
Abstract
Quorum sensing is the process by which bacterial cells can communicate by producing substances to regulate viable processes such as gene expression, virulence, and biofilm formation. Gram-positive bacteria such as Staphylococcus aureus and Enterococcus faecalis have specific enzymes (autoinducers) that control the quorum sensing system. Sortase A is a surface protein that regulates virulence and cell‒cell communication in Gram-positive bacteria. To interfere with this system and reduce virulence and cell‒cell communication, quorum sensing inhibitors are used, which are nonantibiotic substances. In this study, we aimed to use Food and Drug Administration-approved drugs (analgesics and antipsychotics) and investigate their activity using molecular docking and microbiological assays against both quorum sensing in Gram-positive S. aureus and E. faecalis. This study investigated the quorum sensing inhibitors acetylsalicylic acid and trifluoperazine and evaluated their affinity to the active site of SrtA (PDB:1t2w) using AutoDock Vina software. Agar diffusion and minimum inhibitory concentration tests were performed to experimentally validate the quorum sensing inhibitor activity of acetylsalicylic acid and trifluoperazine. Molecular docking illustrated that acetylsalicylic acid and trifluoperazine have high affinity as quorum sensing inhibitors in both S. aureus and E. faecalis. However, only acetylsalicylic acid showed inhibition activity at 1000 µg/ml in E. faecalis and at 250 µg/ml by the agar well diffusion method in S. aureus. The high affinity of these quorum sensing inhibitors, as presented by the molecular docking and inhibition of growth experiments, are indications of their ability to act as quorum sensing inhibitors and as promising synergistic with nonantibiotic drugs to treat infection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Multidrug resistance makes clinical therapy more challenging. Based on the concentration of specific signal molecules, bacteria can detect changes in their cells or other bacterial species present in their microenvironment. Bacterial cells can communicate with one another to coordinate gene expression and virulence to adapt as a group to changing environmental conditions. This process is often referred to as bacterial quorum sensing (QS) [1, 2]. The QS system can control gene expression, biofilm formation, and extracellular polysaccharides by secreting and receiving signal molecules. Bacterial cells as a group can collectively adapt to environmental changes, resulting in negative outcomes such as drug resistance and higher virulency [3, 4]. The QS system enables the expression of pathogenicity and virulence in roughly the following manner: (a) the synthesis of QS signal molecules; (b) the discharge of signal molecules into the microenvironment; (c) the binding of signal molecules to membrane receptors; (d) the retrieval of the receptor-signal complex from the cell; and (e) the transcription of pathogenicity-related genes [5].
In Gram-positive bacteria, the signal molecules of the QS system are predominantly oligopeptides acting as autoinducers (AIs). Furthermore, another type of signal molecule is the furanosyl borate diester molecule known as autoinducer 2 (AI-2), which is present in both Gram-positive and Gram-negative bacteria [6]. The QS system controls many biological features, including the release of virulence factors. The QS system upregulates the expression of pathogenic genes; however, QS interference can decrease pathogenicity, assisting the immune system in eliminating infection-related microorganisms [7]. Zhao et al. [8] stated that bacteria can communicate with each other via a vernacular made of small diffusible chemical signals that affect the regulation of genes when there is high cell density. Such systems are integrated into multilayered, complex signal transduction networks, which take on various multicellular behaviours, such as virulence traits and biofilm formation [9]. In Gram-positive bacteria, sortase A (SrtA), a membrane-associated cysteine transpeptidase, is important to assist in attaching surface proteins to the cell wall peptidoglycan. This process is necessary for bacterial virulence and pathogenicity to be regulated [10].
Inhibitors of the QS process, which are also known as quorum quenching (QQ) enzymes or quorum sensing inhibitors (QSIs), have been designed to decrease the virulence of bacteria, impeding bacterial virulence factors without interfering with the growth of bacteria as a whole and reducing bacterial resistance [11]. SrtA is thought to be an excellent target for anti-virulence therapy. ML346 was discovered by Guan et al. [12] as a novel covalent inhibitor of SrtA with anti-virulence properties in Gram-positive bacteria. At low micromolar concentrations, ML346 reduced the transpeptidation of SaSrtAN24 and SpSrtAN81 in vitro but had only mild inhibitory effects on the human cysteine proteases cathepsin B and cathepsin L, signifying that ML346 is a SrtA-specific inhibitor. ML346 inhibited the attachment of the surface protein SpA to the S. aureus cell membrane, as expected [12]. Specifically, it inhibited a critical virulence component involved in Staphylococci pathogenesis. In the presence of ML346, the ability of S. aureus to produce biofilms was also significantly reduced. Optimization of the ML346 covalent attachment could be a great way to develop a more promising SrtA inhibitor with good in vivo efficacy and safety [12]. The discovery of ML346 as a covalent inhibitor of SrtA might provide a new chemical scaffold for the creation of anti-virulence medicines based on structure.
Virtual screening using molecular docking simulations enables the preselection of promising therapeutic candidates from large libraries of compounds, reducing the number of anticipated hits that must be validated in vitro [13]. Furthermore, according to Azimi et al. [14], docking simulation predicts with high-probability the attachment pattern of candidate hits on the designated targets, creating a molecular foundation for their increase in efficiency in terms of binding affinity. The interference between small molecules and virulence processes in growth holds promise as the most crucial option other than common antibiotics. In regard to the formation of resistance, anti-virulence drugs are believed to decrease bacterial virulence and have a lower selective burden. As a result, virulence mechanisms of the QSI are increasingly being used as molecular targets in the expansion of anti-virulence drugs that target infection and the virulence factors of the bacteria rather than the growth of bacteria.
Emerging evidence suggests that ASA's anti-inflammatory properties could extend to modulating bacterial virulence [15]. Trifluoperazine, an FDA-approved antipsychotic medication, possesses inherent properties that make it a promising candidate for QSI research [16, 17]. Its ability to interact with biological membranes and influence cell signalling pathways raises intriguing possibilities for its role in quorum sensing modulation.
Furthermore, we chose sortase A (SrtA) as the target for our investigation due to its central role in Gram-positive bacterial virulence and pathogenicity. Inhibiting SrtA could disrupt this process and reduce bacterial virulence without necessarily affecting bacterial growth, presenting an innovative avenue for combating infections while minimizing the development of resistance [12].
The choice of acetylsalicylic acid (ASA) and trifluoperazine, as well as SrtA, is underscored by the need for novel approaches that circumvent the challenges posed by traditional antibiotic therapies. By elucidating the potential of these Food and Drug Administration (FDA)-approved drugs to interfere with the QS system and target a key virulence regulator such as SrtA, our study aims to contribute to the development of alternative strategies to effectively manage bacterial infections. We believe that the comprehensive exploration of these drug candidates and their interactions with the QS system will demonstrate their potential utility as quorum sensing inhibitors and pave the way for the development of innovative therapeutic interventions against multidrug-resistant infections.
In this study, we explored a novel possible quorum sensing inhibitor via the specialized molecular docking of FDA-approved medications (analgesics and antipsychotics) against the Gram-positive bacteria S. aureus and E. faecalis.
2 Methods
2.1 Molecular docking
The structures of the target compounds in this study were retrieved from the ACD/ChemSketch program and saved as MDL Molfiles (mol). Using the PyRx program, the energies of the ligands were minimized and the files were converted to PDBQT file format. The 3D crystal structure of the target protein, sortase A (PDB ID: 1t2w), was obtained from the Protein Data Bank in PDB format (https://www.rcsb.org/) [18, 19]. The target protein sortase A was in complex with the LPETG peptide (LEU-PRO-GLU-THR-GLY). The macromolecule was prepared by deleting the peptide chain, heteroatoms and water molecules from the protein; polar hydrogen atoms were added using DS software v 2021, and the file was saved as a prepared protein file in PDB format for further analysis. The docking study was performed in a grid box centred on the cocrystalized ligand with x, y and z dimensions of − 33.53, − 17.60 and 9.07 Å, respectively. Autodock Vina software was used for the docking process using an exhaustiveness of 8 [20]. For validation purposes of the docking procedure, the cocrystalized ligand was redocked in the protein, and the RMSD value between the cocrystal and structures was calculated using DockRMSD [20]. An RMSD value of 2 Å was considered acceptable, and the target compounds in the study were docked using the same procedure.
2.1.1 Stock solution preparation
2.1.1.1 Acetylsalicylic acid (ASA) stock solution preparation
For the preparation of the ASA stock solution, 410 mg ASA raw material was purchased as powder (ACROS organics part of Thermo Fisher Scientific, United States of America). ASA was diluted in 10 ml of 2% dimethyl sulfoxide (DMSO) solution; thus, each sample contained 41 mg. To obtain different ASA concentrations of 200, 250, 500, 750 and 1000 μg/ml, 5 µl, 6 µl, 12 µl, 18 µl, and 24 µl were added to tubes and mixed with 10 ml of brain heart infusion broth. The tubes of different concentrations were then sterilized using an autoclave (121 °C under a pressure of 15 psi (pounds per square inch) for 15 min). After sterilization, the solutions of different concentrations were kept cool to be ready for use in well diffusion and MIC laboratory tests.
2.1.1.2 Trifluoperazine stock solution preparation
For the trifluoperazine solution(which was purchased as tablets from the National Unified Procurement Company “NUPCO’, Saudi Arabia), a 200 mg trifluoperazine tablet was crushed and ground using a mortar and pestle and then mixed with 10 ml of distilled water. Different trifluoperazine concentrations were aliquoted at 200, 250, 500, 750 and 1000 μg/ml and mixed with 10 ml of brain heart infusion broth. These concentrations were autoclaved (121 °C under pressure 15 psi (pounds per square inch) for 15 min) and kept cool to be ready for use in well diffusion and MIC laboratory tests.
2.1.2 Agar well diffusion method
To assess the antimicrobial activity of both ASA and Tri against the Gram-positive bacteria S. aureus (ATCC 29213) and E. faecalis (ATCC 29212), we employed an agar well diffusion assay. This method relies on measuring the diameter of the inhibition zone in millimetres as described previously by Gunasekaran et al. [21]. Bacterial colonies of S. aureus and E. faecalis, which were previously cultured on blood agar, were utilized. We prepared a total of twelve agar culture plates. A 100 µl prepared inoculum, equivalent to a 0.05 MacFarland standard, was spread over the surface of six Muller Hinton agar medium plates (MHA, Q-Lab, Quebec, Canada). These agar plates served as the medium for measuring the inhibitory zones. Using a sterile pipette, we created 6 mm diameter wells on the agar surface. Subsequently, each well was filled by pipetting each concentration prepared. Each concentration was tested in triplicate, and the zone of inhibition for each concentration was measured using a metric scale.
2.1.3 Minimum inhibitory concentration (MIC)
The minimum inhibitory concentration (MIC) for both ASA and Tri against the test microbes S. aureus and E. faecalis was determined using a serial dilution method. Initially, 100 µl of the test compounds was transferred from a concentration of 1000 μg/ml, encompassing five consecutive concentrations. This method followed the protocol previously described [22].
In each well of the 96-well plate, 100 µl of the bacterial suspension was mixed with 100 µl of DMSO to serve as the positive control, and this step was repeated in triplicate. The 96-well plate was then incubated at 37 °C for a duration of 18 h. The MIC values were recorded as the lowest concentration of the molecule that effectively inhibited the growth of the test pathogens.
2.1.4 Statistical analysis
The inhibition zones observed in the agar well diffusion method were measured in millimetres. The diameter of the inhibition zones were measured using a ruler or digital calliper. The data were then typically presented as a tabulated form of the mean value of triplicates and included in Table 2 for comparison across different concentrations and bacterial strains. The MIC values for ASA and Tri against S. aureus and E. faecalis were determined based on visual inspection of the 96-well plates after incubation. The lowest concentration of the compounds that inhibited bacterial growth was recorded, and the mean with standard deviation was added to the table. The data are presented as MIC values in µg/ml, providing a quantitative measure of the inhibitory effects of each concentration. Statistical analysis was employed to assess the significance of differences observed between concentrations, compounds, or bacterial strains using a t test to determine the p values.
3 Results
3.1 Molecular docking
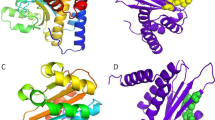
Docking of acetylsalicylic acid in the 1t2w enzyme crystal structure produced an affinity score of − 5 kcal/mol (Table 1). The aromatic ring of acetylsalicylic acid interacts by pi-alkyl bonds with ILE (ILE:199). The carbonyl group and the oxygen atom in the side chain interacted by hydrogen bonding with GLN:178 and GLN:172, respectively, as shown in Fig. 1a. The thiazine ring and the substituted aromatic ring of the tricyclic system in trifluoperazine interacted by pi-cation bonds with ARG:197 (Fig. 1b). The fluorinated carbon atom in trifluoperazine interacted by alkyl bonds with ALA:184, ALA:118, ILE:182 and TRP:194, as shown in Fig. 1b. The substituted aromatic ring interacted by a pi-alkyl bond with ILE (182), and the piperazine ring interacted by a hydrogen bond with PRO (163) in the binding site of the target protein (Fig. 1b).

Ligand interactions of acetylsalicylic acid (a) and trifluoperazine (b) with 1t2w; the hydrogen bond interactions are shown as green dotted lines, and the pi-alkyl interactions are shown as lavender dotted lines
3.2 Microbiological assays
3.2.1 Minimum inhibitory concentration (MIC)
3.2.1.1 Acetylsalicylic acid (ASA)
In our study, the antimicrobial effects of ASA were assessed against both S. aureus and E. faecalis using MIC and biofilm inhibition assays. The results are summarized in Table 2.
At a concentration of 1000 µg/ml, ASA exhibited substantial activity against both bacterial strains. This was reflected in a 10 mm inhibition zone observed in the agar well diffusion method, accompanied by a clear broth solution in the MIC assessment.
However, concentrations of ASA lower than 1000 µg/ml yielded varying results. For S. aureus cultures, concentrations of 250, 500, and 750 µg/ml showed a consistent 10 mm inhibition zone in the well diffusion method. However, for E. faecalis, these concentrations were ineffective, showing no inhibition zone and resulting in turbid solutions. Remarkably, ASA at a concentration of 200 µg/ml exhibited no inhibition against either bacterial strain in the well diffusion and MIC experiments.
3.2.1.2 Trifluoperazine
The antimicrobial activity of trifluoperazine was also evaluated against S. aureus and E. faecalis using MIC and biofilm inhibition assays. These results are summarized in Table 2. In contrast to ASA, trifluoperazine did not demonstrate antimicrobial effects at any tested concentration (1000, 750, 500, 250, and 200 µg/ml) against either bacterial strain. This was evident from the absence of inhibition zones in the agar well diffusion method and turbid broth solutions in the MIC assessment.
Overall, the MIC experiment revealed that only ASA, at a concentration of 1000 µg/ml, exhibited notable antimicrobial activity against both S. aureus and E. faecalis, while trifluoperazine showed no such activity at any tested concentration.
4 Discussion
Drug repurposing (known as drug reprofiling or repositioning) is a drug development method that identifies novel pharmacological uses for already approved pharmaceuticals outside of their original medical applications [23, 24]. As a result, the initial stages of the drug discovery process can be avoided, resulting in significant cost and time savings for pharmaceutical companies [23,24,25]. Even while the expenses of organizing Phase III–IV trials are still substantial, if the new applications for the pharmaceuticals are appropriate, drug companies can still anticipate substantial returns on investment. Historically, drug repurposing was primarily founded on coincidental discoveries from clinical data. Now, there are advantages to evaluating the existing drug pool for off-target effects that may be appropriate for the development of further clinical implementations [23,24,25].
Furthermore, drug repurposing is a potential approach for antibacterial and anti-virulence effects. Many pharmaceutical agents have secondary mechanisms of action that are not fully understood for some drugs, allowing them to be effective against diverse bacteria as directly acting as antibacterial agents or virulence inhibitors [26]. For this reason, there is interest in screening the existing pool of pharmacological compounds for anti-virulence properties; however, there are considerable information gaps in this area [26,27,28].
Docking was performed to predict the potential interactions between the proposed compounds and sortase A (PDB: 1t2w). The docking results of acetylsalicylic acid and trifluoperazine were promising and showed higher affinity than the co-crystalized ligand (Table 1). Docking results of acetylsalicylic acid in the sortase A crystal structure had an affinity score of − 5.0 kcal/mol. Only one similar type of interaction with one amino acid was involved in the interaction of the co-crystalized ligand with the active site of the protein, as shown in the interaction of acetylsalicylic acid with the protein. The oxygen atom in the side chain of acetylsalicylic acid interacted by hydrogen bonding with GLN:172, and the oxygen atom in the co-crystalized ligand interacted with the same key amino acid with the same type of interaction. The docking results of trifluoperazine had high affinity scores of − 6.1 kcal/mol. Interestingly, the tricyclic system in the compound interacted with the same key amino acid (ARG:197) by pi-cation bonds. According to the findings, the antibacterial activity of acetylsalicylic acid and trifluoperazine can be attributed to their ability to bind to the amino acids in the active site of the sortaseA enzyme.
Acetylsalicylic acid (ASA) remains a first-choice treatment for a variety of clinical demands. ASA is primarily used as an analgesic, antipyretic, and antiplatelet agent and may be capable of serving as an anticancer drug, notably for colorectal cancer [29]. Direct and indirect antimicrobial effects of ASA have also been demonstrated but have been ignored for more than 2 decades [15]. Their primary method of action includes the inhibition of cyclooxygenase, which reduces prostaglandin synthesis [30]. At plasma therapeutic levels, ASA showed some reduction in the growth of a number of pathogens, such as Campylobacter pylori, Helicobacter pylori, Klebsiella pneumoniae, Microsporum spp., Trichophyton spp., hepatitis C virus, flavivirus, and the influenza virus [15].
ASA and indomethacin inhibit urease and vacuolating cytotoxin activity in H. pylori [15]. ASA and ibuprofen augment the efficacy of pyrazinamide during the initial phase of tuberculosis treatment in mice. ASA lowers bacterial cell density, bacterial dispersion, and the occurrence of embolic events in rabbits with S. aureus endocarditis [15]. To evaluate the antibacterial impact of ASA against S. aureus and E. faecalis, it was utilized in this investigation. In our study, 1000 mg ASA was active against S. aureus. However, 200 mg of ASA was ineffective upon testing its antimicrobial effect using well diffusion and MIC methods. However, our results contrast with the result from Lass-Florl et al. (2001), in which the S. aureus strain growth was not supressed when cultured in 10 mM ASA, and a rise in biofilm synthesis was found in the strain grown in a medium with 0.3–2 mM ASA [31].
The results of the current study were consistent with those of a study by Chan et al. [32], who demonstrated that ASA had an inhibitory effect against both Gram-positive and Gram-negative bacteria (MIC = 2.5 and 5 mg/mL). This is likely due to the lipopolysaccharide layer of Gram-negative bacteria, which hinders the diffusion of other hydrophilic chemicals relative to aspirin in this study, as is the case with the majority of medications. The outer layer in Gram-negative bacteria forms a barrier that prevents many antibiotics from penetrating the bacteria. Gram-positive bacteria, on the other hand, lack this outer cytoplasmic membrane, making it easier for antimicrobial agents to penetrate the cells [33]. The doses at which these NSAIDs display bactericidal activity are significantly lower than the standard therapeutic dose administered to humans to relieve pain, inflammation, or fever [34]. Both ASA and ibuprofen showed bactericidal effects against MRSA clinical strains [35,36,37]. ASA and ibuprofen showed bacteriostatic and bactericidal activities against S. aureus in these previous studies, suggesting their possible utility as antibacterial adjuvants for MRSA infections [35,36,37]. In addition, a high dose of ASA (324 mg) was related to a lower incidence of S. aureus infection when compared to a low dose (81 mg) [36]. In patients with S. aureus, low-dose ASA was associated with lower short-term mortality [38]. There have been hypotheses that ASA at a dose of 325 mg administered once, along with the prophylactic antibiotic, before dental practices [39] or the daily use of ASA and/or other anti-platelet agents (APA) for medical/surgical indications is beneficial [40]. The usefulness of ASA as an antibacterial agent against Gram-positive cocci and some Gram-negative rods has been well documented [41].
Phenothiazine destroys phagocytosed bacteria by inhibiting the calcium-dependent enzyme systems involved in energy production via ATP hydrolysis, which blocks calcium transport through calcium-binding proteins [41]. Phenothiazines, such as trifluoperzine, have not yet been examined for their in vitro antibacterial activity against S. aureus and E. faecalis to the best of our knowledge. Trifluoperazine's antibacterial effectiveness against this MRSA strain was linked to phenothiazine's ability to inhibit efflux pumps, which are mechanisms responsible for bacterial cell antibiotic resistance. In addition, this class of molecules prevents calcium from binding to certain proteins, including calmodulin-type proteins, and affects verapamil-sensitive efflux pumps [41,42,43]. In addition, it has been found that a lower concentration of phenothiazine has antibacterial activity [43]. Trifluoperazine is an inexpensive, readily administered, previously used antipsychotic medicine with good antibacterial action against MRSA that does not rely on antibiotics [44]. The authors of this study suggested its effectiveness as a preventative agent in the case of postoperative wound infection or as a therapeutic agent in the treatment of MRSA in infective wounds [44].
Rahbar et al. [45] found that the phenothiazines available on the market, including thiethylperazine, increase the effect of vancomycin against clinical isolates of E. faecium resistant to this antibiotic. Checkerboard evaluations and MIC findings indicated that phenothiazines have high (MIC50 lg ml) to moderate (MIC50–400 lg ml) action against vancomycin-resistant enterococci [46]. The antibacterial efficacy of the medicines was unrelated to their other pharmacological properties. Phenothiazines were reported to inhibit Enterococci at concentrations lower than the plasma concentration, and thioridazine enhanced the killing of phagocytosed S. aureus at concentrations below those used for the therapy of psychoses [47]. Therefore, it is speculated that phenothiazines may act in the same way against enterococci. Moreover, in vivo experiments have also demonstrated the potential antibacterial activity of these compounds [48].
Due to government restrictions on purchasing antipsychotic drugs, many of these are not available for use in pharmacies and require further approval to be studied and tested. The present study was only conducted on one antipsychotic drug (trifluoperazine) and one anti-inflammatory drug (ASA), which were shown by docking to have some effect on the quorum sensing mechanisms. However, the microbiological assays showed that only ASA inhibited growth at an MIC equal to 250 μl/mg. This inhibition indicated the effect of the drug on the growth of S. aureus, which is believed to also contribute to the quorum sensing process that is involved in biofilm formation. We also recognize that different assays, including well diffusion, have limitations and advantages. While good diffusion provides a quick assessment of antimicrobial effects, it may not precisely represent the actual concentration achieved in the bacterial environment. Therefore, we incorporated complementary methods, such as minimum inhibitory concentration (MIC) assays, to provide a comprehensive understanding of the drugs' inhibitory effects at different concentrations. Our laboratory investigations, while yielding valuable insights, did not fully mirror the expectations set by docking scores. A possible explanation for this discrepancy could be attributed to the utilization of lower concentrations of the drugs in experimental assays, suggesting that future investigations with expanded concentration ranges could offer a more comprehensive picture. These observations underline the importance of careful consideration when selecting concentrations for antimicrobial studies, balancing clinical relevance with rigorous scientific exploration.
In our study, we aimed to assess the quorum sensing inhibition (QSI) potential of FDA-approved drugs, specifically acetylsalicylic acid (ASA) and trifluoperazine (Tri), against the Gram-positive bacteria S. aureus and E. faecalis. We intentionally selected a range of concentrations to reflect potential physiological relevance and therapeutic feasibility. Our goal was to mimic a scenario where these drugs might be used as adjuncts to existing treatments, aiming for concentrations that are within a plausible range for clinical applications. At the core of our study lies the dual commitment to clinical applicability and scientific robustness. The concentrations chosen were deliberately aligned with realistic therapeutic scenarios, ensuring that our findings retain practical significance. This approach enhances the value of our study by bridging the gap between theoretical potential and real-world utility, emphasizing the translational implications of our results. Looking ahead, future studies could benefit from exploring the potential of higher concentrations of compounds in experimental assays, providing a clearer understanding of their upper limits of inhibitory activity. This direction aligns with the continuous pursuit of refining our comprehension of antimicrobial action.
In conclusion, molecular docking is a reliable method to identify different compound structures that could be used for different purposes. The docking results compellingly showcased enhanced binding scores of the compounds against sortase A, surpassing those of its co-crystallized ligand. This demonstrates the potential of these compounds for interaction with the target protein. However, the convergence of laboratory experiments and computational predictions was not entirely consistent, raising pertinent questions about factors that influence such disparities. Notably, our findings highlight the distinct effectiveness of acetylsalicylic acid over trifluoperazine against S. aureus. This discernible distinction emphasizes the intricate interplay between compound types and bacterial strains. Furthermore, the potential for trifluoperazine to yield better anti-quorum sensing results at higher concentrations is an intriguing avenue that warrants exploration. We believe that our approach allows for a more holistic assessment of the drugs' QSI potential.
Data availability
All data obtained in this study are available upon request.
References
Diggle SP, Crusz SA, Cámara M (2007) Quorum sensing. Curr Biol 17(21):R907–R910
Li Y-H, Tian X (2012) Quorum sensing and bacterial social interactions in biofilms. Sensors 12(3):2519–2538
Dong Y-H et al (2001) Quenching quorum-sensing-dependent bacterial infection by an N-acyl homoserine lactonase. Nature 411(6839):813–817
Miller MB, Bassler BL (2001) Quorum sensing in bacteria. Ann Rev Microbiol 55(1):165–199
Durán N et al (2016) Advances in Chromobacterium violaceum and properties of violacein-its main secondary metabolite: a review. Biotechnol Adv 34(5):1030–1045
Elgaml A, Higaki K, Miyoshi S-I (2014) Effects of temperature, growth phase and luxO-disruption on regulation systems of toxin production in Vibrio vulnificus strain L-180, a human clinical isolate. World J Microbiol Biotechnol 30:681–691
Chen J et al (2019) Quorum sensing inhibitors from marine microorganisms and their synthetic derivatives. Mar Drugs 17(2):80
Zhao X, Yu Z, Ding T (2020) Quorum-sensing regulation of antimicrobial resistance in bacteria. Microorganisms 8(3):425
Bridges AA et al (2022) Signal transduction network principles underlying bacterial collective behaviors. Annu Rev Microbiol 76:235–257
Schneewind O, Missiakas D (2014) Sec-secretion and sortase-mediated anchoring of proteins in Gram-positive bacteria. Biochimica et Biophysica Acta (BBA) Mol Cell Res 1843(8):1687–1697
Maeda T et al (2012) Quorum quenching quandary: resistance to antivirulence compounds. ISME J 6(3):493–501
Guan X-N et al (2022) Covalent sortase A inhibitor ML346 prevents Staphylococcus aureus infection of Galleria mellonella. RSC Med Chem 13(2):138–149
Martins FG, Melo A, Sousa SF (2021) Identification of new potential inhibitors of quorum sensing through a specialized multi-level computational approach. Molecules 26(9):2600
Azimi S et al (2020) Bacterial quorum sensing during infection. Annu Rev Microbiol 74:201–219
Zimmermann P, Curtis N (2017) Antimicrobial effects of antipyretics. Antimicrob Agents Chemother. https://doi.org/10.1128/aac.02268-16
Pacios O et al (2020) Strategies to combat multidrug-resistant and persistent infectious diseases. Antibiotics 9(2):65
Andersson JA et al (2016) New role for FDA-approved drugs in combating antibiotic-resistant bacteria. Antimicrob Agents Chemother 60(6):3717–3729
Dallakyan S, Olson AJ (2015) Small-molecule library screening by docking with PyRx. Chem Biol Methods Protoc. https://doi.org/10.1007/978-1-4939-2269-7_19
Bell EW, Zhang Y (2019) DockRMSD: an open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism. J Cheminform 11(1):1–9
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461
Gunasekaran G, Ramamurthy J, Rajeshkumar S (2020) Evaluation of the antimicrobial activity of solanum nigrum infused selenium nanoparticles-an in vitro study. Int J Pharm Res 12:3159–3168
Zgoda J, Porter J (2001) A convenient microdilution method for screening natural products against bacteria and fungi. Pharm Biol 39(3):221–225
Xue H et al (2018) Review of drug repositioning approaches and resources. Int J Biol Sci 14(10):1232
Pushpakom S et al (2019) Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov 18(1):41–58
Paul SM et al (2010) How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov 9(3):203–214
Miró-Canturri A, Ayerbe-Algaba R, Smani Y (2019) Drug repurposing for the treatment of bacterial and fungal infections. Front Microbiol 10:41
Gatta V et al (2019) Targeting quorum sensing: high-throughput screening to identify novel lsrk inhibitors. Int J Mol Sci 20(12):3112
Soo WC et al (2017) Repurposing of anticancer drugs for the treatment of bacterial infections. Curr Top Med Chem 17(10):1157–1176
Thun MJ, Jacobs EJ, Patrono C (2012) The role of aspirin in cancer prevention. Nat Rev Clin Oncol 9(5):259–267
Dinarello CA (2010) Anti-inflammatory agents: present and future. Cell 140(6):935–950
Lass-Flörl C et al (2001) Antifungal activity against Candida species of the selective serotonin-reuptake inhibitor, sertraline. Clin Infect Dis 33(12):e135–e136
Chan EWL et al (2017) Synergistic effect of non-steroidal anti-inflammatory drugs (NSAIDs) on antibacterial activity of cefuroxime and chloramphenicol against methicillin-resistant Staphylococcus aureus. J Glob Antimicrob Resist 10:70–74
Zhong D et al (2015) Employing carbon dots modified with vancomycin for assaying Gram-positive bacteria like Staphylococcus aureus. Biosens Bioelectron 74:546–553
Ong C et al (2007) An evidence-based update on nonsteroidal anti-inflammatory drugs. Clin Med Res 5(1):19–34
Al-Janabi AAHS (2010) In vitro antibacterial activity of ibuprofen and acetaminophen. J Glob Infect Dis 2(2):105
Sedlacek M et al (2007) Aspirin treatment is associated with a significantly decreased risk of Staphylococcus aureus bacteremia in hemodialysis patients with tunneled catheters. Am J Kidney Dis 49(3):401–408
Riordan JT et al (2011) Alterations in the transcriptome and antibiotic susceptibility of Staphylococcus aureus grown in the presence of diclofenac. Ann Clin Microbiol Antimicrob 10(1):1–11
Osthoff M et al (2016) Low-dose acetylsalicylic acid treatment and impact on short-term mortality in Staphylococcus aureus bloodstream infection: a propensity score-matched cohort study. Crit Care Med 44(4):773–781
Østergaard L et al (2019) Prevalence of infective endocarditis in patients with positive blood cultures: a Danish nationwide study. Eur Heart J 40(39):3237–3244
Dahl A, Bruun NE (2013) Enterococcus faecalis infective endocarditis: focus on clinical aspects. Expert Rev Cardiovasc Ther 11(9):1247–1257
Amaral L, Viveiros M, Molnar J (2004) Antimicrobial activity of phenothiazines. In Vivo 18(6):725–732
García-Díez J, Saraiva C (2021) Use of starter cultures in foods from animal origin to improve their safety. Int J Environ Res Public Health 18(5):2544
Kaatz GW et al (2003) Phenothiazines and thioxanthenes inhibit multidrug efflux pump activity in Staphylococcus aureus. Antimicrob Agents Chemother 47(2):719–726
Ashok S et al (2021) Antibacterial activity of trifluoperazine; in vitro susceptibility of MRSA Staphylococcus aureus, Pseudomonas aeruginosa and E. coli, and in vivo evaluation against methicillin resistant Staphylococcus aureus in surgical wound infection model. JAPS J Anim Plant Sci 11:15
Rahbar M, Mehrgan H, Hadji-nejad S (2010) Enhancement of vancomycin activity by phenothiazines against vancomycin-resistant Enterococcus faecium in vitro. Basic Clin Pharmacol Toxicol 107(2):676–679
Basu LR et al (2005) Antibacterial property of the antipsychotic agent prochlorperazine, and its synergism with methdilazine. Microbiol Res 160(1):95–100
Ordway D et al (2002) Intracellular activity of clinical concentrations of phenothiazines including thioridiazine against phagocytosed Staphylococcus aureus. Int J Antimicrob Agents 20(1):34–43
Mazumder R et al (2001) Trifluoperazine: a broad spectrum bactericide especially active on staphylococci and vibrios. Int J Antimicrob Agents 18(4):403–406
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors have declared that no financial and non-financial competing interest to publish this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Obaid, N.A., Alkhudhir, N.A., Mojally, M. et al. In silico identification of new potential inhibitors of quorum sensing by Gram-positive bacteria through specialized molecular docking. J.Umm Al-Qura Univ. Appll. Sci. 10, 83–90 (2024). https://doi.org/10.1007/s43994-023-00080-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43994-023-00080-3