTA-SA or LPS for 12 h to fix and stain with anti-NF-κB p65, p62/SQSTM1 and LC3. The distributions of these proteins were analyzed by confocal microscopy

Sustained phosphorylation of ERK1/2 by TLR2 signal triggers autophagy to target NF-κB p65. (a) LTA-SA induces sustained phosphorylation of ERK1/2. BMDMs were stimulated with LTA-SA or LPS and the cell lysates were collected at the indicated time. The expressions of phospho-ERK1/2, total ERK, NF-κB p65, and LC3-I/II were analyzed by western blotting. (b) Interference of sustained phosphorylation of ERK1/2 by U0126 prevents NF-κB p65 degradation. BMDMs were treated with LTA-SA and U0126 (5 μM) was added from 0, 3, 6, 12, 18 h post LTA-SA treatment. All total cell lysates were collected at 24 h to detect protein expression of phospho-ERK1/2, total ERK and NF-κB p65 by western blotting. (c) ERK1/2 activation is required to deliver NF-κB p65 to lysosomes by LTA-SA stimulation. BMDMs were pretreated with DMSO or U0126 and then incubated with PBS, LPS, or LTA-SA for 12 h. All cells were fixed and stained with anti-NF-κB p65 and LAMP-1. The distributions of these proteins were analyzed by confocal microscopy
Discussion
Selective autophagy protects cells from metabolic stress and pathogen invasion, and even tumor generation. Here we describe how p62/SQSTM1-mediated selective autophagy limits NF-κB p65 activities in tumor-induced M2 macrophage differentiation. A TLR2-dependent signal from CM or agonists stimulates accumulation of ubiquitinated NF-κB p65 in the cytoplasm, and recruits ubiquitin binding protein p62/SQSTM1 to form ALS in BMDMs. This NF-κB p65-containing ALS is subsequently recognized by autophagosomes, and then degraded through lysosomes. The sustained phosphorylation of ERK1/2 is required for this TLR2-induced NF-κB p65 degradation by promoting autophagosome maturation.
Regulation of NF-κB activity controls various cellular functions, including tumor-derived macrophage differentiation.11 In this study, we noticed that TLR2-related signals from the hepatoma CM rapidly induce NF-κB p65 activation in BMDMs, but the protein level decreases after 24 h post stimulation. Although many different kinds of stimulations can activate NF-κB signaling, little is known about the detailed mechanisms to terminate it. IκBα has been considered to play a crucial role in stopping NF-κB p65 activity by exporting activated-NF-κB p65 from the nucleus.15 Our findings here further demonstrate that these exported NF-κB p65-IκBα complexes form cytosolic ALS, which are recognized and degraded by autophagosomes to terminate NF-κB p65 activation, under TLR2 signaling. However, we also found some NF-κB p65-containing ALS without IκBα incorporation. This might indicate that IκBα will dissociate from NF-κB p65-containing ALS at a later time or that an IκBα-independent pathway can also force this ALS formation, a question which remains for further investigation. Previous studies have demonstrated that ubiquitination of NF-κB by nuclear E3 ligases promotes its nuclear degradation by proteasomes under LPS or TNF-α stimulation.13, 14 However, we surprisingly found cytosolic ubiquitination of NF-κB p65 in LPS, LTA-SA or MCM-stimulated BMDMs, in addition to nuclear ubiquitination, indicating that distinct E3 ligases from COMMD1 and PDLIM2, particularly in the cytoplasm, are responsible for NF-κB p65 ubiquitination, and this therefore needs further investigation. Interestingly, only LTA-SA and MCM, but not LPS, cause ubiquitinated-NF-κB p65 aggregates in the perinucleus of BMDMs, suggesting that different ubiquitin chains are conjugated to NF-κB p65. Although it is not clear why aggregated NF-κB p65 is only formed in TLR2-related ligand stimulation, this aggregated-protein complex will trigger the autophagy process and terminate the NF-κB activity.
p62/SQSTM1 acts as a cargo receptor for selective autophagy to clear intracellular pathogens, aggregated proteins or organelles through its ubiquitin-binding domain.5, 22, 23 This p62/SQSTM1-mediated protein degradation has recently been found to negatively regulate Wnt/β-catenin signaling, which implies that selective autophagy could control cell signaling.24 p62/SQSTM1 can be activated by TLR signals to ubiquitinate TRAF6 and facilitate NF-κB activation.25 However, our results suggest that p62/SQSTM1 can terminate TLR2-induced NF-κB activity by triggering selective autophagy as a feedback regulation on NF-κB. This is the first report to demonstrate that the activity of a transcription factor can be directly regulated by selective autophagy. LPS signaling also causes NF-κB p65 ubiquitination and p62/SQSTM1 recognition, but it will not lead to autophagosome recruitment. Wild et al.9 have reported that these cargo receptors, such as opineurin, need post-translational modification in order to enhance the binding to LC3. We note that TLR2 signaling might induce such a modification in p62/SQSTM1 that is absent in LPS signaling. Apart from this, LPS fails to induce the sustained ERK1/2 phosphorylation that is required to deliver NK-κB p65 to lysosomes, and is shown to facilitate autophogosome maturation.26 These findings might explain why LPS will not induce the autophagy-dependent NF-κB p65 degradation as observed with LTA-SA or MCM. On the other hand, LPS can activate late stage NF-κB to produce IFN-β through an Myd88-independent, but TRIF-dependent, pathway.27 However, the TLR2 signal can only activate NF-κB through a Myd88-dependent pathway and produce less IFN-β and IL-12p40.21, 27 The role of TRIF in TLR-induced autophagy-dependent NF-κB regulation therefore needs further investigation. Recent studies have shown that autophagy limits inflammation responses by controlling NLRP3-inflammasome activation and pro-IL-1β stability.28, 29 Here, our report that selective autophagy negatively regulates TLR2-dependent NF-κB activity provides another pathway to control inflammation responses by the autophagy process.
An identified TLR2 ligand from lung cancer cells, versican, was shown to activate macrophages to cause inflammation and tumor metastasis via TNF-α.17 In this study, we noticed that hepatoma CM did not induce BMDMs to produce inflammatory cytokines, such as TNF-α, in the TLR2-related M2 macrophage differentiation. On the contrary, elevated IL-12p40 and TNF-α were detected in hepatoma CM-stimulated TLR2−/− BMDMs, suggesting that TLR2-related factors from CM negatively regulate inflammatory cytokine production. These factors are distinct from versican and need further identification. Although NF-κB signaling is required for macrophages to initiate a M2-like phenotype, inactivation of NF-κB is always represented in TAMs from well-established tumors, leading to a failure to produce IL-12 to induce effective antitumor immunity.30 It remains unclear how NF-κB activity in TAMs is suppressed. Here we demonstrate that tumor-derived factors induce TLR2-related selective autophagy to degrade NF-κB p65-containing ALS and limit NF-κB activity in M2 macrophages, but not in polarized M1 macrophages. These NF-κB p65-containing ALS are also presented in mice hepatoma-associated macrophages, indicating that this regulation occurs primarily in tumor-associated M2 polarized macrophages. Interruption of the TLR2 signal or the autophagy process can restore the NF-κB protein level and increase IL-12 production. Our findings suggest that tumors may restrict the antitumor activities of TAMs by degrading NF-κB via selective autophagy.
Materials and Methods
Reagents and antibodies
Lipopolysaccharides from Escherichia coli, bafilomycin A1, MG132, 3-methyladenin, U0126, puromycin and Hoechst stain solution were purchased from Sigma-Aldrich (St. Louis, MO, USA). Lipoteichoic acid from Staphylococcus aureus (LTA-SA) was purchased from InvivoGen (San Diego, CA, USA). The antisera, anti-arginase-1 and anti-ubiquitin, were from BD Pharmingen (San Diego, CA, USA), anti-SOCS3 was from Proteintech group (Chicago, IL,USA), anti- NF-κB p65, anti-IκBα, anti-phospho-signal transducer and activator of transcription 1 (STAT1) (Tyr701) and anti-STAT1 were from Cell Signaling (Beverly, MA, USA), antimicrotubule-associated proteins LC3 and anti-p62/sequestosome1 were from MBL (Nagoya, Japan), antilysosomal-associated membrane protein 1 (LAMP 1) was from eBioscience (San Diego, CA, USA), antiautophagy-related gene 5 (Atg5) and NF-κB p50 were from Epitomics Inc. (Burlingame, CA, USA), and anti-β-actin was from Abcam (Cambridge, UK). Fluorescein isothiocyanate (FITC)-conjugated anti-CD204, CD206, CD80, CD86 and F4/80 were purchased from AbD Serotec (Oxford, UK). Mice cytokines were detected by ELISA (R&D systems, Minneapolis, MN, USA).
Cell culture and immunofluorensence staining
BALB/c hepatoma cell line ML-14a cells were adapted from ML-1 cells in BALB/c mice for four generations. Cells were cultured in DMEM (Gibco, Grand Island, NY, USA) supplemented with 10% FBS, L-glutamine and penicillin-streptomycin. Murine bone marrow-derived macrophages (BMDMs) were generated by flushing bone marrow cells from femurs and tibias of 6–10-week old BALB/c mice or C57BL/6 (wild and TLR2 knockout) mice. These cells were cultured for 5–6 days in RPMI (Gibco) containing 10% FBS and 10 ng/ml recombinant mouse M-CSF (Peprotech, Rocky Hill, NJ, USA). RAW-Blue cells that stably express a NF-κB and AP-inducible embryonic alkaline phosphatase (SEAP) secretion were used to determine relative NF-κB activities. These cells were kindly provided by Dr. Kuo-Feng Hua, originally purchased from InvivoGen Corp. (San Diego, CA, USA). The SEAP activities were measured using QUANTI-Blue SEAP detection medium (InvivoGen Corp., San Diego, CA). For immunofluorescence staining, BMDMs were fixed with 4% paraformaldehyde and stained first with anti-LC3, p62, ubiquitin and NF-κB p65 antibodies, followed by secondary antibody conjugated with Alexa Fluor 488 or 594. The cellular distribution of proteins was observed under a confocal fluorescence microscope (Olympus FV 1000, Japan). To observe the autophagic vesicles, the BMDMs were treated with DMEM or MCM for 12 h, then fixed with 4% glutaraldehyde and postfixed in 1% OsO4. The cells were observed under electron microscopy (Hitachi 7000, Japan).
Mice and murine in situ hepatoma model
BALB/c and C57BL/6 mice (female, 8–10 weeks old) were purchased from the National Laboratory Animal Center (Taipei, Taiwan), and TLR2−/− mice were kindly provided by Dr. John T. Kung (Institute of Molecular Biology, Academia Sinica, Taiwan). All mice were maintained in the pathogen-free facility of the Animal Laboratory of National Cheng Kung University. The animals were raised and cared for according to the guidelines set up by the National Science Council, ROC. The mouse experiments were approved by the institutional animal care and use committee. A murine in situ hepatoma model was set up by intrasplenic injection of 1 × 106 viable ML-14a cells into anesthetized mice as previously described.20 The ML-14a cells first colonized in the spleen, and then migrated into the liver forming liver tumor nodules of varied sizes beginning 1 week after injection.
Western blot analysis and immunoprecipitation (IP) assay
Cell lysates were prepared by extracting proteins with lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin). Proteins were separated by SDS-PAGE and transferred to PVDF membranes. The membranes were blocked and incubated with primary antibodies. After incubation with peroxidase-conjugated secondary antibodies, the blots were visualized by enhancing chemiluminescence reagents (PerkinElimer Life Sciences, Boston, MA, USA). In some experiments, the nuclear and cytoplasmic fractions of cells were prepared using the kit ‘NE-PER nuclear and cytoplasmic extraction reagents’ (Pierce, Rockford, IL, USA). For the NF-κB p65 immunoprecipitation assay, cells were extracted by lysis buffer and centrifuged at 1000 g for 20 min at 4 °C to collect the extracted proteins. Anti-NF-κB p65 antibody and protein G-Sepharose beads were added to extracted proteins at 4 °C. The beads were isolated and washed by centrifugation and the bound proteins were determined by western blotting as described above.
Lentivirus-based short hairpin RNA (shRNA) transfection
Atg5 and p62 were knocked down in RAW 264.7 cells by stably expressing lentivirus-based shRNA, which targeted mice Atg5 and p62. The clones were obtained from the National RNAi Core Facility (Institute of Molecular Biology/Genomic Research Center, Academia Sinica, Taiwan) and the target sequence for p62 is TRCN0000098618 5′-GCTATGTCCTATGTGAAAGAT-3′, for Atg5 is TRCN 0000375819 5′-AGCCGAAGCCTTTGCTCAATG-3′, for control luciferase is TRCN0000072247 5′-GAATCGTCGTATGCAGTGAAA-3′. The recombinant lentivirus was produced by cotransfection with two helper vectors, pCMVdeltaR8.91 and pMD.G, and a target vector pLKO.1-puro-shRNA to 293T cells. RAW 264.7 cells were then infected with recombinant lentivirus for 24 h, and stably expressed cells were selected by puromycin. The knockdown efficiency of target proteins were determined by western blotting as described above.
Abbreviations
- NF-κB:
-
nuclear factor-κB
- p62/SQSTM1:
-
p62/sequestosome-1
- TAM:
-
tumor-associated macrophage
- ERK1/2:
-
extracellular signal-regulated kinase 1/2
References
Mizushima N, Levine B, Cuervo AM, Klionsky DJ . Autophagy fights disease through cellular self-digestion. Nature 2008; 451: 1069–1075.
Wang RC, Levine B . Autophagy in cellular growth control. FEBS Lett 2010; 584: 1417–1426.
Kraft C, Peter M, Hofmann K . Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 2010; 12: 836–841.
Komatsu M, Ichimura Y . Selective autophagy regulates various cellular functions. Genes Cells 2010; 15: 923–933.
Komatsu M, Ichimura Y . Physiological significance of selective degradation of p62 by autophagy. FEBS Lett 2010; 584: 1374–1378.
Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J 2010; 29: 969–980.
Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 2009; 33: 505–516.
Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F . The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol 2009; 10: 1215–1221.
Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011; 333: 228–233.
Biswas SK, Lewis CE . NF-kappaB as a central regulator of macrophage function in tumors. J Leukoc Biol 2010; 88: 877–884.
Mancino A, Lawrence T . Nuclear factor-kappaB and tumor-associated macrophages. Clin Cancer Res 2010; 16: 784–789.
Verstrepen L, Bekaert T, Chau TL, Tavernier J, Chariot A, Beyaert R . TLR-4, IL-1R and TNF-R signaling to NF-kappaB: variations on a common theme. Cell Mol Life Sci 2008; 65: 2964–2978.
Maine GN, Mao X, Komarck CM, Burstein E . COMMD1 promotes the ubiquitination of NF-kappaB subunits through a cullin-containing ubiquitin ligase. EMBO J 2007; 26: 436–447.
Tanaka T, Grusby MJ, Kaisho T . PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol 2007; 8: 584–591.
Arenzana-Seisdedos F, Thompson J, Rodriguez MS, Bachelerie F, Thomas D, Hay RT . Inducible nuclear expression of newly synthesized I kappa B alpha negatively regulates DNA-binding and transcriptional activities of NF-kappa B. Mol Cell Biol 1995; 15: 2689–2696.
Bortoluci KR, Medzhitov R . Control of infection by pyroptosis and autophagy: role of TLR and NLR. Cell Mol Life Sci 2010; 67: 1643–1651.
Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009; 457: 102–106.
Mantovani A, Sica A . Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol 2010; 22: 231–237.
Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG et al. Macrophage polarization in tumour progression. Sem Cancer Biol 2008; 18: 349–355.
Chang CP, Yang MC, Liu HS, Lin YS, Lei HY . Concanavalin A induces autophagy in hepatoma cells and has a therapeutic effect in a murine in situ hepatoma model. Hepatology 2007; 45: 286–296.
Agrawal S, Agrawal A, Doughty B, Gerwitz A, Blenis J, Van Dyke T et al. Cutting edge: different Toll-like receptor agonists instruct dendritic cells to induce distinct Th responses via differential modulation of extracellular signal-regulated kinase-mitogen-activated protein kinase and c-Fos. J Immunol 2003; 171: 4984–4989.
Ichimura Y, Komatsu M . Selective degradation of p62 by autophagy. Semin Immunopathol 2010; 32: 431–436.
Kirkin V, McEwan DG, Novak I, Dikic I . A role for ubiquitin in selective autophagy. Mol Cell 2009; 34: 259–269.
Gao C, Cao W, Bao L, Zuo W, Xie G, Cai T et al. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat Cell Biol 2010; 12: 781–790.
Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008; 13: 343–354.
Corcelle E, Nebout M, Bekri S, Gauthier N, Hofman P, Poujeol P et al. Disruption of autophagy at the maturation step by the carcinogen lindane is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Cancer Res 2006; 66: 6861–6870.
Takeda K, Akira S . TLR signaling pathways. Semin Immunol 2004; 16: 3–9.
Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011; 12: 222–230.
Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA et al. Autophagy Controls IL-1{beta} Secretion by Targeting Pro-IL-1{beta} for Degradation. J Biol Chem 2011; 286: 9587–9597.
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A . Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 2002; 23: 549–555.
Acknowledgements
We thank Dr. Kuo-Feng Hua for kindly providing RAW-Blue cells. This work was supported by grant NSC-98-2320-B-006-031-MY2 from the National Science Council, Taiwan, ROC and program from Center of Infectious Disease and Signaling Research, National Cheng Kung University.
Author contributions
In memoriam: Dr. Huan-Yao Lei (1955-2012), a leader, educator and immunologist, has made important contributions to this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by H-U Simon.
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Chang, CP., Su, YC., Hu, CW. et al. TLR2-dependent selective autophagy regulates NF-κB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death Differ 20, 515–523 (2013). https://doi.org/10.1038/cdd.2012.146
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2012.146
