
Abstract
Apoptosis induced by most stimuli proceeds through the mitochondrial pathway. One such stimulus is nutrient deprivation. In this study we studied death induced by glucose deprivation in cells deficient in Bax and Bak. These cells cannot undergo mitochondrial outer membrane permeabilization (MOMP) during apoptosis, but they undergo necrosis when treated with MOMP-dependent apoptotic stimuli. We find in these cells that glucose deprivation, rather than inducing necrosis, triggered apoptosis. Cell death required caspase activation as inhibition of caspases with peptidic inhibitors prevented death. Glucose deprivation-induced death displayed many hallmarks of apoptosis, such as caspase cleavage and activity, phosphatidyl-serine exposure and cleavage of caspase substrates. Neither overexpression of Bcl-xL nor knockdown of caspase-9 prevented death. However, transient or stable knockdown of caspase-8 or overexpression of CrmA inhibited apoptosis. Cell death was not inhibited by preventing death receptor–ligand interactions, by overexpression of c-FLIP or by knockdown of RIPK1. Glucose deprivation induced apoptosis in the human tumor cell line HeLa, which was prevented by knockdown of caspase-8. Thus, we have found that glucose deprivation can induce a death receptor-independent, caspase-8-driven apoptosis, which is engaged to kill cells that cannot undergo MOMP.
Similar content being viewed by others


Main
Apoptosis is a form of cell death required for homeostasis of human tissues. Apoptotic cells display several morphological and biochemical changes, which are a consequence of the activity of caspase proteases.1 Caspases are normally inactive in the cytosol, but they become activated by dimerization and/or proteolysis by other caspases. ‘Executioner’ caspases such as caspase-3 are activated through cleavage by ‘initiator’ or ‘apical’ caspases, of which the best characterized are caspases-8 and -9. Caspase-8 is activated by its recruitment to the multimeric DISC (death-inducing signaling complex) in response to extracellular ligands such as Fas/CD95-ligand, TRAIL or TNF;2 this is the death receptor, or extrinsic pathway of apoptosis. Caspase-9 is activated by dimerization after recruitment to the apoptosome, a complex of APAF1 proteins formed in response to the release of the mitochondrial protein cytochrome c into the cytosol.3 This caspase activation cascade is called the mitochondrial or intrinsic pathway of apoptosis.
The mitochondrial pathway of apoptosis is regulated by proteins of the Bcl-2 family, which regulate the integrity of the mitochondrial outer membrane, thereby controlling cytochrome c and subsequent caspase activation.4 Impairment of the mitochondrial pathway is common in human tumors. Cancer cells frequently overexpress the antiapoptotic proteins Bcl-2, Bcl-xL and Mcl-1, or they lack functional proapoptotic Bcl-2 family members, including Bax, Bak or both.5 The overexpression of antiapoptotic Bcl-2 proteins not only blocks the morphological features of apoptosis, but also enables clonogenic cell survival.6 For these reasons, cells from Bax, Bak double-knockout (DKO) mice are a suitable model to identify non-mitochondrial pathways of cell death, which may be targeted for therapy.
Bax/Bak-deficient murine embryonic fibroblasts (MEFs) are resistant to apoptosis induced by stimuli that normally kill cells through the mitochondrial pathway.7, 8 However, some drugs such as staurosporine, etoposide, thapsigargin and tunicamycin, which kill wild-type (WT) cells in a mitochondria-dependent manner, can kill Bax-, Bak-deficient cells by a slower, non-apoptotic mechanism.9, 10 We analyzed cell death induced by glucose deprivation in these cells, as this stimulus has been shown to kill some cell lines through necrosis.11, 12 We report here that glucose deprivation can induce death of Bax-, Bak-deficient MEFs. Surprisingly, in these cells glucose deprivation induces apoptosis that proceeds through an unconventional pathway requiring caspase-8. Our results uncover a pathway for activation of caspase-8 different than death receptor ligation, and provide new insight into the mechanism of cell death induced by nutrient deprivation.
Results
Glucose deprivation kills Bax-, Bak-deficient cells by apoptosis
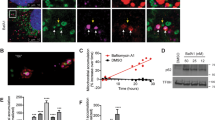
Glucose deprivation has been shown to kill cells either by necrosis11, 12 or through the mitochondrial or ‘intrinsic’ pathway of apoptosis.13, 14, 15, 16, 17 We reasoned that cells deficient in Bax and Bak, which cannot die through the intrinsic pathway,8 would undergo a rapid necrotic death when deprived of glucose, as glucose deprivation kills most cells in 24–48 h. Alternatively, it was possible that these cells could remain atrophic but alive for weeks, as it was described that Bax-, Bak-deficient hematopoietic cells, when deprived of growth factors, downregulate glucose transporter 1 but do not die for several weeks.18 We tested whether glucose deprivation provoked cytostatic or cytotoxic effects in SV40-transformed, Bax, Bak DKO MEFs.8 As assessed by morphology and propidium iodide (PI) incorporation, about 10% of cells appeared dead by 24 h, with approximately 75% dead cells within 72 h (Figure 1a and b). Their WT counterparts died more rapidly, and were all dead by 48 h (Figure 1b), suggesting that these cells die through the mitochondrial pathway.

Glucose removal triggers cell death in Bax, Bak DKO cells with mixed features of apoptosis and necrosis. (a) Bax, Bak DKO MEF were subjected to glucose deprivation in the absence or presence of 20 μM Q-VD for 72 h, or treated with thapsigargin 10 μM or TNF for 24 h. (b) Glucose deprivation induces DNA degradation. WT and Bax, Bak DKO MEFs were treated as described in Materials and Methods section and collected for subG1 analysis or propidium iodide (PI) incorporation at times shown. Data show average and S.D. of at least three (PI) or four (subG1) experiments. (c) WT and DKO cells display cleavage of caspase-3 and PARP upon glucose deprivation. Cells were subjected to western blot (WB) analysis at times shown (h). Note the different kinetics among cell lines. (d) Glucose deprivation induces phosphatidyl-serine (PS) externalization measured by Annexin V incorporation in DKO cells. Q-VD inhibits both events. A representative experiment is shown. (e, f) DKO cells undergo atypical nuclear condensation after glucose deprivation. Bax, Bak DKO MEFs were transfected transiently with GFP-Histone 2A plasmid in chamber slides. Cells were treated for 72 h without glucose (in the presence or absence of Q-VD) or with TNF or thapsigargin. Nuclei were visualized by confocal microscopy (e) or electron microscopy (f). Representative images are shown. In (f), only dead (floating) cells were collected after being deprived of glucose for 48 h without Q-VD. Note the absence of massive cytosolic vacuolation which characterizes necrotic/autophagic cell death in Bax-, Bak-deficient MEFs9, 10, 19
Bax, Bak DKO MEFs can die by a non-apoptotic form of cell death in response to staurosporine, etoposide,9, 19 alkylating DNA agents20 and ER stressors.10 We determined whether the form of cell death induced by glucose deprivation was apoptosis or necrosis. The morphology of dead DKO cells was different from the classical necrotic death induced by thapsigargin in these cells,10 and was more similar to the morphology of cells dying by apoptosis induced by the combination of TNF and cycloheximide (CHX) (Figure 1a): cells rounded up and detached from the culture plate, and they showed a reduction of volume (shrinkage). Moreover, when we incubated glucose-deprived cells in the presence of the pan-caspase inhibitor Q-VD-OPH (Q-VD), we observed a reduction in the number of dead cells as measured by morphology and PI incorporation, thus indicating that death is caspase-dependent (Figure 1a and b). In addition, we observed that cells displayed sub-G1 DNA fragmentation, a marker of apoptosis, with similar kinetics to that of plasma membrane disruption (PI uptake) (Figure 1b). Z-VAD-FMK (Z-VAD), an alternative pan-caspase inhibitor, inhibited both subG1 DNA fragmentation and PI incorporation. Other markers of apoptosis such as caspase-3 and PARP cleavage were observed in Bax-, Bak-deficient cells, albeit with slower kinetics than in WT cells (Figure 1c). We stained cells with annexin V, which detects phosphatidyl-serine exposure on the surface of apoptotic cells. Glucose-deprived cells displayed annexin V binding before they became PI-positive, another indication of apoptosis (Figure 1d). Moreover, phosphatidyl-serine exposure was inhibited by Q-VD.
We observed chromatin condensation after glucose deprivation (Figure 1e and f). Nuclei had a different appearance to the classical apoptotic morphology observed after treatment with TNF, or to the relaxed appearance of necrotic cells treated with thapsigargin. After glucose deprivation, DNA was condensed around the periphery of the nuclei. However, this condensation may not be directly related to caspase activation as it occurred in glucose-deprived cells treated with Q-VD. Thus, apoptosis induced by glucose deprivation in Bax-, Bak-deficient cells displayed all of the hallmarks of apoptosis analyzed, with the exception of the characteristic chromatin condensation.
Glucose deprivation-induced death is caspase-dependent and does not proceed through the mitochondrial pathway
We had observed that Q-VD and Z-VAD inhibited features of cell death induced by glucose deprivation. To confirm that caspases were active in glucose-deprived DKO cells, we analyzed the cleavage of a synthetic DEVD peptide in cell extracts. Glucose deprivation induced DEVD-ase activity, which was inhibited by Q-VD (Figure 2a). In addition, we analyzed cleavage of the effector caspase-3, which is a marker of its activation. We observed cleavage of caspase-3 in DKO cells after glucose withdrawal that was inhibited by ZVAD (Figure 2b). Moreover, we verified that caspases were active by analyzing the cleavage of substrates of effector caspases: co-chaperone p23 and PARP. These proteins were cleaved in a Z-VAD-inhibitable manner (Figure 2b).

Glucose deprivation induces caspase activation in Bax, Bak DKO cells. (a) Glucose deprivation induces caspase activity. Bax, Bak DKO MEFs were cultured for the indicated times without glucose, in the presence or absence of Q-VD. Cell lysates were incubated with AC-DEVD-amc as described in Materials and Methods section. Results show average and S.E.M. of three experiments. (b) Glucose deprivation induces cleavage of procaspase-3 and the caspase substrates co-chaperone p23 and PARP, which are inhibited by Z-VAD. Cell lysates were prepared as in (a) and proteins were resolved by SDS-PAGE and immunoblot. (c, d) Caspase inhibitors block apoptotic features in glucose-deprived Bax, Bak DKO cells. Bax, Bak DKO MEFs were grown in glucose-free medium in the presence of caspase inhibitors or DMSO as vehicle control. Inhibitors and DMSO were replenished every 24 h. Cells were collected and subjected to subG1 (c) or PI uptake (d) analysis. Data represent average of two experiments
To exclude a possible effect of Z-VAD-fmk or Q-VD-OPH on proteases other than caspases, we studied the inhibition of death by other peptidic inhibitors. We observed that a panel of caspase inhibitors prevented both subG1 DNA fragmentation (Figure 2c) and PI incorporation (Figure 2d). These inhibitors were more efficient in inhibiting DNA cleavage than PI uptake, suggesting that the intracellular concentrations of these compounds can inhibit some apoptotic features better than others, or that inhibition of caspases switches the form of death to necrosis in a small percent of the cells. Consistent with data reported in Jurkat cells,21 VDVAD-fmk and LEHD-fmk were less efficient than IETD-fmk, Z-VAD-fmk and Q-VD-OPH in preventing death.
Bax and Bak are required for mitochondrial outer membrane permeabilization (MOMP) during apoptosis, and cells lacking these proteins cannot release cytochrome c or undergo apoptosis in response to many stimuli, including microinjection of tBid.8 However, it was possible that glucose deprivation activates the mitochondrial pathway through an unidentified, alternative mechanism. Therefore, we treated cells with shRNA for caspase-9 (Figure 3a). This treatment did not affect any of the parameters of cell death tested (Figure 3b). Furthermore, we generated DKO cells stably overexpressing Bcl-xL. Bcl-xL inhibits MOMP during apoptosis.22 Bulk-transduced cells (pool) and four clones were analyzed. As shown in Figure 3c, Bax, Bak DKO MEFs overexpressing Bcl-xL (Figure 3d) were not protected from death when compared with the parental line.

Glucose deprivation-induced apoptosis in Bax, Bak DKO MEF is not dependent on the classical mitochondrial pathway of apoptosis. (a) DKO cells were infected with retroviruses encoding two different shRNA against caspase-9, or the empty vector. Expression of caspase-9 is shown. (b) The same pools were collected after 72 h of glucose withdrawal and analyzed for subG1 DNA content. DKO cells treated with Q-VD-OPH are shown as a control for caspase inhibition. Results show average and S.E.M. of five experiments. (c) DKO cells (shown as ‘parent’) were stably transduced with Bcl-xL. Several clones were generated by serial dilution and blotted for expression of Bcl-xL or actin. (d) The clones, bulk population and cells from the parent cell line described in (c) were deprived of glucose and collected at 40 and 72 h for analysis of subG1 DNA content. Results show average and S.E.M. of three experiments
Intriguingly, although caspase inhibitors effectively blocked glucose deprivation-induced cell death in Bax, Bak DKO MEF, we did not observe this effect in WT MEF transduced with Bcl-xL (data not shown). It is possible that Bcl-xL promotes necrosis in MEF in response to glucose deprivation, similar to what has been observed in response to etoposide.9 Alternatively, it is possible that the adaptation to the Bax, Bak double-deficient condition primes cells for a novel caspase activation mechanism. We therefore sought to identify the caspase involved in initiating cell death in this setting.
Caspase-8 is engaged in death induced by glucose deprivation in Bax, Bak DKO MEFs and HeLa cells
In the absence of a mitochondrial pathway of apoptosis, a likely candidate for initiating the caspase cascade is caspase-8, required for death receptor-induced23 and other forms of apoptosis.11, 24, 25 We therefore asked whether caspase-8 was the apical caspase in glucose starvation-induced apoptosis in these cells. We generated a stable cell line expressing short hairpin RNA (shRNA) targeting caspase-8, and selected individual clones expressing low levels of caspase-8 (Figure 4a). As expected, these clones and the parental pool displayed decreased sensitivity to TNF when compared with cells transfected with the empty vector (Figure 4b). When incubated in the absence of glucose, we observed that these clones and the parental pool were less sensitive than their mock-transfected counterparts to glucose deprivation (Figure 4c). To exclude the possibility of an off-target effect of this construct, or a compensation effect of the stable downregulation of caspase-8, we transiently transfected two different siRNA oligonucleotides against caspase-8. We were able to achieve a moderate reduction in caspase-8 levels (Figure 4d). This reduction conferred a mild but reproducible protection from death ligands (data not shown) and glucose deprivation (Figure 4e). Knockdown of caspase-8 reduced glucose deprivation-induced cleavage of caspase-3 (Figure 4f).

Caspase-8 is required for death induced by glucose deprivation in Bax, Bak DKO MEF. (a) DKO cells were infected with retroviruses encoding shRNA against caspase-8 (shown as ‘pool’), or the empty vector. Expression of caspase-8 by immunoblot is shown for the pool population, empty vector-transfected cells and two clones that were selected for low caspase-8 expression (A8 and D5). (b) Cells described in (a) were treated with TNF for 24 h and collected for subG1 analysis. Results show average and S.E.M. of four experiments. (c) Same cells as in (a) were cultured without glucose for 72 h. Apoptosis was measured by subG1 analysis. Average and S.E.M. of five experiments are shown. (d) Dowregulation of caspase-8 protein with small interferent RNA. Cells were transiently transfected with siRNA against caspase-8 (8-1) or a control oligonucleotide (pBlue). Protein was collected 24 h after transfection. (e) Cells were transfected with a control oligonucleotide or siRNA against caspase-8. Forty-eight hours after transfection, they were deprived of glucose for 72 h and collected for subG1 analysis. The average of three experiments is shown. (f) Clone A8 described in (a) and cells transfected with the empty vector were starved of glucose and collected for western blot of Bid or caspase-3 at times shown. A mixture of antibodies against the procaspase and the cleaved forms was used. (g) DKO MEFs or cells transfected with a CrmA-encoding vector were treated with TNF or anti-Fas antibody. Cell death was measured by PI exclusion. Results show average and range of two experiments. (h) DKO or CrmA-transfected MEFs were starved of glucose in the presence of DMSO or Q-VD, and collected at 24 or 48 h for subG1 analysis. Results show average and S.E.M. of five experiments
We had observed that the peptidic caspase inhibitor IETD-fmk inhibited glucose deprivation-induced death (Figure 2c and d), suggesting that the enzymatic activity of caspase-8 is required for death. However, although IETD-fmk is widely used to block caspase-8 activity, it can also inhibit effector caspases.21 In order to test whether the enzymatic activity of mature caspase-8 is required for apoptosis in this system, we transfected DKO cells with the viral caspase inhibitor CrmA, which preferentially inhibits caspase-8, although it can also inhibit the inflammatory caspase-1.2, 26 Cells transfected with CrmA were protected from death receptor-induced apoptosis (Figure 4g). CrmA effectively inhibited death induced by glucose deprivation in Bax, Bak DKO MEF (Figure 4h).
To test whether caspase-8 dependency was exclusive of Bax, Bak DKO MEF, we used siRNA to knockdown caspase-8 in established tumor lines. Although we observed no effect on glucose deprivation-induced death in the MCF7 breast cancer line (not shown), we observed an effect in HeLa cells. Knockdown of caspase-8 in HeLa cells reduced caspase-8 levels (Figure 5a), and prevented death induced by TNF (Figure 5b). Strikingly, this decrease in caspase-8 reduced glucose deprivation-induced cell death to the same extent than with incubation with Q-VD (Figure 5b).

Downregulation of caspase-8 prevents apoptosis in HeLa cells. (a) Caspase-8 knockdown in HeLa cells using siRNA. Seventy-two hours after transfection, cells were subjected to glucose deprivation in the presence or absence of Q-VD as indicated, and collected at times shown for FACS analysis (b). As a positive control, cells were treated with TNF. Results show average and range of three experiments
The extrinsic pathway is not involved in glucose starvation-induced apoptosis in Bax, Bak DKO cells
Caspase-8 is activated in response to death receptor ligation. It has been shown that some drugs can induce caspase-8-mediated apoptosis through the induction of death ligands such as FasL, TNF or TRAIL.27, 28, 29 Thus, one possibility is that glucose deprivation is activating the extrinsic pathway by inducing the synthesis and/or secretion of death ligands, which might kill the cells in an autocrine manner. To explore this possibility, we analyzed by RT-PCR the mRNA levels of death ligands and used blocking antibodies against death ligands and receptors. We could not detect the expression of Fas ligand mRNA in these cells (not shown). Furthermore, we starved cells from glucose in the presence of neutralizing antibodies for Fas-ligand or Fas-Fc (Figure 6a), which did not prevent death. TRAIL expression did not change after glucose withdrawal (Supplementary Figure S1A). Moreover, we could not detect toxicity of TRAIL in these cells, even in the presence of CHX or upon glucose deprivation, which has been shown to sensitize human tumor cells to TRAIL30 (Supplementary Figure S1B). In addition, we incubated cells in the presence of anti-TRAIL or TRAIL-R-FC (Supplementary Figure S1C and D), which did not have any effect on cell death induced by glucose deprivation.

Glucose deprivation-induced death in Bax, Bak DKO cells is not dependent on death receptor–ligand interactions. (a) Blocking Fas/Fas ligand interaction does not inhibit glucose deprivation-induced cell death. Cells were subjected to glucose deprivation for 72 h in the presence of anti-FasL or Fas-FC at concentrations shown (in μg/ml). (b) TNF mRNA is slightly induced upon glucose removal. DKO cells were deprived of glucose and collected at indicated times. RT-PCR of TNF and a loading control (HMBS) are shown. (c) TNF was added 24 h after treatment with glucose deprivation. CHX (0.2 μM) was added 30 min before TNF. Cells were collected after 48 h of glucose withdrawal for subG1 analysis. Data represents average and S.E.M. of three experiments. (d) Blockage of TNF/TNF-R interactions does not prevent death by glucose starvation. Cells were incubated without glucose for 24 h and then anti-TNF-R1 was added at the indicated concentrations (left panel), expressed in μg/ml. Antibody was readded 24 h later. Cells were collected 72 h after glucose removal to measure PI exclusion. Right panel: cells were incubated for 72 h without glucose in the presence of TNF-R-Fc and they were collected for subG1 analysis. Bars show average and range of two experiments. Positive controls for blocking reagents are shown in Supplementary Figure S1
A slight increase in TNF mRNA levels was detected (Figure 6b). Although TNF did not kill these cells unless they were co-incubated with CHX (Figure 6c), the possibility remained that glucose deprivation sensitizes cells to TNF.30 To explore this possibility, we preincubated cells without glucose and then added TNF. We observed that glucose deprivation did not sensitize these cells to TNF, as the combination of glucose deprivation and TNF caused the same amount of death than glucose deprivation alone. The combination of glucose deprivation plus CHX and TNF caused additive effects on cell death numbers (Figure 6c). In addition, we incubated glucose-deprived cells in the presence of neutralizing antibodies against TNF and TNF-R1, which did not prevent death (Figure 6d).
In another approach to examining the possible role of the death receptor pathway in glucose deprivation-induced apoptosis in Bax, Bak DKO cells, we examined the effects of c-FLIP. Overexpression of c-FLIP inhibits caspase-8 activation at the DISC.31 We generated Bax, Bak DKO MEF that stably overexpress c-FLIPL, and as expected, these cells were protected from TNF or anti-Fas-induced apoptosis (Figure 7a). However, they were not protected from glucose deprivation-induced apoptosis, suggesting that caspase-8 is activated independently of a conventional death receptor-induced DISC (Figure 7b).

cFLIPL overexpression or RIPK1 downregulation do not protect from death induced by glucose deprivation in Bax, Bak DKO MEF. (a) DKO cells stably overexpressing cFLIPL were treated with anti-Fas or TNF-á for 24 h and collected for analysis of PI incorporation. (b) Same cells were deprived of glucose for indicated times and collected for subG1 DNA analysis. Graphs show average and S.E.M. of three (a) or four (b) experiments. (c) Cells were deprived of glucose for 48 h in the presence of necrostatin-1 50 μM or Q-VD. PI uptake was measured. (d–f) Cells were transfected with control siRNA or siRNA against RIPK1 (labeled as ‘RIP’) for 48 h and subjected to: western blot (d), glucose deprivation for indicated times (h) (e) or treatment with TNF 10 ng/ml and the IAP antagonist Compound A (500 nM) for 24 h (f)
Recently, a novel form of caspase-8- and FADD-mediated apoptosis has been described.32 When TNF is combined with Smac mimetics, TNF induces the formation of a caspase-8 activation complex formed by RIPK1, FADD and caspase-8, which leads to apoptosis. Cell death mediated by this complex is poorly inhibited by FLIP. In order to test a role for RIPK1 in our system, we incubated DKO cells in the presence of Necrostatin-1 (Nec-1). Nec-1 is an allosteric inhibitor of RIPK1 kinase activity, and it inhibits RIPK1-mediated necroptosis (death receptor-mediated necrosis). As RIPK1 kinase activity is required for apoptosis induced by combination of TNF and Smac mimetics, we deprived cells of glucose in the presence of Nec-1. Incubation with Nec-1 did not prevent glucose deprivation-induced cell death (Figure 7c). To exclude a function of RIPK1, which could not be inhibited by Nec-1, we used siRNA to knockdown RIPK1 (Figure 7d). The resultant decrease in RIPK1 was sufficient to inhibit death induced by TNF in the presence of the Smac mimetic Compound A (Figure 7f), but did not influence glucose deprivation-induced cell death (Figure 7e and f). Thus, neither the classical DISC nor the second, RIPK1-dependent caspase-8 activating complex appear to be involved in glucose deprivation-induced apoptosis.
Discussion
Bax and Bak are essential mediators of apoptosis induced by most stimuli.8 Because the mitochondrial pathway is frequently inactivated in tumor cells, it may be clinically relevant to find means to induce necrosis or mitochondria-independent apoptosis. Glucose deprivation has been shown to induce necrosis in some cancer cell lines. We show here that SV-40-transformed MEFs with an impairment of the mitochondrial pathway can die by apoptosis upon glucose deprivation, and that this form of apoptosis is mediated by caspase-8.
Inhibition of cell metabolism by nutrient deprivation has been extensively studied as a means to preferentially kill tumor cells, because these cells display numerous alterations in metabolic pathways, including abnormal glucose and glutamine uptake.33 Indeed, a non-metabolizable glucose analog, 2-deoxyglucose, is currently in clinical trials as an antitumor agent. Moreover, growth factors upregulate glucose uptake and metabolism to induce prosurvival and antiapoptotic effects.13 Glucose metabolism interferes with apoptosis at different steps, regulating both the extrinsic and the intrinsic pathways in tumor cells.17, 30, 34 Therefore, deciphering the death pathways engaged by glucose deprivation will be helpful for rational design of clinical trials with antimetabolites and molecules which inhibit growth factor signaling. Our results suggest that tumors deficient in caspase-8 may be resistant to glucose deprivation or non-metabolizable glucose analogs, opening the possibility that this protease may be a prognostic marker of success of anti-glycolytic treatments. Caspase-8 expression is frequently impaired in tumors, especially those of the brain and childhood tumors.35, 36 An intriguing possibility raised by our findings is that perhaps tumor cells with diminished caspase-8 expression are selected for their ability to resist conditions of low blood flow and glucose supply, which are commonly found in the tumor microenvironment.
In this work, we show that glucose deprivation induces an atypical form of caspase-8 activation in Bax-, Bak-deficient MEFs. Caspase-8 is usually activated in a DISC complex formed upon ligation of death receptors. It is worth noting that caspase-8-dependent death has been described in response to stimuli other than death receptors,11, 24, 25 which suggests that caspase-8 can be activated in ‘alternative’ platforms. It is unlikely that the classical death ligand-induced DISC is formed upon glucose deprivation, as blocking interactions between death receptors and their ligands did not block death. Moreover, FLIP overexpression, which inhibits death receptor-induced apoptosis, did not inhibit apoptosis induced by glucose removal.
Caspase-8 has been described to be activated in an FADD-dependent manner with some stimuli, in the absence of death receptor–ligand interactions.37, 38 We have not been able to obtain conclusive results regarding the involvement of FADD as the adapter protein for caspase-8 activation after glucose deprivation. We cannot exclude that, upon glucose removal, endogenous FADD may be aggregated intracellularly, interacting with caspase-8 and promoting its activation. However, as FLIP overexpression did not prevent apoptosis in our system, we studied the possible role of RIPK1. This protein contains a death domain and forms a complex with FADD and caspase-8 upon treatment with TNF and Smac mimetics.32 Although knockdown of RIPK1 prevented the latter form of cell death, it did not prevent apoptosis in our system, suggesting that a novel caspase-8 activating complex is formed upon glucose deprivation. One stimulus that has been shown to promote activation of caspase-8 in the cytosol is the accumulation of polyglutamine repeats. Expression of ataxin-3 with an expanded polyglutamine tract is toxic to neurons. Sánchez et al.39 observed that cell death is caspase-8-dependent in this system. Caspase-8 and FADD were shown to be recruited to polyglutamine inclusions, which could serve as an aggregation platform for caspase-8. As glucose is required for protein glycosylation, and impairment of glycosylation leads to accumulation of misfolded proteins, it can be speculated that glucose deprivation could promote the generation of protein inclusions, which could recruit and activate caspase-8.
The results presented here, together with other reports, indicate that glucose deprivation can kill cells in three different ways: necrosis, mitochondrial apoptosis or caspase-8-dependent apoptosis. Why some cells undergo ‘uncontrolled’ necrotic death while other cells undergo apoptosis is still not clear. In our system, caspase inhibitors did not support clonogenicity of glucose-deprived cells (Supplementary Figure 2), indicating that this stimulus can induce caspase-independent cell death in the same cells. We show here that HeLa cells, which are able to undergo apoptosis through the mitochondrial pathway, die in a caspase-8-dependent manner upon glucose deprivation. This suggests that the caspase-8-dependent pathway of apoptosis described here could be engaged in cells with an intact mitochondrial pathway as well as in Bcl-2 or Bcl-xL-overexpressing tumor cells. This pathway may be relevant for pathologies that involve conditions of low glucose, such as ischemia and cancer.
Materials and Methods
Cell culture and treatments
Bax, Bak DKO MEFs immortalized with V-408 were maintained in high-glucose pyruvate-free DMEM (Invitrogen, Prat de Llobregat, Spain) supplemented with 2 mM L-glutamine, 200 mg/ml penicillin, 100 mg/ml streptomycin sulfate and 10% FBS (Invitrogen).
For treatments, cells were plated at a concentration of 150 000/ml. and treated 24 h later, when they reached the concentration of 500 000/ml. Glucose deprivation was performed by rinsing the cells twice with glucose-free medium (Gibco/Invitrogen) and incubating them in glucose-free medium with freshly added 2 mM glutamine and antibiotics, plus 10% FBS dialyzed against PBS. Unless specified, caspase inhibitors were added at the time of addition of glucose-free medium. Q-VD-OPH (SM Biochemicals LLC, Anaheim, CA, USA) was used at 20 μM. Other caspase inhibitors (ICN/MP) were used at 50 μM. An equal amount of DMSO (solvent) was added to the controls. Unspecified reagents were from Sigma-Aldrich (Madrid, Spain).
For treatment with death ligands TRAIL (Peprotech, Rocky Hill, NJ, USA), Fas ligand (Enzo Life Sciences, Lausen, Switzerland), TNF and death receptor agonistic antibodies, cells were preincubated with 5 μM CHX (Sigma-Aldrich) for 30 min. HeLa cells were treated with 10 ng/ml human TNF (Peprotech), and DKO MEF with 1 ng/ml TNF (Peprotech) or 0.1 μg/ml anti-murine Fas agonist antibody Jo2 (BD Pharmingen, San Diego, CA, USA, cat. no. 554254) for 20 h unless specified otherwise in figure legends. The following blocking reagents were used: anti-TNF receptor type I/p55 (clone 55R-170), anti-Fas ligand clone MFL3, and Fas-FC cat. no. 556578 (all from BD Pharmingen), anti-TRAIL N2B2 (gift by Edith Janssen), TRAIL-R-FC (R&D Systems, Minneapolis, MN, USA) and TNF-R-FC (Enbrel/etanercept from Wyeth, Immunex Corporation, Thousand Oaks, CA, USA).
Measurement of cell death
For analysis of viability, detached and attached cells were collected by trypsinization and subjected to FACS analysis to detect incorporation of PI 1 μg/ml (10 min incubation in PBS). For subG1 analysis, cells were washed in PBS, fixed in 70% cold ethanol while vortexing and incubated for 1–10 days at 4°C. They were further washed, resuspended in PBS with 40 μg/ml PI and 100 μg/ml RNAse A (Sigma) and incubated for 30 min at 37°C before FACS analysis. Percent of dead HeLa cells was defined as the population with low FSC/SSC compared with untreated cells. Staining with Annexin V-FITC (Immunostep, Salamanca, Spain) was performed according to the manufacturer's instructions in the presence of 0.5 μg/ml PI.
Caspase activity was measured as DEVD-ase activity. Cells treated in six-well plates were lysed in lysis buffer (5 mM Tris-HCl pH: 8, 20 mM EDTA, 0.5% Triton-X 100). Twenty μg of protein were incubated with 20 μM AC-DEVD-amc (BD Pharmingen) in a reaction buffer (HEPES 1 M pH: 7.5, glycerol 87%, DTT 1 M) for 2 h. Fluorescence values (arbitrary units) are divided by the value of fluorescence of untreated cells (used as value ‘1’).
Western blots
Cells were treated as indicated in 10 cm plates. They were trypsinizated, rinsed and lysed by resuspending them in Laemmli buffer (5 mM Tris-ClH/2% SDS) without bromophenol blue (Figures 2 and 4f) or freezing (rest of figures) in the presence of Complete antiprotease cocktail (Roche, Barcelona, Spain). After sonication, protein concentration was measured with BCA (Pierce/Thermo Scientific, Rockford, IL, USA). Equal amounts of protein were mixed with Laemmli loading buffer. After electrophoresis, protein was transferred to a PVDF membrane (Millipore, Madrid, Spain). Membrane was blocked with 5% nonfat dry milk in TBS-Tween (0.1%). Secondary antibodies (1/5000) were HRP conjugated (Zymax, Escondido, CA, USA) and detected with ECL reagent (Pierce). Antibodies used for western blot were as follows: actin (ICN clone C4), Bid (R&D Systems af860), murine caspase-8 (Alexis/Enzo Lifesciences, clone 3B10), human Caspase-8 (MBL M032-3), Caspase-3 (Cell Signaling, Danvers, MA, USA, cat. 9661 and 9662); co-chaperone p23 (Affinity Bioreagents-MA3-414, Lafayette, CO, USA), Bcl-xL (BD Pharmingen, cat. no. 556361), Fadd: Immunotech (MBL, Marseille, France), RIP-1 (BD Bioscience, 610458), PARP (Cell Signaling, 9542).
Plasmids and RT-PCR
pBABE-Bcl-xL was made by insertion of the sequence of Bcl-xL into pBABE-puro. The plasmid encoding histone 2B-green fluorescent protein (GFP) was a kind gift of Dr. R Agami (The Netherlands Cancer Institute, Amsterdam, The Netherlands).
LMP vectors (Open Biosystems, Huntsville, AL, USA) encoding shRNA against murine caspases were generated by following the manufacturer's instructions. Sense sequences against caspase-9 were (5′ to 3′): I – CGCCAGAGGTTCTCAGACCAGA, II – CGGCCACTGCCTCATCATCAAC, and against caspase-8: AACGACTGCACTGCAAATGAAA.
For RT-PCR, 1.5 × 106 cells were collected, and their RNA was extracted with RNeasy kit from Qiagen (Madrid, Spain). RNA from 50 000 cells (∼500 ng) was reverse-transcripted to cDNA with the high capacity cDNA reverse transcription kit from Applied Biosystems (Madrid, Spain). 15% of the resulting cDNA was used as a template in a PCR (25 cycles). The following primers were used: Fasl, 5′-GCAAATAGCCAACCCCAGTACAC-3′ and 5′-GCCACCTTTCTTATACTTCACTCCAG-3′; TRAIL, 5′-CCTCTCGGAAAGGGCATTC-3′ and 5′-TCCTGCTCGATGACCAGCT-3′; TNF, 5′-ATCCGCGACGTGGAACTG-3′ and 5′-ACCGCCTGGAGTTCTGGAA-3′; HMBS (loading control), 5′-TTGCACGATCCTGAAACTCT-3′ and 5′-TCTTGGAATGTTACGGGCAGT-3′.
Transfections and generation of cell lines
Stable cell lines were generated by retroviral infection with viruses produced in Phoenix-Eco cells. Cells infected with pMX-IRES-GFP with CrmA cDNA (kindly provided by JP Medema), with LZRS-MS-IRES-eGFP encoding c-FLIPL,40 or with shRNA against caspases were selected by sorting for GFP expression. For transient transfections, cells were rinsed with DMEM without FBS or antibiotics and incubated for 6 h with a mix of 2 μl/ml lipofectamine 2000 (Invitrogen) and plasmidic Histone-GFP DNA (1 μg) or siRNA at 100 nM except where indicated. RIPK1 and human caspase-8 were downregulated using Dharmacon (Lafayette, CO, USA) On-Target smart pool (cat. no. L-040150 and cat. no. L-003466, respectively) with Dharmafect1. Dharmacon ON-Target plus non-targeting pool was used as a control. Sense sequence of siRNA duplexes against murine caspase-8 was 5′-CAGACAAAGUUUACCAAAU[dT][dT] (Sigma). As a control, an irrelevant siRNA oligonucleotide (pBlue, sense strain: 5′-GUAAGACACGACUUAUCGC-3′) was used. The same sequence coupled to a fluorochrome was used to measure transfection efficiency (pBlue-FITC), which was around 80% in all cases.
Microscopy
For analysis of nuclear morphology by visualization of Histone H2A-GFP, cells were plated and treated in four-well chamber slides (Mattek, Ashland, MA, USA), which had been previously coated with fibronectin (1 μg/ml in PBS). Confocal microscopy was performed in non-fixed cells in culture medium at room temperature using a spinning disk confocal microscope (Marianas SDC imaging system, Intelligent Imaging Innovations/3i, Denver, CO, USA) with a 473 nm laser. Images were acquired using a Neofluar × 63 1.4 NA DIC objective on a CascadeII 512 EMCCD (Photometrics, Tucson, AZ, USA) using SlideBook 4.2 software (3i). For analysis of cell morphology, cells were treated in six-well plates and visualized in an Olympus (Center Valley, PA, USA) IX70 inverted microscope with a × 20 objective. Brightfield pictures were taken with an Olympus C-5060 camera. One tenth of the whole field is shown.
For electron microscopy, cells were fixed with 2.5% glutaraldehyde in 0.1 M pH 7.4 buffer at 4°C. After fixation, samples were placed in 1% osmium tetroxide in 0.1 M pH 7.4 buffer, dehydrated in a graded series of ethyl alcohol and embedded in Spurr resin.
Abbreviations
- CHX:
-
cycloheximide
- GFP:
-
green fluorescent protein
- DISC:
-
death-inducing signaling complex
- DKO:
-
SV40-transformed Bax-, Bak-deficient MEFs
- MEF:
-
murine embryonic fibroblast
- MOMP:
-
mitochondrial outer membrane permeabilization
- PI:
-
propidium iodide
- Q-VD:
-
quinoline-Val-Asp-CH2-difluorophenoxy (Q-VD-OPH)
- Z-VAD:
-
Z-Val-Ala-Asp(OMe)-CH2F (Z-VAD-FMK)
References
Taylor RC, Cullen SP, Martin SJ . Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 2008; 9: 231–241.
Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH et al. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J 1997; 16: 2794–2804.
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997; 91: 479.
Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD . The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 1997; 275: 1132–1136.
Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A . BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 2007; 12: 171–185.
Ekert PG, Read SH, Silke J, Marsden VS, Kaufmann H, Hawkins CJ et al. Apaf-1 and caspase-9 accelerate apoptosis, but do not determine whether factor-deprived or drug-treated cells die. J Cell Biol 2004; 165: 835–842.
Lindsten T, Ross AJ, King A, Zong W-X, Rathmell JC, Shiels HA et al. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol Cell 2000; 6: 1389.
Wei MC, Zong W-X, Cheng EHY, Lindsten T, Panoutsakopoulou V, Ross AJ et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292: 727–730.
Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 2004; 6: 1221–1228.
Ullman E, Fan Y, Stawowczyk M, Chen HM, Yue Z, Zong WX . Autophagy promotes necrosis in apoptosis-deficient cells in response to ER stress. Cell Death Differ 2007; 15: 422.
Suzuki A, Kusakai G, Kishimoto A, Lu J, Ogura T, Esumi H . ARK5 suppresses the cell death induced by nutrient starvation and death receptors via inhibition of caspase 8 activation, but not by chemotherapeutic agents or UV irradiation. Oncogene 2003; 22: 6177–6182.
Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y . Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol 2007; 178: 93–105.
Vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH, Thompson CB . Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol Cell Biol 2001; 21: 5899–5912.
Gonin-Giraud S, Mathieu AL, Diocou S, Tomkowiak M, Delorme G, Marvel J . Decreased glycolytic metabolism contributes to but is not the inducer of apoptosis following IL-3-starvation. Cell Death Differ 2002; 9: 1147–1157.
Lee YJ, Galoforo SS, Berns CM, Tong WP, Kim HR, Corry PM . Glucose deprivation-induced cytotoxicity in drug resistant human breast carcinoma MCF-7/ADR cells: role of c-myc and bcl-2 in apoptotic cell death. J Cell Sci 1997; 110: 681–686.
Alves NL, Derks IA, Berk E, Spijker R, van Lier RA, Eldering E . The Noxa/Mcl-1 axis regulates susceptibility to apoptosis under glucose limitation in dividing T cells. Immunity 2006; 24: 703–716.
Zhao Y, Coloff JL, Ferguson EC, Jacobs SR, Cui K, Rathmell JC . Glucose metabolism attenuates p53 and puma-dependent cell death upon growth factor deprivation. J Biol Chem 2008; 283: 36344–36353: M803580200.
Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005; 120: 237.
Tu HC, Ren D, Wang GX, Chen DY, Westergard TD, Kim H et al. The p53-cathepsin axis cooperates with ROS to activate programmed necrotic death upon DNA damage. Proc Natl Acad Sci USA 2009; 106: 1093–1098.
Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB . Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev 2004; 18: 1272–1282.
McStay GP, Salvesen GS, Green DR . Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ 2007; 15: 322–331.
Duckett CS, Li F, Wang Y, Tomaselli KJ, Thompson CB, Armstrong RC . Human IAP-like protein regulates programmed cell death downstream of Bcl-xL and cytochrome c. Mol Cell Biol 1998; 18: 608–615.
Boldin MP, Goncharov TM, Goltseve YV, Wallach D . Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell 1996; 85: 803.
Stupack DG, Puente XS, Boutsaboualoy S, Storgard CM, Cheresh DA . Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. J Cell Biol 2001; 155: 459–470.
Wrasidlo W, Mielgo A, Torres VA, Barbero S, Stoletov K, Suyama TL et al. The marine lipopeptide somocystinamide A triggers apoptosis via caspase 8. Proc Natl Acad Sci USA 2008; 105: 2313–2318.
Zhou Q, Snipas S, Orth K, Muzio M, Dixit VM, Salvesen GS . Target protease specificity of the viral serpin CrmA. ANALYSIS OF FIVE CASPASES. J Biol Chem 1997; 272: 7797–7800.
Kasibhatla S, Brunner T, Genestier L, Echeverri F, Mahboubi A, Green DR . DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-[kappa]B and AP-1. Mol Cell 1998; 1: 543.
Gong B, Almasan A . Apo2 ligand/TNF-related apoptosis-inducing ligand and death receptor 5 mediate the apoptotic signaling induced by ionizing radiation in leukemic cells. Cancer Res 2000; 60: 5754–5760.
Vince JE, Wong WW-L, Khan N, Feltham R, Chau D, Ahmed AU et al. IAP antagonists target cIAP1 to induce TNF[alpha]-dependent apoptosis. Cell 2007; 131: 682.
Munoz-Pinedo C, Ruiz-Ruiz C, Ruiz de Almodovar C, Palacios C, Lopez-Rivas A . Inhibition of glucose metabolism sensitizes tumor cells to death receptor-triggered apoptosis through enhancement of death-inducing signaling complex formation and apical procaspase-8 processing. J Biol Chem 2003; 278: 12759–12768.
Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997; 388: 190.
Wang L, Du F, Wang X . TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008; 133: 693–703.
DeBerardinis RJ, Sayed N, Ditsworth D, Thompson CB . Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Develop 2008; 18: 54.
Munoz-Pinedo C, Robledo G, Lopez-Rivas A . Thymidylate synthase inhibition triggers glucose-dependent apoptosis in p53-negative leukemic cells. FEBS Lett 2004; 570: 205–210.
Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA et al. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat Med 2000; 6: 529–535.
Harada K, Toyooka S, Shivapurkar N, Maitra A, Reddy JL, Matta H et al. Deregulation of caspase 8 and 10 expression in pediatric tumors and cell lines. Cancer Res 2002; 62: 5897–5901.
Gröbner S, Adkins I, Schulz S, Richter K, Borgmann S, Wesselborg S et al. Catalytically active Yersinia outer protein P induces cleavage of RIP and caspase-8 at the level of the DISC independently of death receptors in dendritic cells. Apoptosis 2007; 12: 1813.
Rebe C, Cathelin S, Launay S, Filomenko R, Prevotat L, L’Ollivier C et al. Caspase-8 prevents sustained activation of NF-{kappa}B in monocytes undergoing macrophagic differentiation. Blood 2007; 109: 1442–1450.
Sanchez I, Xu CJ, Juo P, Kakizaka A, Blenis J, Yuan J . Caspase-8 is required for cell death induced by expanded polyglutamine repeats. Neuron 1999; 22: 623–633.
Werner AB, de Vries E, Tait SWG, Bontjer I, Borst J . TRAIL receptor and CD95 signal to mitochondria via FADD, caspase-8/10, Bid, and Bax but differentially regulate events downstream from truncated Bid. J Biol Chem 2002; 277: 40760–40767.
Acknowledgements
We thank Josh Goldstein for the pBABE-Bcl-xL plasmid, Seamus Martin for antibodies, John Silke and Joan Gil for discussions and Pat Fitzgerald, Marina Noriega, Ana Yrla and Marcela Saenz for technical assistance. We also thank TetraLogic Pharmaceuticals for supplying Compound A for studies carried out in John Silke's laboratory in La Trobe University. This work was supported by grants from the U.S. National Institutes of Health (to DRG), by Association pour la Recherche sur le Cancer and l’Agence Nationale de la Recherche (ref ANR-09-JCJC-0003-01) (to JER), and by the Fondo de Investigaciones Sanitarias-ISCIII (Grants CP05/0036, PI071027 and RTICC RD06/0020 to CM-P).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by RA Knight
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Caro-Maldonado, A., Tait, S., Ramírez-Peinado, S. et al. Glucose deprivation induces an atypical form of apoptosis mediated by caspase-8 in Bax-, Bak-deficient cells. Cell Death Differ 17, 1335–1344 (2010). https://doi.org/10.1038/cdd.2010.21
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2010.21
Keywords
This article is cited by
-
Limiting glutamine utilization activates a GCN2/TRAIL-R2/Caspase-8 apoptotic pathway in glutamine-addicted tumor cells
Cell Death & Disease (2022)
-
A role for caspase-8 and TRAIL-R2/DR5 in ER-stress-induced apoptosis
Cell Death & Differentiation (2018)
-
Central role of lactate and proton in cancer cell resistance to glucose deprivation and its clinical translation
Signal Transduction and Targeted Therapy (2017)
-
Glucose deprivation promotes activation of mTOR signaling pathway and protein synthesis in rat skeletal muscle cells
Pflügers Archiv - European Journal of Physiology (2015)
-
A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3
Nature (2014)
