
Abstract
Large-scale genomic analyses of cutaneous melanoma have revealed insights into the aetiology and heterogeneity of this disease, as well as opportunities to further personalise treatment for patients with targeted and immune therapies. Herein, we review the proposed genomic classification of cutaneous melanoma from large-scale next-generation sequencing studies, including the largest integrative analysis of melanoma from The Cancer Genome Atlas (TCGA) Network. We examine studies that have identified molecular features of melanomas linked to immune checkpoint inhibitor response. In addition, we draw attention to low-frequency actionable mutations and highlight frequent non-coding mutations in melanoma where little is known about their biological function that may provide novel avenues for the development of treatment strategies for melanoma patients.
Similar content being viewed by others


Main
Cutaneous melanoma is the deadliest form of skin cancer that originates in melanocytes. Early-stage melanoma is generally curable with early detection and surgery. Patient prognosis significantly decreases for patients with late-stage metastatic disease (Balch et al, 2009). Historically, metastatic melanoma has been considered an untreatable disease, where previous standard of care treatment regimens of either dacarbazine, interferon-α-2b (in the adjuvant setting) and high-dose IL-2 (HD IL-2) for advanced melanoma produce variable benefits to patients, with few patients exhibiting durable responses (reviewed in Girotti et al, 2014). Following the discovery of the BRAF V600 mutation in melanoma and with the advances in immune checkpoint inhibitors, melanoma has emerged as the poster child for both targeted and immune therapy.
Hotspot mutations in the V600 codon of BRAF in ∼50% of melanomas (Davies et al, 2002) and the G12, G13 and Q61 codons of NRAS in ∼25% of cases (Sekiya et al, 1984; Albino et al, 1989; van't Veer et al, 1989) were identified before the era of next-generation sequencing. These findings led to the development of BRAF inhibitors (e.g., vemurafenib and dabrafenib) and MEK kinase inhibitors (e.g., trametinib). Unfortunately, although the antitumour responses to MAPK inhibitors have been marked, resistance remains a clinical challenge. Conversely, response to immune checkpoint antibodies, such as anti-CTLA4 (e.g., ipilimumab) and anti-PD-1 (e.g., nivolumab and pembrolizumab), have produced more durable responses in the treatment of metastatic melanoma patients; however, not all patients respond. As a result, understanding the molecular mechanisms that modulate the response to immunotherapies are critical.
Herein, we review the latest large-scale genomic studies of cutaneous melanoma, including the largest integrative analysis from The Cancer Genome Atlas (TCGA) Network (Cancer Genome Atlas Network, 2015), and present how such information has been proposed to inform clinical decision-making (Table 1). We briefly provide clinical updates on MAPK inhibitors and immunotherapies and highlight findings from biomarker analyses from immune checkpoint inhibitor studies in melanoma. In addition, we draw attention to low-frequency actionable mutations that could inform on potential rare combination strategies to further personalise treatment options for patients. Finally, we examine the discoveries of non-coding mutations that occur in a large fraction of patients where little is known about their biology that provide new avenues of investigation in melanoma.
The challenge of identifying significantly mutated genes in melanoma
Cutaneous melanoma has one of the highest mutation burdens of any cancer sequenced to date (Hodis et al, 2012; Krauthammer et al, 2012), with the latest estimate of ∼17 mutations per Mb calculated by TCGA from whole-exome sequencing (WES) of 318 primary and metastatic melanomas originating from non-glabrous (hair-bearing) skin (Cancer Genome Atlas Network, 2015). Over 75% of the cutaneous melanoma samples sequenced in the TCGA cohort possessed a UV signature, defined by having C>T transitions at dipyrimidine sites accounting for >60%, or CC>TT mutations in >5%, of the total mutation burden (Brash, 2015). Ultraviolet A and UVB are the two wavelengths humans are exposed to from solar radiation. Ultraviolet B-induced DNA damage results in the formation of cyclobutane pyrimidine dimers (CPDs). The most widely accepted mutational mechanism leading to the generation of the UV signature involves the deamination of cytosines or 5-methyl-cytosines in the CPDs to uracils or thymidines, which are then replicated in an error-free manner to produce C>T transitions at dipyrimines (Taylor, 2015). Experimental systems have shown G>T substitutions as being the major mutations resulting from UVA-mediated by oxidative damage (Brash, 2015). However, recent studies have shown that UVA can induce C>T signature mutations through a reactive oxygen species (ROS)-dependent mechanism involving both the the photoprotective black eumelanin and the phototoxic red pheomelanin (Noonan et al, 2012; Premi et al, 2015). Furthermore, pheomelanin is thought to be phototoxic itself through the generation of ROS, which has shown to increase incidence of melanoma in mice even in the absence of UV exposure (Mitra et al, 2012). As a result, identifying driver mutations in melanoma is challenging given the high mutation burden, especially in the case of tumour suppressors and infrequently mutated oncogenes.
This problem was first raised in the analyses of over 100 melanoma exomes in 2012 (Hodis et al, 2012; Krauthammer et al, 2012). For example, more than 500 genes were mutated in at least 10% of the 121 melanomas sequenced – many of which were not expressed in melanocytes and melanoma cell lines. To address this problem, sophisticated statistical tools were developed, including InVEx, which uses sequencing data from intronic and untranslated regions (UTRs) to infer gene-specific mutation burdens (Hodis et al, 2012). Using this tool, genes that were found to be significantly mutated included well-established melanoma oncogenes and tumour suppressors, BRAF, NRAS, MAP2K1, PTEN and CDKN2A, as well as novel significantly mutated genes (SMGs) containing hotspot and loss-of-function mutations that were expressed in melanoma cell lines. These novel SMGs that possessed hotspot mutations caused by C>T transitions were the Rho GTPase, RAC1 (p. P29S), the catalytic subunit of the heterotrimeric PP6 protein phosphatase complex, PPP6C (p. R301C), and the serine threonine kinase, STK19 (p. D89N). Novel SMGs possessing loss-of-function mutations included ARID2, which encodes a component of the SWI/SNF chromatin-remodeling complex. These novel SMGs were concurrently identified in an exome study of 147 melanomas using an alternative approach to define SMGs, which included consideration of gene expression (Krauthammer et al, 2012). This represented an advance to the field as hotspot mutations caused by C>T transitions in oncogenes were not well known.
Algorithms developed before InVEx, such as MutSig, have evolved over the years to take into consideration factors shown to affect regional mutation rates (e.g., gene size, replication time and expression). A pancancer study in which MutSigCV was used to identify SMGs across many cancers estimated that to identify genes possessing driver mutations in ⩾2% of melanoma patients, up to 5300 samples would need to be sequenced given the high mutation burden of this cancer (Lawrence et al, 2014). Ensuing studies have sequenced a fraction of this sample size (TCGA consortium, n=318; Yale cohort, n=213), revealing additional SMGs possessing coding mutations in melanoma, which include NF1, RB1 and IDH1 (Cancer Genome Atlas Network, 2015; Krauthammer et al, 2015). These genes were previously implicated in melanoma through focused sequencing or functional studies, but were found for the first time to be significantly mutated. Almost all IDH1 mutations encoded the p.R132C amino-acid substitution with an incidence of ∼5% and ∼2% in samples of the TCGA and Yale cohorts, respectively. Furthermore, DDX3X, a novel SMG was identified by InVEx in ∼6% of samples (Cancer Genome Atlas Network, 2015).
Genomic classification of melanoma: BRAF, RAS, NF1 and Triple wild-type
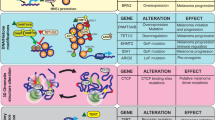
Following BRAF and NRAS, the next most frequently SMG possessing coding mutations was NF1, found in ∼15% of patients (Figure 1). Germline mutations in NF1 are known to cause an inherited multisystem genetic disorder, neurofibromatosis type 1, which is characterised by changes in skin colouring pigmentation (e.g., café-au-lait spots) and the growth of both benign and malignant tumours (Andersen et al, 1993). NF1 encodes for neurofibromin, a RAS-GTPase-activating protein, which negatively regulates RAS signalling by facilitating hydrolysis of RAS-GTP to the RAS-GDP-inactive form. Both the Cancer Genome Atlas Network (2015) and Krauthammer et al (2015) observed that close to 40–50% of melanomas that lacked a hotspot mutation in BRAF (p.V600 or K601E) or NRAS (p. G12, G13 or Q61) possessed loss-of-function mutations in NF1. Previous studies have clearly demonstrated the mutual exclusivity of hotspot BRAF and NRAS mutations in melanoma; however, NF1 mutations were also significantly anticorrelated with hotspot BRAF, but not NRAS, mutations. Altogether, these results supported the categorisation of cutaneous melanoma into four genetic subgroups by MAPK driver mutations: BRAF, RAS (N-H-K), NF1 (lacking a BRAF p.V600 or RAS p.G12, G13 and Q61 hotspot mutation) and Triple wild-type (WT) melanomas (Table 1). NF1 subtype patients were older, had a higher mutation burden (Cancer Genome Atlas Network, 2015) and were reported to possess significant co-occurring mutations in additional RASopathy genes (e.g., RASA1, RASA2, PTPN11 and SOS1) (Arafeh et al, 2015; Krauthammer et al, 2015). Furthermore, WES analysis of desmoplastic melanoma, a rare form of melanoma with sarcomatous histology that occur in chronically sun-exposed skin, revealed significant loss-of-function mutations in NF1 in over 50% of samples (Shain et al, 2015a). Notably, BRAF p.V600E and NRAS p.Q61L/R mutations were not found, providing strong genetic evidence of a role for NF1 in melanomagenesis.

MAPK pathway genetic alterations that constitute the genomic subtypes. Approximate frequency of single-nucleotide variants (SNVs) of SMGs in BRAF, RAS and NF1 are shown. In the latest TCGA study, the estimate of NRAS SNVs in melanomas is ∼30%, with the majority encoding amino-acid Q61 changes, although low-frequency SNVs encoding for alterations in amino acids G12, G13 and Q61 in HRAS and KRAS were also found in ∼2% of samples. Significant amplification of the 4q12 amplicon as well as recurrent KIT mutations were found more frequently in melanomas lacking MAPK mutations (Triple WT). Of note, NF1 is either mutated or deleted (DEL) in ∼55% of desmoplastic melanomas, where no hotspot BRAF and few hotspot RAS mutations have been reported.
A number of molecular and clinical features were associated with the genomic subtypes. For example, only ∼30% of Triple WT samples harboured a UV signature compared with over 90% for the other three subtypes (Cancer Genome Atlas Network, 2015). Somatic copy number analysis revealed Triple WT melanomas had a higher fraction of samples possessing significant copy number amplifications consistent with other studies (e.g., Curtin et al, 2005). Amplicons included the 4q12 minimal common region containing KIT, PDGFRA and KDR (also known as VEGFR2), as well as amplifications in loci encompassing TERT, CDK4 and CCND1. Fusion analysis from RNA sequencing (seq) and whole-genome sequencing (WGS) data revealed that Triple WT melanomas had more complex structural rearrangements and candidate fusion drivers (although few recurrent fusions were identified). Furthermore, Triple WT melanomas had more frequent recurrent KIT mutations (Cancer Genome Atlas Network, 2015). We suspect that the established genomic framework based on somatic alterations including MAPK mutations can aid in personalised therapeutic decision-making in melanoma (Table 1).
Melanoma heterogeneity
A key question in melanoma pathogenesis concerns the temporal acquisition of driving genetic alterations. To address this problem, a unique study sequenced over 290 cancer-associated genes in 150 areas of 37 primary melanomas alongside neighbouring precursor lesions. The various pathogenic stages of melanoma (benign, intermediate, intraepidermal and invasive) were identified through independent histopathological scoring from eight dermatopathologists (Shain et al, 2015b). This group found benign lesions possessed BRAF (p.V600E) mutations exclusively. Intermediate lesions harboured BRAF (p.V600K/K601E) and NRAS mutations, as well as additional driver events that included TERT promoter mutations, which were commonly found in both intermediate lesions and melanomas in situ. Biallelic inactivation of CDKN2A and mutations in chromatin remodellers (e.g., ARID2) were found in invasive melanomas. In addition, PTEN and TP53 mutations were found in advanced tumours.
Exome sequencing of matched primary and metastatic samples from the same patients have revealed important insights into the processes involved in metastatic spread. Specifically, a recent study provided evidence that primary tumours metastasise in parallel to different anatomical sites, often from a common parental sub-population, rather than sequentially from one site to the next (Sanborn et al, 2015). Conversely, individual metastases were sometimes founded by more than one cell population in the primary tumour, which could explain why specific resistance variants are found in different sites after initial response to therapy. Single-cell RNA sequencing analysis of bulk melanomas provided further evidence of tumour heterogeneity at the transcriptional level, revealing that different malignant cells within the same tumour can possess distinct transcriptional states that are associated with either sensitivity or resistance to MAPK targeted therapies (Tirosh et al, 2016).
MAPK inhibitors in BRAF mutant melanomas
In the United States between 2011 and 2013, the FDA approved BRAF inhibitors, vemurafenib (Chapman et al, 2011; McArthur et al, 2014) and dabrafenib (Hauschild et al, 2012), as well as the MEK inhibitor, trametinib (Flaherty et al, 2012), for the treatment of unresectable or metastatic melanoma after the demonstration of improved PFS (progression-free survival) and/or OS (overall survival) in comparison with dacarbazine in BRAF mutant melanoma patients. BRAF inhibitor treatment causes a number of side effects, which include photosensitivity, pyrexia and secondary cutaneous squamous cell carcinomas (SCCs), as well as keratoacanthomas (reviewed in Carlino et al, 2015). BRAF inhibitor-induced SCCs and keratoacanthomas are thought to be induced by the well-characterised paradoxical MAPK activation, which is caused by BRAF inhibitor-mediated homo- and heterodimerisation of non-mutant RAF isoforms leading to the activation of MAPK pathway in BRAF wild-type cells (reviewed in Girotti et al, 2014). Furthermore, the development of resistance emerges in ∼50% of patients within 7 months of treatment (Chapman et al, 2011; Hauschild et al, 2012).
Subsequent combined BRAF and MEK inhibitor phase III clinical trials for patients with a BRAF (p.V600E or p.V600K) mutation were shown to increase PFS and/or OS compared with BRAF inhibitor monotherapy (Larkin et al, 2014; Long et al, 2014; Robert et al, 2015). Inhibition of the MAPK pathway targeting two nodes resulted in a reduction in BRAF inhibitor resistance in ∼25% of patients and a decrease in secondary SCCs and keratoacanthomas, presumably by preventing paradoxical MAPK activation. However, resistance is still a problem, and combination immune and targeted therapy strategies are next steps in treating BRAF mutant melanoma patients (reviewed in Prieto et al, 2016). Clinical trials to date of ipilimumab in combination with BRAF and MEK inhibitors have shown an increase in severe adverse effects, such as hepatotoxicity and bowel perforation. Nonetheless, different combinations of BRAF inhibitors and ipilimumab are well tolerated (e.g., dabrafenib and ipilimumab) (Ribas et al, 2013; Puzanov et al, 2014). Furthermore, combination therapies with anti-PD-1 and anti-PD-L1 appear to be more promising given their reduced toxicity. Studies have demonstrated that BRAF inhibitor treatment can produce a more favourable microenvironment leading to increased CD8+ T-cell infiltration and a decrease in immunosuppressive cytokines (Boni et al, 2010). Some caution has been suggested regarding use the of MEK inhibitors with immune therapy because of their effects on T-cell function; however, studies using mouse models have demonstrated the feasibility of combination BRAF and MEK inhibitors with anti-PD-1 therapeutics (Hu-Lieskovan et al, 2015). Interestingly, transcriptomic analyses of melanomas acquiring MAPK inhibitor resistance demonstrated in half of disease progressing melanomas potential cross-resistance mechanisms to salvage anti-PD-1/PD-L1 immunotherapy through T-cell exhaustion and loss of antigen presentation (Hugo et al, 2015). As a result, identifying the most optimal treatment regimens for combination targeted and immune therapy remains an important avenue of investigation (Grob et al, 2015).
A number of preclinical studies and characterisation of patient samples treated with BRAF inhibitors have identified numerous mechanisms that lead to resistance, which will not be covered here and have been reviewed elsewhere (e.g., Carlino et al, 2015). However, the majority of these mechanisms involve reactivation of the MAPK pathway, including mutations of NRAS, promotion of the splice variant, p61 BRAF p.V600, and BRAF p.V600E amplification, which all lead to increased BRAF dimerisation and constitutive signalling (Carlino et al, 2015). Recent studies of RAF inhibitors able to inhibit both BRAF V600E and BRAF mutant dimers may have more clinical promise and may reduce incidence of resistance (Girotti et al, 2015; Yao et al, 2015; Zhang et al, 2015).
Genomic classification: implications for clinical management of patients with targeted and immune therapies
Given the clinical utility of MAPK targeted therapy and the anticorrelative relationship of hotspot BRAF, RAS and loss-of-function NF1 mutations, we suspect that the framework generated by the TCGA study will be useful to guide personalised therapeutic decisions for cutaneous melanoma (Table 1). Currently, ongoing clinical trials of MEK and CDK4 inhibitors have shown clinical promise in treating patients with NRAS mutant melanomas (Sosman et al, 2014), which were based on preclinical studies that demonstrated MEK inhibitors induce low-level apoptosis in melanoma, but not cell cycle arrest in a NRAS-driven mouse model (Kwong et al, 2012). The discovery of the NF1 subtype in cutaneous and desmoplastic melanomas provides a potential new subclass of melanomas to be targeted with MAPK inhibitors (Cancer Genome Atlas Network, 2015; Krauthammer et al, 2015; Shain et al, 2015a). Early, preclinical studies have demonstrated that NF1-null melanomas activate RAS leading to MEK kinase dependence as well as sensitivity to MAPK inhibitor treatment (Maertens et al, 2013; Nissan et al, 2014). Other studies have shown not all NF1 mutant melanomas respond to MEK inhibitors (Krauthammer et al, 2015; Ranzani et al, 2015). Further investigation is required to determine the mechanisms that mediate MEK inhibitor response in NF1 mutant melanomas, which may aid in further stratifying patients who could benefit from such therapies in combination with immune checkpoint inhibitors given the co-occurring high mutation burden of this subtype (see below and Table 1). In addition, other studies have suggested that pan-RAF and MEK inhibitor combinations may be a useful strategy to target NF1 mutant cancers (Whittaker et al, 2013; Whittaker et al, 2015). Combination therapy targeting a third node of the MAPK pathway may also be useful for all three subtypes in the form of ERK inhibitors (Carlino et al, 2015). Finally, Triple WT melanomas were found to possess more frequent KIT recurrent mutations as well as amplification of the 4q12 amplicon containing KIT, PDGFRA and KDR (Cancer Genome Atlas Network, 2015). Clinical trials for patients harbouring KIT aberrations with tyrosine kinase inhibitors, imatinib and dasatinib, have produced responses in a subset of patients (Hodi et al, 2008; Woodman et al, 2009). The observation of coamplification of PDGFRA and KDR support consideration of combination therapies with receptor tyrosine kinase inhibitors, sorafenib, crenolanib, regunafenib and pasopanib, in KIT-amplified melanomas.
Integrative analysis of the genomic subtypes in combination with clustering data from additional data platforms including mRNA, reverse phase protein array (RPPA) and histopathological analysis revealed a subset of samples with evidence of lymphocytic infiltration associated with improved patient survival, which were equally distributed across the genomic subtypes (Cancer Genome Atlas Network, 2015) (Table 1). Clustering mRNA-seq data revealed three subgroups of melanoma enriched for genes involved in immune function (‘immune’ subgroup), skin and neuronal development (‘keratin’ subgroup) and low expression of the melanocytic lineage-specific transcription factor, MITF (‘MITF-low’ subgroup). The immune subgroup also correlated with lymphocytic infiltration by pathology, and high LCK protein expression from RPPA data. Approximately 50% of samples within each subtype possessed an immune signature, which included PD-L1. Presumably, these are potential subset of patients who will respond to immune therapies.
Molecular features linked to immune therapy response in melanoma
The importance of immunotherapy in melanoma has long been recognised, which is demonstrated, in part, by the fact that HD IL-2 treatment can produce long-term remissions in ∼6–8% of patients (Girotti et al, 2014). Immune therapies to treat melanoma can be broadly characterised into categories that include cytokines/chemokines (e.g., HD IL-2), cancer vaccine therapies, oncolytic virus therapies (e.g., T-VEC), immune checkpoint inhibitors (e.g., anti-CTLA-4 and anti-PD-1) and adoptive cell transfer (ACT) (Sharma and Allison, 2015).
The biggest clinical success in melanoma has come from the immune checkpoint antibodies targeting PD-1 (the programmed cell death-1) and CTLA-4 (cytotoxic T lymphocyte antigen-4). Programmed cell death-1 and CTLA-4 are coinhibitory T-cell receptors that drive a negative signal to diminish T-cell activation and induce immune tolerance. Programmed cell death-1 has two ligands, PD-L1 that is expressed in many cell types including tumours cells, and PD-L2, which is predominately expressed in antigen-presenting cells (Sharma and Allison, 2015). Anti-PD-1 (e.g., nivolumab and pembrolizumab), anti-PD-L1 and anti-CTLA-4 (e.g., ipilimumab) antibodies can reverse immune suppression and activate T cells, triggering an antitumour response. Ipilimumab was the first agent to improve OS in advanced melanoma patients (Hodi et al, 2010), and in combination with dacarbazine improved OS to 11.2 months compared with 9.1 months with higher survival rates at 3 years to 20.8% vs 12.2% (hazard ratio for death, 0.72; P<0.001) (Robert et al, 2011) compared with dacarbazine and placebo. Longer-term follow-up studies demonstrated a ‘plateauing’ effect of the OS curve with 20% survival beginning at 3 years (Schadendorf et al, 2015).
Early-phase anti-PD-1 clinical trials reported higher response rates and fewer immune-related toxicities produced compared with anti-CTLA-4 (reviewed in Girotti et al, 2014; Prieto et al, 2016). Combination anti-PD-1 and anti-CTLA-4 studies revealed further impressive results (Wolchok et al, 2013; Larkin et al, 2015; Postow et al, 2015). However, more severe adverse effects (grade 3 or 4) were observed. In the phase III Checkmate 067 study that evaluated monotherapies of either nivolumab or ipilimumab compared with combination therapy (nivolumab and ipilimumab), stratifying patients by BRAF mutation, disease stage and PD-L1 expression (PD-L1 positivity cutoff ⩾5% on tumour cells with any staining of any intensity on the cell surface) (Larkin et al, 2015), revealed an overall response rate (ORR) of 57.6% for the combination group vs 43.7% and 19% for monotherapies nivolumab and ipilimumab, respectively. Notably, the PD-L1-positive cohort reported median PFS of 14 months (combination or nivolumab therapy) vs 3.9 months (ipilimumab monotherapy), whereas the PD-L1-negative cohort displayed increased success with combination therapies with a PFS of 11.2 months over both nivolumab (5.2 months) and ipilimumab (2.8 months) monotherapies (Larkin et al, 2015). One could interpret these results that given the increased toxicity of dual checkpoint inhibitor treatment, combination anti-PD-1 and anti-CTLA-4 could be reserved for PD-L1-negative patients who have shown improved ORR and PFS compared with monotherapy (Larkin et al, 2015). Whereas PD-L1-positive patients would receive anti-PD-1 monotherapy as a result of having similar benefit compared with combination treatment. The major caveat for this proposition being that overall survival results have not been reported yet, which is the best indicator for immune therapy response (Ascierto et al, 2015). In contrast, it should be noted that the Checkmate 069 study demonstrated the superiority of combination therapy (ipilimumab and nivolumab) over ipilimumab monotherapy with an ORR of 61% compared with 11% in BRAF wild-type patients (similar results found in BRAF mutant patients) (Postow et al, 2015), and also that the ORR was independent of PD-L1 status with similar response rates in PD-L1-positive and -negative tumours (Postow et al, 2015). Quantitative immune profiling studies that examined not only immune cell presence but also density and PD-1/PD-L1 proximity within melanoma samples revealed that pre-existing CD8-positive T cells negatively regulated by the PD-1/PD-L1 axis are predictive of tumour regression to anti-PD1 therapy (Tumeh et al, 2014). PD-L1 as a biomarker for response is challenged by the fact that PD-L1 expression is dynamic and heterogeneous, both within different portions of the tumour and immune cells. Furthermore, detection methods, time of measurement (e.g., pre-treatment vs on treatment) and determining appropriate cutoffs for positive staining complicates standardisation between studies (Schalper et al, 2016).
To date, a number of studies have revealed molecular features of melanoma that modulate response to immune therapy, which include: mutational load, intrinsic transcriptomic states and intestinal microbiome, as well as the composition of peripheral immune cells in patients and melanoma immune infiltrates. For example, WES data has been used to identify neoantigens with implications for immunotherapy in a number of cancers (reviewed in Schumacher and Schreiber, 2015), including melanoma (van Rooij et al, 2013; Snyder et al, 2014; Van Allen et al, 2015). Neoantigens can be defined as ‘non-self’ peptides produced from somatic mutations that are presented on major histocompatibility complex class I molecule that are recognised by the adaptive immune system. The earliest multipatient melanoma exome sequencing study with anti-CTLA-4 blockade response data revealed that a high mutational burden was associated with increased survival (Snyder et al, 2014). The authors also observed a number of four amino-acid (tetrapeptide) motifs within predicted neoantigens that were shared by patients with long-term clinical benefit, but absent in patients that had minimal or no clinical benefit (Snyder et al, 2014). Other studies have not observe this tetrapeptide signature in larger patient cohorts (Schumacher et al, 2015; Van Allen et al, 2015). A study of 110 patients with complete WES and a subset of RNA-seq data determined mutational and neoantigen load, as well as expression of cytolytic markers in the microenvironment were associated with clinical benefit to anti-CTLA4 treatment (Van Allen et al, 2015). Specifically, granzyme A (GZMA), perforin (PRF1), PD-L2 and CTLA-4 were all significantly upregulated in the cohort that showed clinical benefit. In addition, studies performing integrative analysis from publicly available TCGA data revealed a WNT/β-catenin signature associated with an absence of a T-cell expression signature (Spranger et al, 2015). Follow-up functional studies in preclinical melanoma mouse models demonstrated that activation of the WNT/β-catenin pathway produces resistance to anti-PD1 therapy. Furthermore, transcriptomic analysis of pre-treatment melanoma biopsies with anti-PD-1 checkpoint inhibitor response data identified increased expression of genes involved in mesenchymal transition, cell adhesion, extracellular matrix remodeling, angiogenesis and wound healing as barriers to anti-PD-1 response (Hugo et al, 2016). Expression markers of the CD8 T-cell cytolytic score, CTLA-4, and PD-L2 that correlated with response to anti-CTLA4 (Van Allen et al, 2015) were not upregulated in anti-PD-1-responsive melanomas, suggesting differential response mechanisms between the two checkpoint inhibitors (although sample data sets are still relatively small). Although few specific gene mutations have been found to modulate response to immune therapy, mutations in BRCA2 have been linked to improved response (Hugo et al, 2016) and PTEN mutations/deletions in resistance to anti-PD-1 checkpoint inhibitors (Peng et al, 2016). A number of environmental factors have been linked to response to immune therapy including the presence of cytokines, chemokines and ratios of peripheral and intratumoral immune cells. For example, clinical studies have reported a high peripheral neutrophil-to-lymphocyte ratio measured before ipilimumab treatment to be correlated with reduced OS (Ferrucci et al, 2015; Zaragoza et al, 2016). An increased eosinophil count detected early in ipilimumab-treated patients was associated with improved OS, whereas elevated levels of monocytic myeloid-derived suppressor cells were found in non-responders (Delyon et al, 2013; Gebhardt et al, 2015). Work from mouse models have shown activation of intratumoral antigen-presenting CD103+ dendritic cells that increase CD8+ T-cell priming improve response in mice to BRAF and PD-L1 blockade (Salmon et al, 2016). Interestingly, two preclinical studies recently demonstrated intestinal microbiota of mice influence the response to anti-CTLA-4 and anti-PD-L1 checkpoint inhibitors, suggesting that manipulating the microbiota in human patients could be used as a strategy to improve response to immunotherapy (Sivan et al, 2015; Vetizou et al, 2015).
One of the best examples of personalised medicine in the melanoma arena is ACT therapy whereby tumour-infiltrating T-lymphocytes are isolated from a patient’s tumour, expanded and activated ex vivo and reinfused back into the patient (Rosenberg and Restifo, 2015). This strategy has been shown to lead to complete tumour regressions in metastatic melanoma patients. Combining this strategy with WES analysis, neoantigen prediction, followed by expansion of T cells obtained from the tumour or peripheral blood that react to predicted neoantigens, has produced impressive improvements to personalise ACT response (Cohen et al, 2015). One of the benefits of this strategy being that this approach leads to the targeting of neoantigens solely in the tumour-limiting immune-related side effects. Combining ACT strategies with dendritic cell immunisation (Carreno et al, 2015) and immune checkpoint inhibitor treatment, as well as targeted therapy, may provide opportunities to increase long-term durable responses in metastatic melanoma patients.
Potential low-frequency actionable mutations
In an attempt to identify low-frequency actionable and driver mutations without sequencing the estimated 5300 samples required to identify driver mutations in ⩾2% of melanoma patients (Lawrence et al, 2014), studies have cross-referenced the COSMIC mutation database that compiles mutation data from all next-generation sequencing (NGS) cancer studies. Such analyses has revealed low frequency actionable mutations in EZH2, AKT3 and PIK3CA, as well as driver mutations frequently found in other types of melanoma, such as GNAQ and GNA11 (Hodis et al, 2012; Cancer Genome Atlas Network, 2015). Therapeutic strategies to target these frequently mutated G proteins in uveal melanomas include combination MAPK- (e.g., PD0325901 or MEK162) and PKC- (e.g., AEB071) targeted therapies (Chen et al, 2014). In addition, hotspot mutations in epigenetic regulators, such as IDH1 (p. R132), and less the frequently mutated EZH2 (p.Y641), indicate another potential mode of action for targeted therapy in melanoma. IDH1 inhibitors (e.g., AG-120) are currently in phase 1/2 clinical studies for patients with glioma and acute myeloid leukaemia (Popovici-Muller et al, 2012), and recent preclinical studies have demonstrated the clinical utility of EZH2 inhibitors in melanoma (Tiffen et al, 2015). Furthermore, AKT1/3 and PIK3CA mutations observed in BRAF and RAS mutant melanomas may be useful biomarkers for possible combination therapy utilising MAPK and PI(3)K/AKT/mTOR inhibitors (Cancer Genome Atlas Network, 2015). Understanding the relationships of these low-frequency actionable mutations within the different genomic subtypes may inform clinicians on rare therapeutic combinations that may potentially lead to durable response in combination with MAPK and immune targeted therapies (Table 1). In contrast, a number of studies have identified frequent non-coding mutations in genes where the biological significance is still unclear, but affect druggable pathways that may lead to new therapeutic approaches.
Dark matter: non-coding mutations in melanoma
In the era of next-generation sequencing, some of the most novel genomic insights in cutaneous melanoma aetiology have come from the analyses of non-genic portions of the genome. For example, telomerase activity is upregulated in almost all cancers, including melanoma. However, the mechanisms that mediate these processes were not entirely clear. In 2013, two concurrent studies identified two mutually exclusive recurrent TERT promoter mutations found in ∼70% of cutaneous melanoma (Horn et al, 2013; Huang et al, 2013). These hotspot mutations were discovered through interrogation of familial melanomas (Horn et al, 2013) and the analysis of WGS data (Huang et al, 2013). In the familial study, linkage analysis and high-throughput sequencing of a melanoma prone family revealed a disease-segregating germline mutation in the TERT promoter (Horn et al, 2013). Subsequent sequencing of sporadic melanomas did not identify the same TERT promoter mutation, but instead identified 74% of metastatic melanoma cell lines, 85% of metastatic tissue and 33% of primary melanomas that possessed TERT promoter mutations predominantly at two positions, chr 5: 1 295 228 C>T (C228T) and chr 5: 1 295 250 C>T (C250T), in a mutually exclusive manner (Table 2). These mutants produce a new ETS/TCF binding motif that led to increased transcriptional activity and TERT expression (Horn et al, 2013; Huang et al, 2013). A number of telomerase inhibitor strategies have been developed, and are currently in clinical trials for the treatment of polycythemia vera and multiple myeloma treatment (Ruden and Puri, 2013). In addition to TERT, six genes harbouring recurrent non-coding mutations at frequencies between 5 and 10% in cutaneous melanoma have been discovered (Table 2).
Sequencing analyses of 20 desmoplastic melanomas identified hotspot promoter mutations in NFKBIE in ∼15% of cases (Table 2), altering the binding motifs for several transcription factors (e.g., ELF1; Shain et al, 2015a). The protein, IκBɛ encoded by NFKBIE, inhibits the NF-κB signalling pathway via the cytoplasmic retention of transcription factors. Remarkably, these mutations were concurrently identified in six non-desmoplastic melanomas. In addition, a number of frequent non-coding mutations causing C>T transitions have been found in cutaneous melanoma, including the mutation in the 5′-UTR of RPS27 identified in ∼10% of samples by combining data sets generated from four NGS studies with unpublished data (Dutton-Regester et al, 2014) (Table 2). RPS27 is a component of the 40S subunit of ribosomes whose aberrant expression has been linked to a number of cancers, including melanoma (Santa Cruz et al, 1997). The RPS27 mutation is thought to increase its expression by expanding the 5′TOP element (Dutton-Regester et al, 2014), a motif known to control mRNA translation regulated through the PI(3)K/AKT and mTOR pathways. Additional genes whose expression is positively regulated by non-coding mutations include a bi-directional promoter mutation in ∼10% of melanomas in two genes, diphthamide biosynthesis 3 (DPH3) (Fredriksson et al, 2014) and oxidoreductase NAD-binding domain containing 1 (OXNAD1) genes (Denisova et al, 2015) (Table 2). DPH3 is required for the generation of a unique post-translational modification in the elongation factor-2, which is essential for protein synthesis. Although reporter assays have suggested that the recurrent mutations in DPH3 and OXNAD1 results in a significant increase in promoter activity, increased mRNA expression of these genes in tumours was not observed (Denisova et al, 2015).
In contrast, WGS studies revealed promoter mutations that reduce expression in two genes, which included the succinate dehydrogenase complex, subunit D (SDHD) (Weinhold et al, 2014) and the NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 9 (NUDFB9) (Poulos et al, 2015) (Table 2). A pancancer WGS analysis first revealed the SDHD promoter mutations, which disrupt ETS binding sites, resulting in decreased expression (Weinhold et al, 2014). SDHD is one of the four subunits of the succinate dehydrogenase (SDH) enzyme whose function is to anchor SDH to the matrix of the mitochondrial inner membrane and has an important function as it is involved in both the citric acid cycle and oxidative phosphorylation energy conversion pathways. Decreased SDHD expression caused by the promoter mutations is consistent with its tumour suppressor function (Weinhold et al, 2014). The recurrent NDUFB9 promoter mutation was discovered from the first sequencing of a melanoma cell line, COLO-829, and was subsequently validated with TCGA data and functional studies, which determined that ∼5% of melanomas possess this mutation. Luciferase assays demonstrated modest effects on the promoter activity, and the expression of the corresponding gene in tumour data is not significant between mutant and wild-type samples. This may be due to issues related to tumour purity, but still requires further investigation. In the TCGA study, MutSig CV determined RPS27, NDUFB9 and the mitochondrial ribosomal protein S31 (MRPS31) possessing a novel 5′-UTR mutation in ∼6% of samples to be significantly mutated (Cancer Genome Atlas Network, 2015). However, the biological function of the majority of these newly discovered genes in melanoma with non-coding mutations is unclear.
future studies
We still have much to learn about the biology and accompanying biomarkers that influence response to immune therapy in melanoma. With the long-term benefits and durable responses produced by immune checkpoint inhibitors, combination targeted and immune therapy treatment strategies are evident next steps in the evolution of melanoma therapies. Identifying biomarkers that predict response and the mechanisms of resistance to combination targeted and immune therapy will be vital. Although there is a growing list of molecular determinants of immune therapy response in melanoma, analysis using additional data platforms and comprehensive examination of multiple patient biopsies during the course of treatment in large cohorts will likely reveal new factors that influence immune checkpoint inhibitors response. Furthermore, genomic studies to date have revealed actionable, low-frequency driver mutations in melanoma. This raises the possibility of the use of additional combination strategies with MAPK inhibitors and immune therapies with drugs targeting low-frequency driver events, to further personalise treatment options for melanoma patients. It is clear that preclinical studies are needed to determine efficacy of such treatment strategies to target less well-understood mutations in melanoma, including NF1 mutant melanomas. Finally, understanding the biological significance of novel recurrent non-coding mutations in genes with poorly understood function that occur in a relatively large fraction of patients may provide new therapeutic strategies to treat melanoma.
References
Albino AP, Nanus DM, Mentle IR, Cordon-Cardo C, McNutt NS, Bressler J, Andreeff M (1989) Analysis of ras oncogenes in malignant melanoma and precursor lesions: correlation of point mutations with differentiation phenotype. Oncogene 4 (11): 1363–1374.
Andersen LB, Fountain JW, Gutmann DH, Tarle SA, Glover TW, Dracopoli NC, Housman DE, Collins FS (1993) Mutations in the neurofibromatosis 1 gene in sporadic malignant melanoma cell lines. Nat Genet 3 (2): 118–121.
Arafeh R, Qutob N, Emmanuel R, Keren-Paz A, Madore J, Elkahloun A, Wilmott JS, Gartner JJ, Di Pizio A, Winograd-Katz S, Sindiri S, Rotkopf R, Dutton-Regester K, Johansson P, Pritchard AL, Waddell N, Hill VK, Lin JC, Hevroni Y, Rosenberg SA, Khan J, Ben-Dor S, Niv MY, Ulitsky I, Mann GJ, Scolyer RA, Hayward NK, Samuels Y (2015) Recurrent inactivating RASA2 mutations in melanoma. Nat Genet 47 (12): 1408–1410.
Ascierto PA, Marincola FM, Atkins MB (2015) What’s new in melanoma? Combination!. J Transl Med 13: 213.
Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, Buzaid AC, Cochran AJ, Coit DG, Ding S, Eggermont AM, Flaherty KT, Gimotty PA, Kirkwood JM, McMasters KM, Mihm MC Jr, Morton DL, Ross MI, Sober AJ, Sondak VK (2009) Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 27 (36): 6199–6206.
Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, Ferrone CR, Flaherty KT, Lawrence DP, Fisher DE, Tsao H, Wargo JA (2010) Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res 70 (13): 5213–5219.
Brash DE (2015) UV signature mutations. Photochem Photobiol 91 (1): 15–26.
Cancer Genome Atlas Network (2015) Genomic classification of cutaneous melanoma. Cell 161 (7): 1681–1696.
Carlino MS, Long GV, Kefford RF, Rizos H (2015) Targeting oncogenic BRAF and aberrant MAPK activation in the treatment of cutaneous melanoma. Crit Rev Oncol Hematol 96 (3): 385–398.
Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, Linette GP (2015) Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348 (6236): 803–808.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O'Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA, Group B-S (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364 (26): 2507–2516.
Chen X, Wu Q, Tan L, Porter D, Jager MJ, Emery C, Bastian BC (2014) Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene 33 (39): 4724–4734.
Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, Parkhurst MR, Ankri C, Prickett TD, Crystal JS, Li YF, El-Gamil M, Rosenberg SA, Robbins PF (2015) Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest 125 (10): 3981–3991.
Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Brocker EB, LeBoit PE, Pinkel D, Bastian BC (2005) Distinct sets of genetic alterations in melanoma. N Engl J Med 353 (20): 2135–2147.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA (2002) Mutations of the BRAF gene in human cancer. Nature 417 (6892): 949–954.
Delyon J, Mateus C, Lefeuvre D, Lanoy E, Zitvogel L, Chaput N, Roy S, Eggermont AM, Routier E, Robert C (2013) Experience in daily practice with ipilimumab for the treatment of patients with metastatic melanoma: an early increase in lymphocyte and eosinophil counts is associated with improved survival. Ann Oncol 24 (6): 1697–1703.
Denisova E, Heidenreich B, Nagore E, Rachakonda PS, Hosen I, Akrap I, Traves V, Garcia-Casado Z, Lopez-Guerrero JA, Requena C, Sanmartin O, Serra-Guillen C, Llombart B, Guillen C, Ferrando J, Gimeno E, Nordheim A, Hemminki K, Kumar R (2015) Frequent DPH3 promoter mutations in skin cancers. Oncotarget 6 (34): 35922–35930.
Dutton-Regester K, Gartner JJ, Emmanuel R, Qutob N, Davies MA, Gershenwald JE, Robinson W, Robinson S, Rosenberg SA, Scolyer RA, Mann GJ, Thompson JF, Hayward NK, Samuels Y (2014) A highly recurrent RPS27 5′-UTR mutation in melanoma. Oncotarget 5 (10): 2912–2917.
Ferrucci PF, Gandini S, Battaglia A, Alfieri S, Di Giacomo AM, Giannarelli D, Cappellini GC, De Galitiis F, Marchetti P, Amato G, Lazzeri A, Pala L, Cocorocchio E, Martinoli C (2015) Baseline neutrophil-to-lymphocyte ratio is associated with outcome of ipilimumab-treated metastatic melanoma patients. Br J Cancer 112 (12): 1904–1910.
Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, Dummer R, Trefzer U, Larkin JM, Utikal J, Dreno B, Nyakas M, Middleton MR, Becker JC, Casey M, Sherman LJ, Wu FS, Ouellet D, Martin AM, Patel K, Schadendorf D, Group MS (2012) Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 367 (2): 107–114.
Fredriksson NJ, Ny L, Nilsson JA, Larsson E (2014) Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat Genet 46 (12): 1258–1263.
Gebhardt C, Sevko A, Jiang H, Lichtenberger R, Reith M, Tarnanidis K, Holland-Letz T, Umansky L, Beckhove P, Sucker A, Schadendorf D, Utikal J, Umansky V (2015) Myeloid cells and related chronic inflammatory factors as novel predictive markers in melanoma treatment with ipilimumab. Clin Cancer Res 21 (24): 5453–5459.
Girotti MR, Lopes F, Preece N, Niculescu-Duvaz D, Zambon A, Davies L, Whittaker S, Saturno G, Viros A, Pedersen M, Suijkerbuijk BM, Menard D, McLeary R, Johnson L, Fish L, Ejiama S, Sanchez-Laorden B, Hohloch J, Carragher N, Macleod K, Ashton G, Marusiak AA, Fusi A, Brognard J, Frame M, Lorigan P, Marais R, Springer C (2015) Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell 27 (1): 85–96.
Girotti MR, Saturno G, Lorigan P, Marais R (2014) No longer an untreatable disease: how targeted and immunotherapies have changed the management of melanoma patients. Mol Oncol 8 (6): 1140–1158.
Grob JJ, Long GV, Schadendorf D, Flaherty K (2015) Disease kinetics for decision-making in advanced melanoma: a call for scenario-driven strategy trials. Lancet Oncol 16 (13): e522–e526.
Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr, Kaempgen E, Martin-Algarra S, Karaszewska B, Mauch C, Chiarion-Sileni V, Martin AM, Swann S, Haney P, Mirakhur B, Guckert ME, Goodman V, Chapman PB (2012) Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380 (9839): 358–365.
Hodi FS, Friedlander P, Corless CL, Heinrich MC, Mac Rae S, Kruse A, Jagannathan J, Van den Abbeele AD, Velazquez EF, Demetri GD, Fisher DE (2008) Major response to imatinib mesylate in KIT-mutated melanoma. J Clin Oncol 26 (12): 2046–2051.
Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363 (8): 711–723.
Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, Cibulskis K, Sivachenko A, Voet D, Saksena G, Stransky N, Onofrio RC, Winckler W, Ardlie K, Wagle N, Wargo J, Chong K, Morton DL, Stemke-Hale K, Chen G, Noble M, Meyerson M, Ladbury JE, Davies MA, Gershenwald JE, Wagner SN, Hoon DS, Schadendorf D, Lander ES, Gabriel SB, Getz G, Garraway LA, Chin L (2012) A landscape of driver mutations in melanoma. Cell 150 (2): 251–263.
Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, Schadendorf D, Kumar R (2013) TERT promoter mutations in familial and sporadic melanoma. Science 339 (6122): 959–961.
Hu-Lieskovan S, Mok S, Moreno BH, Tsoi J, Robert L, Goedert L, Pinheiro EM, Koya RC, Graeber TG, Comin-Anduix B (2015) Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAFV600E melanoma. Sci Transl Med 7 (279): 279ra41.
Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA (2013) Highly recurrent TERT promoter mutations in human melanoma. Science 339 (6122): 957–959.
Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, Moriceau G, Hong A, Dahlman KB, Johnson DB, Sosman JA, Ribas A, Lo RS (2015) Non-genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell 162 (6): 1271–1285.
Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, Seja E, Lomeli S, Kong X, Kelley MC, Sosman JA, Johnson DB, Ribas A, Lo RS (2016) Genomic and transcriptomic features of response to anti-PD-1 Therapy in metastatic melanoma. Cell 165 (1): 35–44.
Krauthammer M, Kong Y, Bacchiocchi A, Evans P, Pornputtapong N, Wu C, McCusker JP, Ma S, Cheng E, Straub R, Serin M, Bosenberg M, Ariyan S, Narayan D, Sznol M, Kluger HM, Mane S, Schlessinger J, Lifton RP, Halaban R (2015) Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat Genet 47 (9): 996–1002.
Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, Ariyan S, Narayan D, Dutton-Regester K, Capatana A, Holman EC, Bosenberg M, Sznol M, Kluger HM, Brash DE, Stern DF, Materin MA, Lo RS, Mane S, Ma S, Kidd KK, Hayward NK, Lifton RP, Schlessinger J, Boggon TJ, Halaban R (2012) Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 44 (9): 1006–1014.
Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, Langsdorf AE, Jakubosky D, Genovese G, Muller FL, Jeong JH, Bender RP, Chu GC, Flaherty KT, Wargo JA, Collins JJ, Chin L (2012) Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med 18 (10): 1503–1510.
Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, Mandala M, Demidov L, Stroyakovskiy D, Thomas L, de la Cruz-Merino L, Dutriaux C, Garbe C, Sovak MA, Chang I, Choong N, Hack SP, McArthur GA, Ribas A (2014) Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 371 (20): 1867–1876.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD (2015) Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 373 (1): 23–34.
Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G (2014) Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505 (7484): 495–501.
Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, Chiarion Sileni V, Lebbe C, Mandala M, Millward M, Arance A, Bondarenko I, Haanen JB, Hansson J, Utikal J, Ferraresi V, Kovalenko N, Mohr P, Probachai V, Schadendorf D, Nathan P, Robert C, Ribas A, DeMarini DJ, Irani JG, Casey M, Ouellet D, Martin AM, Le N, Patel K, Flaherty K (2014) Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 371 (20): 1877–1888.
Maertens O, Johnson B, Hollstein P, Frederick DT, Cooper ZA, Messiaen L, Bronson RT, McMahon M, Granter S, Flaherty K, Wargo JA, Marais R, Cichowski K (2013) Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov 3 (3): 338–349.
McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, Ribas A, Hogg D, Hamid O, Ascierto PA, Garbe C, Testori A, Maio M, Lorigan P, Lebbe C, Jouary T, Schadendorf D, O'Day SJ, Kirkwood JM, Eggermont AM, Dreno B, Sosman JA, Flaherty KT, Yin M, Caro I, Cheng S, Trunzer K, Hauschild A (2014) Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol 15 (3): 323–332.
Mitra D, Luo X, Morgan A, Wang J, Hoang MP, Lo J, Guerrero CR, Lennerz JK, Mihm MC, Wargo JA, Robinson KC, Devi SP, Vanover JC, D'Orazio JA, McMahon M, Bosenberg MW, Haigis KM, Haber DA, Wang Y, Fisher DE (2012) An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature 491 (7424): 449–453.
Nissan MH, Pratilas CA, Jones AM, Ramirez R, Won H, Liu C, Tiwari S, Kong L, Hanrahan AJ, Yao Z, Merghoub T, Ribas A, Chapman PB, Yaeger R, Taylor BS, Schultz N, Berger MF, Rosen N, Solit DB (2014) Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res 74 (8): 2340–2350.
Noonan FP, Zaidi MR, Wolnicka-Glubisz A, Anver MR, Bahn J, Wielgus A, Cadet J, Douki T, Mouret S, Tucker MA, Popratiloff A, Merlino G, De Fabo EC (2012) Melanoma induction by ultraviolet A but not ultraviolet B radiation requires melanin pigment. Nat Commun 3: 884.
Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, Williams LJ, Deng W, Chen G, Mbofung R, Lazar AJ, Torres-Cabala CA, Cooper ZA, Chen PL, Tieu TN, Spranger S, Yu X, Bernatchez C, Forget MA, Haymaker C, Amaria R, McQuade JL, Glitza IC, Cascone T, Li HS, Kwong LN, Heffernan TP, Hu J, Bassett RL Jr, Bosenberg MW, Woodman SE, Overwijk WW, Lizee G, Roszik J, Gajewski TF, Wargo JA, Gershenwald JE, Radvanyi L, Davies MA, Hwu P (2016) Loss of pten promotes resistance to T cell-mediated immunotherapy. Cancer Discov 6 (2): 202–216.
Popovici-Muller J, Saunders JO, Salituro FG, Travins JM, Yan S, Zhao F, Gross S, Dang L, Yen KE, Yang H, Straley KS, Jin S, Kunii K, Fantin VR, Zhang S, Pan Q, Shi D, Biller SA, Su SM (2012) Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HG in vivo. ACS Med Chem Lett 3 (10): 850–855.
Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, Shaheen M, Ernstoff MS, Minor D, Salama AK, Taylor M, Ott PA, Rollin LM, Horak C, Gagnier P, Wolchok JD, Hodi FS (2015) Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 372 (21): 2006–2017.
Poulos RC, Thoms JA, Shah A, Beck D, Pimanda JE, Wong JW (2015) Systematic screening of promoter regions pinpoints functional cis-regulatory mutations in a cutaneous melanoma genome. Mol Cancer Res 13 (8): 1218–1226.
Premi S, Wallisch S, Mano CM, Weiner AB, Bacchiocchi A, Wakamatsu K, Bechara EJ, Halaban R, Douki T, Brash DE (2015) Photochemistry. Chemiexcitation of melanin derivatives induces DNA photoproducts long after UV exposure. Science 347 (6224): 842–847.
Prieto PA, Reuben A, Cooper ZA, Wargo JA (2016) Targeted therapies combined with immune checkpoint therapy. Cancer J 22 (2): 138–146.
Puzanov I, Callahan MK, Linette GP, Patel SP, Luke JJ, Sosman JA, Wolchok JD, Hamid O, Minor DR, Orford KW (2014) Phase 1 study of the BRAF inhibitor dabrafenib (D) with or without the MEK inhibitor trametinib (T) in combination with ipilimumab (Ipi) for V600E/K mutation-positive unresectable or metastatic melanoma (MM). ASCO Annu Meet Proc 32 (15_Suppl): 2511.
Ranzani M, Alifrangis C, Perna D, Dutton-Regester K, Pritchard A, Wong K, Rashid M, Robles-Espinoza CD, Hayward NK, McDermott U, Garnett M, Adams DJ (2015) BRAF/NRAS wild-type melanoma, NF1 status and sensitivity to trametinib. Pigment Cell Melanoma Res 28 (1): 117–119.
Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J (2013) Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med 368 (14): 1365–1366.
Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, Chiarion-Sileni V, Drucis K, Krajsova I, Hauschild A, Lorigan P, Wolter P, Long GV, Flaherty K, Nathan P, Ribas A, Martin AM, Sun P, Crist W, Legos J, Rubin SD, Little SM, Schadendorf D (2015) Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372 (1): 30–39.
Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, Davidson N, Richards J, Maio M, Hauschild A, Miller WH Jr, Gascon P, Lotem M, Harmankaya K, Ibrahim R, Francis S, Chen TT, Humphrey R, Hoos A, Wolchok JD (2011) Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 364 (26): 2517–2526.
Rosenberg SA, Restifo NP (2015) Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348 (6230): 62–68.
Ruden M, Puri N (2013) Novel anticancer therapeutics targeting telomerase. Cancer Treat Rev 39 (5): 444–456.
Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, Chakarov S, Rivera C, Hogstad B, Bosenberg M, Hashimoto D, Gnjatic S, Bhardwaj N, Palucka AK, Brown BD, Brody J, Ginhoux F, Merad M (2016) Expansion and activation of CD103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity 44 (4): 924–938.
Sanborn JZ, Chung J, Purdom E, Wang NJ, Kakavand H, Wilmott JS, Butler T, Thompson JF, Mann GJ, Haydu LE, Saw RP, Busam KJ, Lo RS, Collisson EA, Hur JS, Spellman PT, Cleaver JE, Gray JW, Huh N, Murali R, Scolyer RA, Bastian BC, Cho RJ (2015) Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc Natl Acad Sci USA 112 (35): 10995–11000.
Santa Cruz DJ, Hamilton PD, Klos DJ, Fernandez-Pol JA (1997) Differential expression of metallopanstimulin/S27 ribosomal protein in melanocytic lesions of the skin. J Cutan Pathol 24 (9): 533–542.
Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM, Wolchok JD (2015) Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 33 (17): 1889–1894.
Schalper KA, Kaftan E, Herbst RS (2016) Predictive biomarkers for PD-1 axis therapies: the hidden treasure or a call for research. Clin Cancer Res 22 (9): 2102–2104.
Schumacher TN, Kesmir C, van Buuren MM (2015) Biomarkers in cancer immunotherapy. Cancer Cell 27 (1): 12–14.
Schumacher TN, Schreiber RD (2015) Neoantigens in cancer immunotherapy. Science 348 (6230): 69–74.
Sekiya T, Fushimi M, Hori H, Hirohashi S, Nishimura S, Sugimura T (1984) Molecular cloning and the total nucleotide sequence of the human c-Ha-ras-1 gene activated in a melanoma from a Japanese patient. Proc Natl Acad Sci USA 81 (15): 4771–4775.
Shain AH, Garrido M, Botton T, Talevich E, Yeh I, Sanborn JZ, Chung J, Wang NJ, Kakavand H, Mann GJ, Thompson JF, Wiesner T, Roy R, Olshen AB, Gagnon A, Gray JW, Huh N, Hur JS, Busam KJ, Scolyer RA, Cho RJ, Murali R, Bastian BC (2015a) Exome sequencing of desmoplastic melanoma identifies recurrent NFKBIE promoter mutations and diverse activating mutations in the MAPK pathway. Nat Genet 47 (10): 1194–1199.
Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, Dummer R, North J, Pincus L, Ruben B, Rickaby W, D'Arrigo C, Robson A, Bastian BC (2015b) The genetic evolution of melanoma from precursor lesions. N Engl J Med 373 (20): 1926–1936.
Sharma P, Allison JP (2015) Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161 (2): 205–214.
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, Chang EB, Gajewski TF (2015) Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350 (6264): 1084–1089.
Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, Li Y, Elipenahli C, Liu C, Harbison CT, Wang L, Ribas A, Wolchok JD, Chan TA (2014) Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 371 (23): 2189–2199.
Sosman JA, Kittaneh M, Lolkema MP, Postow MA, Schwartz G, Franklin C, Matano A, Bhansali S, Parasuraman S, Kim K (2014) A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS-mutant melanoma: early encouraging clinical activity. ASCO Annu Meet Proceed 32 (15_Suppl): 9009.
Spranger S, Bao R, Gajewski TF (2015) Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523 (7559): 231–235.
Taylor JS (2015) Biomolecules. The dark side of sunlight and melanoma. Science 347 (6224): 824.
Tiffen JC, Gunatilake D, Gallagher SJ, Gowrishankar K, Heinemann A, Cullinane C, Dutton-Regester K, Pupo GM, Strbenac D, Yang JY, Madore J, Mann GJ, Hayward NK, McArthur GA, Filipp FV, Hersey P (2015) Targeting activating mutations of EZH2 leads to potent cell growth inhibition in human melanoma by derepression of tumor suppressor genes. Oncotarget 6 (29): 27023–27036.
Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, Fallahi-Sichani M, Dutton-Regester K, Lin JR, Cohen O, Shah P, Lu D, Genshaft AS, Hughes TK, Ziegler CG, Kazer SW, Gaillard A, Kolb KE, Villani AC, Johannessen CM, Andreev AY, Van Allen EM, Bertagnolli M, Sorger PK, Sullivan RJ, Flaherty KT, Frederick DT, Jane-Valbuena J, Yoon CH, Rozenblatt-Rosen O, Shalek AK, Regev A, Garraway LA (2016) Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352 (6282): 189–196.
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515 (7528): 568–571.
van 't Veer LJ, Burgering BM, Versteeg R, Boot AJ, Ruiter DJ, Osanto S, Schrier PI, Bos JL (1989) N-ras mutations in human cutaneous melanoma from sun-exposed body sites. Mol Cell Biol 9 (7): 3114–3116.
Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MH, Goldinger SM, Utikal J, Hassel JC, Weide B, Kaehler KC, Loquai C, Mohr P, Gutzmer R, Dummer R, Gabriel S, Wu CJ, Schadendorf D, Garraway LA (2015) Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350 (6257): 207–211.
van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJ, Behjati S, Hilkmann H, El Atmioui D, Nieuwland M, Stratton MR, Kerkhoven RM, Kesmir C, Haanen JB, Kvistborg P, Schumacher TN (2013) Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol 31 (32): e439–e442.
Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CP, Poirier-Colame V, Roux A, Becharef S, Formenti S, Golden E, Cording S, Eberl G, Schlitzer A, Ginhoux F, Mani S, Yamazaki T, Jacquelot N, Enot DP, Berard M, Nigou J, Opolon P, Eggermont A, Woerther PL, Chachaty E, Chaput N, Robert C, Mateus C, Kroemer G, Raoult D, Boneca IG, Carbonnel F, Chamaillard M, Zitvogel L (2015) Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350 (6264): 1079–1084.
Weinhold N, Jacobsen A, Schultz N, Sander C, Lee W (2014) Genome-wide analysis of noncoding regulatory mutations in cancer. Nat Genet 46 (11): 1160–1165.
Whittaker SR, Cowley GS, Wagner S, Luo F, Root DE, Garraway LA (2015) Combined pan-RAF and MEK inhibition overcomes multiple resistance mechanisms to selective RAF inhibitors. Mol Cancer Ther 14 (12): 2700–2711.
Whittaker SR, Theurillat JP, Van Allen E, Wagle N, Hsiao J, Cowley GS, Schadendorf D, Root DE, Garraway LA (2013) A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov 3 (3): 350–362.
Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, Inzunza HD, Feely W, Horak CE, Hong Q, Korman AJ, Wigginton JM, Gupta A, Sznol M (2013) Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 369 (2): 122–133.
Woodman SE, Trent JC, Stemke-Hale K, Lazar AJ, Pricl S, Pavan GM, Fermeglia M, Gopal YN, Yang D, Podoloff DA, Ivan D, Kim KB, Papadopoulos N, Hwu P, Mills GB, Davies MA (2009) Activity of dasatinib against L576P KIT mutant melanoma: molecular, cellular, and clinical correlates. Mol Cancer Ther 8 (8): 2079–2085.
Yao Z, Torres NM, Tao A, Gao Y, Luo L, Li Q, de Stanchina E, Abdel-Wahab O, Solit DB, Poulikakos PI, Rosen N (2015) BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell 28 (3): 370–383.
Zaragoza J, Caille A, Beneton N, Bens G, Christiann F, Maillard H, Machet L (2016) High neutrophil to lymphocyte ratio measured before starting ipilimumab treatment is associated with reduced overall survival in patients with melanoma. Br J Dermatol 174 (1): 146–151.
Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, Zhang J, Lin J, Ewing T, Matusow B, Tsang G, Marimuthu A, Cho H, Wu G, Wang W, Fong D, Nguyen H, Shi S, Womack P, Nespi M, Shellooe R, Carias H, Powell B, Light E, Sanftner L, Walters J, Tsai J, West BL, Visor G, Rezaei H, Lin PS, Nolop K, Ibrahim PN, Hirth P, Bollag G (2015) RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 526 (7574): 583–586.
Acknowledgements
We thank Dr Pei-Ling Chen from the MD Anderson Cancer Centre for helpful comments and suggestions for this review. Shivshankari Rajkumar is a recipient of the 2015 Canderel Studentship. Ian Watson is a recipient of the 2016 Melanoma Research Alliance Young Investigator Award.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Rajkumar, S., Watson, I. Molecular characterisation of cutaneous melanoma: creating a framework for targeted and immune therapies. Br J Cancer 115, 145–155 (2016). https://doi.org/10.1038/bjc.2016.195
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.195
