
Abstract
Elderly individuals with chronic disorders tend to develop inflammaging, a condition associated with elevated levels of blood inflammatory markers, and increased susceptibility to chronic disease progression. Native and adaptive immunity are both involved in immune system senescence, kidney fibrosis and aging. The innate immune system is characterized by a limited number of receptors, constantly challenged by self and non-self stimuli. Circulating and kidney resident myeloid and lymphoid cells are all equipped with pattern recognition receptors (PRRs). Recent reports on PRRs show kidney overexpression of toll-like receptors (TLRs) in inflammaging autoimmune renal diseases, vasculitis, acute kidney injury and kidney transplant rejection. TLR upregulation leads to proinflammatory cytokine induction, fibrosis, and chronic kidney disease progression. TLR2 blockade in a murine model of renal ischemia reperfusion injury prevented the escape of natural killer cells and neutrophils by inflammaging kidney injury. Tumor necrosis factor-α blockade in endothelial cells with senescence-associated secretory phenotype significantly reduced interleukin-6 release. These findings should encourage experimental and translational clinical trials aimed at modulating renal inflammaging by native immunity blockade.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inflammaging is an age-related long-term result of premature immune system senescence during a persistent, low-grade, non-resolving inflammatory state. It is associated with chronic stimulation of the innate and adaptive immune systems, driven by aging, in the course of metabolic syndrome [1], uremia [2], rheumatic and neurodegenerative disorders [3, 4], viral infections [5, 6] and malignancy [7].
The term “Inflamm-aging” was first conceived by Franceschi and his group, who suggested that up-regulation of the inflammatory response at an older age results from a low-grade, chronic, systemic pro-inflammatory state, and that this state contributes to age-associated diseases [8]. Aging is a process involving both the innate and the adaptive immune system [9]. Senescence of the adaptive immune system is characterized by the loss of naive T cells; as the memory T cell pool increases, it results in a limited T-cell receptor (TCR) repertoire [10]. The innate immune system also contributes to aging [9]. Native immune cells are the first line of host defense, constantly challenged by exogenous and endogenous stimuli. Innate immunity absorbs several immunological stimuli by a limited number of immune receptors, resulting in a multitude of effector molecules [11]. Innate myeloid cells, such as tissue-resident macrophages, are the first line of defense against invading microbial infection and removing cellular debris [12]. Myeloid cells are equipped with pattern recognition receptors.
(PRRs) sense pathogen- or danger-associated molecular patterns (PAMP or DAMP, respectively), that initiate the proinflammatory cascade [13]. Toll-like receptors (TLRs) are localized either on the cellular membrane (TLRs 1,2,4,5,6,10) or within the lysosomal compartment (TLRs 3,7,9) and recognize defined proteins, lipids, DNA or RNA [14]. TLRs signal, via the myeloid differentiation primary response protein 88/TIR-domain-containing adapter-inducing interferon-β (MyD88/TRIF), to activate the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) -related target genes [15]. Interleukin-6 (IL-6) as well as tumor necrosis factor (TNF) are part of the NFκB-induced secretome suggesting the involvement of TLR signaling during the aging process [16]. NOD-like receptors (NLRs) are relevant pro-inflammatory native immunity receptors with the ability to form multi-protein complexes or inflammasomes that assemble with the apoptosis-associated speck-like (ASC) protein and recruit the effector caspase-1, thus enabling the cleavage of IL-1β and IL-18 [17]. Inflammasomes have been associated with immunosenescence and inflammaging [18]. Abnormal complement activation is related to native kidney glomerular disorders as well as kidney transplant rejection [19].
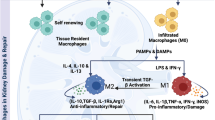
We will further discuss renal inflammaging, resulting from accelerated immunosenescence induced by the chronic activation of native immunity and its involvement in the pathogenesis of common age-related renal pathologies (Fig. 1). Recent literature on the involvement of innate immune system receptors and cells in the progression of chronic inflammation and aging in renal diseases has been reviewed.

PRRs and IL-6 blockade as potential treatment to modulate inflammaging and accelerated renal aging. PRRs pattern recognition receptors, TLRs toll-like receptors, CTLRs C-type lectin receptors, NLRs NOD-like receptors, RLRs RIG-I-like helicase receptors, ROS reactive oxygen species, IL-6 interleukin-6
Innate immune system receptors in chronic kidney inflammation and accelerated renal aging
Pattern recognition receptors and proteolysis
PRRs are soluble cell-associated receptors specialized in virus, bacteria, fungi recognition of PAMPs and DAMPs. These receptors elicit an immune response resulting in NFκB activation and cytokine secretion [20]. PRRs are divided into five classes (TLRs, C-type lectin receptors [CTLRs], NLRs, RIG-I-like helicase receptors [RLRs], Scavenger receptors [SRs]) and are expressed by dendritic cells (DCs), natural killer cells (NKs), neutrophils, epithelial cells and cells of the adaptive immune system. Soluble PAMPs bind microorganisms by soluble PRR molecules leading to pathogen proteolytic lysis [21]. PRR cell-associated phagocytosis of PAMP infectious pathogens proceed in cytokine release and amplification of the immune response [22]. DAMPs are released during uncontrolled cell death or necrosis and are removed by macrophages or other cells of the innate immune system [23].
Pattern recognition receptors in sterile inflammation
DAMP sterile inflammation is characterized by the release of IL-1 family cytokines that enhance NK cytotoxicity and macrophage phagocytosis function by activating naive T-cells [24]. The role of these receptors is not only in pathogen recognition but also in eradication of dead cells from the host. The complement cascade and the C-reactive protein (CRP) are soluble PRRs. When CRP is complexed with residues of bacteria, it has the property of activating the classical and not the alternative complement pathway, resulting in microorganism opsonisation by adherence to phagocytes [25]. Microbe phagocytosis and lysis are instead facilitated by the alternative complement pathway. Over the course of metabolic diseases like diabetes mellitus, animal studies on diabetic kidney disease (DKD) have shown that renal inflammation is mainly characterized by neutrophil inflammation, release of proinflammatory cytokines, PRRs, and native immunity receptor expression. In such experimental models, worsening kidney injury has been observed with and without arterial hypertension, suggesting that inflammation is a factor required for DKD progression to end-stage renal disease (ESRD) [26].
Evidence for a role of toll-like receptors in inflammaging
Toll-like receptors initiating intracellular signaling cascade
Ten TLRs have been identified in humans [27]. They are displayed on the cell surface (TLR1-2, TLR4–6, TLR10) or in intracellular endosomes (TLR3, TLR7–9) [28]. TLR ligands include bacterial lipopeptides, lipopolysaccharides (LPS), dsDNA, viral ssRNA, and flagellin. After ligand engagement and signal transduction promoted by the cytoplasm protein MyD88, TLRs activate NFκB and members of the Interferon regulatory factor family (IRF) [29,30,31,32] regulating type I interferon (IFN) genes (e.g. INF-α) [31].
Toll-like receptor expression in human kidney diseases
TLR1–6 and 9 mRNA expression has been reported in immortalized human podocytes [33]. Human tubular epithelial cell lines express TLR1–4 and 6, suggesting a role in the activation of the native immune response in tubulointerstitial injury (e.g. bacterial pyelonephritis, sepsis, and transplant nephropathy) [34]. Human kidney proximal tubule cells express TLR2, 4 and 6. Increased detection of such TLRs has been described after exposure to multiple myeloma free light chains via the damage-associated molecular pattern high-mobility group box 1 (HMGB1) [35]. HMGB1 is a nuclear protein that regulates DNA transcription. In the course of anti-neutrophil cytoplasmic antibody- (ANCA) associated vasculitis, increased expression of TLR2 and TLR4 on glomerular endothelial cells and podocytes has been detected. TLR2 and TLR4 overexpression correlated with glomerular segmental necrosis and cellular crescents [36]. In ANCA-associated vasculitis TLR2 and TLR4 tubulointerstitial immunofluorescence was significantly higher when compared to controls. Glomerular TLR4 expression was also associated with the severity of renal injury [37]. MyD88 gene upregulation in renal tissue and increased levels of TLR4 mRNA in peripheral blood mononuclear cells (PBMCs) have been reported as being suggestive of cell-mediated rejection [38] or graft dysfunction in kidney transplantation [39]. TLR2 and TLR4 have been identified as proinflammatory and fibrotic receptors in peritoneal dialysis (PD), after microbial- and sterile inflammation-induced responses [40]. A more intense TLR4 expression was observed in lupus nephritis (LN), glomeruli and tubules when compared with controls [41]. Activation of the TLR3/IFN pathway appears to play a pivotal role in chronic kidney disease (CKD). Cylindromatosis stimulation, a ubiquitin-specific protease, in TLR3-activated cultured human mesangial cells downregulates NFκB thus inhibiting post-transcriptional regulation of retinoic acid-inducible gene-I (RIG-I) and melanoma differentiation associated gene 5 (MDA5) [42]. PBMC TLR3, TLR4 mRNA expression levels have been reported as being higher in patients with steroid-sensitive nephrotic syndrome (NS) than in those with steroid-resistant NS [43]. TLR3 and TLR9 have been found overexpressed in glomerular and tubulointerstitial biopsies of patients with LN. TLR7 expression was restricted into the tubulointerstitium of LN biopsies [44]. A negative correlation between PBMC, CD4+ T cell and B cell expression of TLR3, TLR8 and proteinuria in children with idiopathic NS was instead observed [45]. In a mouse model of septic acute kidney injury (AKI), LPS binding to TLR4 induced proximal tubular paracellular leakage of the glomerular filtrate, and oliguria [46]. In vitro evidence also indicated long-term protection by NFκB inhibition providing insights for novel therapeutic strategies for oliguria by protecting proximal tubular tight junctions [46, 47]. Weak TLR5 expression has been detected in LN glomeruli, but Polymorphonuclear leukocytes (PMN) TLR7 is not upregulated in ESRD [48]. TLR9 is predominantly expressed by vasculitis podocytes [36], whereas an increase of TLR9 expression has been described in the glomeruli [49] and tubular cells [50] of LN biopsies. These findings suggest that TLRs are involved in humoral and cellular immune responses during renal autoimmune diseases [34]. TLR10 is an important innate immune sensor of influenza virus infection leading to proinflammatory cytokine and interferon induction [51]. TLR renal expression in inflammatory diseases is summarized in Table 1.
Toll-like receptor immune aging in experimental renal diseases
Although data on immune aging associated with human TLR2 are not available, murine models show that TLR2 is a key mediator of senescence by promoting cell cycle arrest and regulating the induction of acute-phase serum amyloids A1 and A2 that can function as DAMPs [52]. As apoptosis regulates cell death, necroptosis is a process regulating necrosis that requires protein receptor interacting protein kinase-3 (RIPK3) and mixed lineage kinase like (MLKL) and is induced by death receptors, interferons, TLRs and other mediators. Activated TLR3 forms an endosome platform, which is involved in NFκB activation and induction of a type I IFN, allowing interaction with RIPK3. TLR3 and TLR4 stimulation may induce TRIF-mediated necroptosis. Interesting data reported that RIPK3 deficiency ameliorates induced renal ischemia reperfusion injury (IRI) and inflammation, prolonging kidney allograft survival. It might suggest that graft cell necroptosis triggering inflammation could contribute to transplant rejection. If apoptosis is considered a weak inducer of inflammation, necroptosis is characterised by a massive DAMP release, inducing relevant tissue inflammation [53]. As reported in several human studies TLR overexpression has been identified in acute and chronic renal pathologies leading to fibrosis. Animal studies have found that changes in TLR expression is also associated with renal aging. Furthermore, higher levels of TLR1-5 found in elderly rats are associated with NFκB signaling activation, thus suggesting a role in both renal chronic inflammation and renal aging [54].
Evidence for a role of C-type lectin receptors in inflammaging
C-type lectin receptors are mainly expressed by myeloid cells acting as antigen-uptake receptors via their carbohydrate-recognition domain for internalization and subsequent presentation to T-cells. CTLRs recognize self-antigens such as DAMPs released from dead cells, leading to the production of inflammatory mediators. These receptors promoting extent, duration and outcome of the immune response can also balance out inflammatory and resolution pathways. Several authors suggest that CTLR dysregulation leads to the development of various pathologies such as autoimmune diseases, allergy and cancer [55, 56]. In kidney transplanted patients with acute cytomegalovirus (CMV) infection, increased CTLR expression on NKs and decreased secretion of IFN-γ were observed [57]. CTLR renal expression in inflammatory human diseases is summarized in Table 1.
Evidence for a role of NOD-like receptors in inflammaging
Inflammasome-dependent and -independent cell death
NOD-like receptors are cytoplasmic soluble proteins acting as PAMP receptors. Among the NLRs identified to date, NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) is also known as the inflammasome and is an intracellular NLR complex which is activated by the cleavage of pro-caspase-1 to active caspase-1. It is followed by the activation of pro-inflammatory cytokines IL-1β and IL-18 and induction of pyroptosis, an inflammatory form of apoptosis resulting in immune and epithelial cell infection by intracellular pathogens [58]. NLRP3 inflammasome-dependent pyroptosis is characterized by rapid cell lysis, associated with the release of cytoplasmic IL-1β and IL-18 triggering necroinflammation. NLRP3 has also been implicated in inflammasome-independent functions, such as in response to TNF/CD95 binding, when the expression of NLRP3, ASC, and caspase-8 to apoptotic cell death [59]. The NLRP3 inflammasome has been implicated in the pathogenesis of several renal conditions including AKI, CKD, DKD and crystal-related nephropathy. Interesting data show that NLRP3 is critical for the development of metabolic diseases like NLRP3 deficiency which resulted in decreased systemic inflammation, reduced immune cell activation and improved insulin resistance. NLRP3 activation is induced by a variety of endogenous triggers including uric acid, damaged mitochondria and reactive oxygen species (ROS), aggregated proteins and lipids, resulting in crystal-related nephropathies and CKD [59, 60]. Inflammaging is associated with the accumulation of damage-related molecules, suggesting that NLRP3 activation might be the result of several triggers, rather than a consequence of a single toxic ligand [18].
Inflammasome expression in human kidney diseases
Several studies have shown NLRP3 expression in human podocytes, mesangial and intercalated cells. In patients with AKI urinary IL-18 is associated with tubular inflammation, early acute tubular necrosis and increased disease severity [51]. Tissues from human renal biopsies of a wide variety of fibrotic kidney diseases showed that NLRP3 increased mRNA expression, which was associated with a decline of renal function. These data suggest that NLRP3 drives progression of CKD in both inflammasome-dependent and -independent manners [59]. NLR and inflammasome renal expression in inflammatory human kidney diseases are summarized in Table 1.
Evidence for a role of RIG-I-like helicase receptors in inflammaging
RIG-I-like helicase receptors are cytoplasmic sensors, activated by viral dsRNA via type-I IFN production and DC activation [61]. HCV RNA–protein complexes released into the circulation can reach the glomerular mesangium, and are often associated with glomerulonephritis. Immunostaining of kidney biopsies from patients with active LN, identified RLRs in mesangial areas, and the intensity staining correlated with disease activity [62]. RLR renal expression in inflammatory human kidney diseases is summarized in Table 1.
Evidence for a role of scavenger receptors in human renal inflammaging
Scavenger receptors are cell surface receptors that promote the removal of non-self or altered-self targets by endocytosis, phagocytosis, adhesion, and signaling. Ten classes of human SRs (A-L) are currently recognized [63]. CD36 (SR-B2) is a membrane glycoprotein identified on platelets, mononuclear phagocytes, adipocytes, hepatocytes, myocytes, epithelial cells, and microvascular endothelial cells. (see comment As cellular receptor binds oxidized lipids and phospholipids, advanced oxidation protein and glycation end products. It has a role in inflammatory signalling, apoptosis and kidney fibrosis). Renal CD36 is mainly expressed in tubular epithelial cells, podocytes, and mesangial cells, and is upregulated in CKD [64]. Soluble CD163 (sCD163) is a SR-B for the hemoglobin-haptoglobin complex or for hemoglobin alone. Authors found a correlation between plasma sCD163 and renal dysfunction suggesting that sCD163 may play a role in the development of AKI in patients with hemorrhagic fever infected by Hantaan virus [65]. Interesting data indicated that glomeruli from patients with small vessel vasculitis had higher levels of CD163 mRNA than did those with LN, DKD, or NS. Vasculitis patients expressed CD163 in glomeruli and interstitium, and high urinary CD163 levels were also measured. Urinary sCD163 levels were associated with active renal vasculitis [66]. Chemokine (C-X-C motif) ligand 16 (CXCL16) is a soluble chemokine, an adhesion molecule and a cell surface SR regulating inflammation, tissue injury and fibrosis. In normal human kidney the C-X-C motif chemokine receptor 6 (CXCR6, a CXCL16 receptor) is expressed by proximal tubules, but not by distal tubules or glomeruli. Reports on CXCL16 in human diseases have associated its increased expression in distal and collecting tubules with renal transplant acute tubular necrosis (ATN) [67]. SR renal expression in inflammatory human kidney diseases is summarized in Table 1.
Evidence for a role of the complement system in human renal inflammaging
The complement cascade is classified into three pathways: the alternative, lectin, and classical pathways (AP, LP and CP, respectively), merging at the level of C3 cleavage. Abnormal complement activation and glomerular deposition of circulating active fragments have been associated with several glomerular disorders such as LN, C3 glomerulopathy, IgAN, ANCA-associated vasculitis, aHUS, renal transplant rejection, and in AKI-to-CKD transition (Table 1). Anomalous complement activation causes self-cell lysis, but also triggers oxidative stress, neutrophil infiltration, and the cytosol NLRP3 inflammasome, inducing aging by tubular chronic inflammation and interstitial fibrosis. Several studies suggest that in IRI-AKI the complement is involved in accelerated renal senescence, leading to CKD progression [68].
Evidence for a role of innate immune system cells in human renal inflammaging
The kidney mononuclear phagocyte (MNP) compartment of tissue-resident immune cells is characterized by two monocyte-derived macrophage populations with antibacterial function. A smaller, alternatively activated macrophage- (M2) enriched population, similar to fetal kidney macrophages has also been observed [69].
Myeloid cells
Dendritic cells and macrophages
DCs mediate immune surveillance and antigen presentation through the initiation of either immune effector-cell functions or the development of tolerance. Macrophages are also innate immune cells, predominantly acting by phagocytosis and production of toxic metabolites. However, renal DCs and macrophage phenotype and functions are often indistinguishable within the kidney, where they overlap surface markers and functional capabilities. Mouse studies showed the presence of renal DCs within the interstitium, glomeruli, and low numbers in the mesangial matrix. Little is known about the role of DCs in human renal diseases. However, experimental models of glomerulonephritis, LN, ureteral obstruction, IRI-induced AKI, and renal transplantation suggest that intrarenal antigen presentation by DCs to T cells and B cells promotes chronic, immune-cell-mediated inflammatory responses [70]. Macrophages are present in the normal kidney, and in increasing numbers, in kidney diseases. Their action is relevant in renal injury, inflammation, and fibrosis. Macrophage functions are defined by two main phenotypes referred to as classically activated macrophages (M1) produced by exposure to LPS or IFN-γ and M2, produced by Th2 cytokines such as IL-4 and IL-10. M1 macrophages produce pro-inflammatory mediators and mediate antimicrobial defense and antineoplastic immunity [71]. M2 macrophages have anti-inflammatory functions and are involved in parasite containment, wound healing, and fibrosis [72]. M2 macrophages can be further divided into three subgroups: M2a wound-healing macrophages induced by IL-4 and/or IL-13, M2b macrophages induced by immune complexes with LPS or IL-1β that are involved in immunoregulation, and Th2 activation and M2c macrophages or regulatory macrophages that are involved in immunoregulation and tissue remodeling [73]. During AKI, neutrophils and NKs are recruited from tissue injury. Inflammatory monocytes infiltrate to the site of tissue injury shortly after neutrophils, where they differentiate and polarize into M1 macrophages by inflammatory mediators, such as IFN-γ released by neutrophils, NKs, and T effector cells (e.g. Th1/17). Activated M1 macrophages can exacerbate tissue inflammation and cause tissue damage. Subsequently, Th2 and regulatory T cells (Tregs) are recruited into the kidney and switch macrophages to an M2 anti-inflammatory phenotype contributing to inflammation resolution and tissue repair. Fibrosis may occur, depending on the severity of injury and whether pro-inflammatory factors continue to be expressed [74]. In CKD, M1 macrophages are increased in kidney tissue following neutrophil, NKs, and Th1/17 early injury and inflammation infiltration. Due to progressive injury and persistent inflammation, renal fibrosis in CKD is associated with persistent inflammatory and fibrotic factors [75]. Fibrosis occurs when epithelial healing is incomplete or insufficient, such as in the cases of sustained injury and unresolved renal inflammation. Macrophages promote renal fibrosis through several mechanisms: M2 macrophages produce pro-fibrotic factors, e.g. TGF-β1, fibroblast growth factor-2 (FGF-2) and platelet-derived growth factor (PDGF), which promote myofibroblast proliferation, and overproduction of extracellular matrix (ECM). Macrophages also produce fibronectin and collagen in response to a pro-fibrotic microenvironment, or transdifferentiate into collagen-producing fibrocytes. Activated macrophages damage glomerular and peritubular capillaries, promoting hypoxia-driven fibrosis [76]. M2a and M2b macrophages have been reported as predominant sub-populations in kidney tissues of IgAN. M2a macrophages appeared to be mainly distributed in the tubulointerstitium where segmental glomerulosclerosis and tubular atrophy/interstitial fibrosis were found, while M2c macrophages were observed in glomeruli with minor lesions. This suggests that M2a and M2c macrophages are associated with the persistent systemic inflammatory pathogenesis of IgAN [77, 78]. In LN, M2 macrophages have also been identified. It is still not clear whether the macrophage transition from a more inflammatory to a less inflammatory/reparative phenotype is associated with excessive repair and fibrosis [79]. A report on chronic kidney allograft injury showed the association between CD68+ CD163+ (SR-B) kidney tissue immunostaining of M2 interstitial macrophages or elevated urine CD163+ levels, and renal function associated with interstitial fibrosis progression [80]. In IRI, tubular cell necrosis following ROS production leads to sterile inflammation, with the release of DAMPs from dying cells. The shift from M1 to M2 macrophage phenotype may be induced during later phases of IRI-induced AKI. Although the pathophysiological mechanism is unclear [81], macrophages rather than DCs appear to drive renal fibrosis [67]. Several authors suggest that translational research on kidney-specific DC and/or macrophage ablation should be implemented to obtain renal specific data in acute and chronic inflammatory kidney diseases, leading to renal aging, immunosenescence and inflammaging.
Neutrophils
Formation of neutrophil extracellular traps (NETs) is the mechanism of neutrophil pathogen killing. NETs are chromatin extruded into the extracellular space, associated with nuclear, granule, and cytoplasmic proteins that facilitate pathogen immobilization and killing. NETs are involved in infectious diseases and in sterile tissue injuries such as thrombosis, tissue necrosis, inflammation, and autoimmunity. In ANCA-associated kidney vasculitis. Vascular necrosis induced by neutrophil release of antimicrobial proteins and cytotoxic agents along the glomerular endothelium is a process associated with NETs formation. In such patients NET components trigger ANCA production activating the TLR signaling pathway in both B cells and DCs. It is common opinion that NETs are a main mechanism of microvascular necrosis leading to crescent formation in crescentic glomerulonephritis [82].
Tubular cell necrosis in ischemic AKI is characterized by necroinflammation, and following the release of DAMPs which trigger the release of cytokine and chemokines, initiates neutrophil infiltration into the kidney. Infiltrating the neutrophils is subject to NET formation, and can be associated with neutrophil death by necroptosis and histone release, eliciting direct cytotoxic effects on endothelial cells [83, 84]. Renal myeloid cell involvement in inflammatory human kidney diseases is summarized in Table 2.
Evidence for a role of lymphoid cells in human renal inflammaging
Natural killer cells
Human NKs are CD3−/CD56+/CD335 (NKp46+) mononuclear cells further classified according to the expression levels of CD56 (NCAM) into low density (CD56dim) and high density (CD56bright) subsets. CD56dim NKs are the dominant subset in peripheral blood, acting as cytotoxic effector cells. CD56bright NKs have been mainly identified in lymphoid and peripheral tissues. CD56bright NKs mediate immune responses by secreting pro-inflammatory cytokines like IFN-γ and TNF-α [85].
Several studies suggest that IRI and immune responses to transplants are the leading cause of delayed graft function (DGF) and with an increased incidence in acute renal rejection [86, 87]. Renal IRI triggers an inflammatory response mediated by the innate immune system. Recent murine studies reported that around 20% of kidney NKs belong to a CD49a+DX5− tissue-resident NK population already described in the liver, uterus and pancreas. Tissue-resident NKs appeared to contribute to IRI renal injury [88, 89], and tubular epithelial cells are potential targets [90] or activators [91] of NK organ cytotoxicity in IRI kidney transplant outcomes. Renal lymphoid cell involvement in inflammatory kidney diseases is summarized in Table 2.
Evidence for a role of native immunity in hemodialysis
Persistent systemic inflammation has been recognized as a major contributor to the long-term complications of CKD and hemodialysis (HD) (e.g. cardiovascular disease, osteoporosis and frailty) and a predictor of cardiovascular and total mortality [92, 93]. Indoleamine 2,3-dioxygenase-1 (IDO1) activity has a relevant function on T cell immunosuppression, Treg activation, and inhibition of NKs. Elevated nitric oxide (NO) levels are associated with hyperactivation of macrophage activity. IDO1 activity and NO formation are increased in patients with CKD or under chronic HD treatment. Data on chronic HD patients treated with more biocompatible vitamin E-loaded hemofilters showed reduced IDO1 activity and NO formation when compared to chronic HD patients treated with polysulfone membrane dialyzers. These data suggest that vitamin E-loaded dialyzers might lower HD inflammaging [2] (Table 3). Calcification occurring within the media of blood vessel atherosclerotic plaques is the leading cause of cardiovascular mortality and morbidity in CKD and ESRD. Vascular calcifications have been found in stage 5 CKD children under conservative management or chronic dialysis treatment [94]. In HD children, IL-6 dialysis levels were significantly higher when compared to the stage 5 CKD group, and both versus healthy control children. IL-6 levels rose with increased time on dialysis. It suggested that CKD and the dialysis environment is associated with increased oxidative DNA damage and premature senescence of human vascular smooth muscle cells (hVSMCs) in vivo. It is worth mentioning that such results were not stratified according to dialysis modality, and that PD treatment has better biocompatibility when compared to HD [95].
Promoters of vascular calcification, such as serum parathyroid hormone (PTH) [96], CRP [97], TGF-β [98] have been associated with increased IL-6 levels in CKD, HD and PD. In a systematic review IL-6, not PTH was significantly associated with increased ESRD mortality [97]. The role of high levels of serum IL-6 as a predictor of poor outcome in HD patients appears even more relevant in patients with malnutrition-inflammation-cachexia syndrome (MICS) usually showing suppressed PTH serum levels and severe atherosclerotic cardiovascular disease [99]. The relation between IL-6 and PTH serum levels is also matter of investigation in the ESRD older candidates to renal transplantation suffering frailty and sarcopenia [100].
Loss of mineralization inhibitors like fetuin-A, matrix Gla protein has been demonstrated on heart valves and atherosclerotic plaques in CKD, representing key mechanisms in vascular calcification [101, 102]. However, there is a lack of clinical evidence of a close association between kidney disease inflammation markers like IL-6 and such inhibitors of calcification. Experimental reports have instead shown that sIL-6 can induce the transformation of hVSMCs into an osteoblast phenotype potentially linking chronic inflammation and vascular calcifications [103]. It raises the question of whether IL-6 inhibitors, like IL-6R antagonists as tocilizumab and sarilumab, really slow human vascular atherosclerotic calcifications, helping to eventually prevent inflammaging and the risk of cardiovascular events and mortality in CKD and HD patients.
Evidence for a role of native immunity in COVID-19 kidney injury
A recent observational report on five patients with SARS-CoV-2 pneumonia and fluid overload treated by CytoSorb cartridge hemoperfusion showed better clinical outcome when compared to controls [104]. One out of five CytoSorb-treated patients died; while only 2 of them were intubated, all controls required intubation or died. Lymphocytopenia worsened in control patients but not in the CytoSorb group. IL-6, IL-8 and TNF-α decreased after CytoSorb cartridge hemoperfusion. A prospective study on 701 in-hospital patients with COVID-19 showed that the prevalence of AKI was 5.1%. Patients with elevated baseline creatinine levels had significantly higher in-hospital mortality [105]. Kudose et al. [106] investigated the mechanisms underlying COVID-19-associated kidney injury, evaluating renal histological changes in 14 native and 3 allograft kidney biopsies from COVID-19 patients who developed AKI or nephrotic-range proteinuria. Various glomerular and tubular changes had taken place, including collapsing glomerulopathy, minimal change disease, membranous nephropathy (MN), anti-GBM nephritis, acute tubular injury, exacerbation of pre-existing autoimmune GN and allograft rejection. No definitive evidence of SARS-CoV-2 in renal biopsy samples was found by in situ hybridization, immunohistochemistry or electron microscopy, leading to hypothesize that direct viral infection of the kidney is an unlikely underlying pathology. These findings suggest that viral infection, influencing innate or adaptive immune responses, could trigger glomerular diseases such as podocytopathies or anti-GBM nephritis, facilitating renal aging and immunosenescence [107]. In the case of COVID-19 this was instead associated with pre-existing autoimmune or alloimmune conditions (LN, MN, allograft rejection), further kidney injury might result from an already developing immunosenescence and inflammaging [108]. SARS-CoV-2 innate immune system evasion strategies have been reported; this virus infects airway epithelial cells by the receptor-binding domain of the spike glycoprotein that binds to the tip of subdomain I of the trans-membrane enzyme angiotensin-converting enzyme 2 (ACE2). While RNA viruses usually activate TLR3 and/or 7 in intracellular endosomes and RNA sensors RIG-I and MDA5, SARS-CoV-2 effectively suppresses the activation of TNF receptor-associated factors (TRAFs) limiting the activation of NFκB and IRFs and inhibiting early pro-inflammatory responses by IFN or pro-inflammatory cytokines like IL-1, IL-6 and TNF-α [109]. Immune complexes consisting of ineffective SARS-CoV-2 antibodies may also be phagocytized by macrophages causing their infection. In infected macrophages, SARS-CoV-2 can inhibit IFN signaling, allowing pro-inflammatory IL-1, IL-6 and TNF-α expression contributing to hyperinflammation and the cytokine storm syndrome [110], eventually accelerating renal fibrosis.
Summary
The volume and value of data associating chronic stimulation of native immunity with accelerated immune system senescence and inflammaging is on the rise [1]. Overexpression of PRRs and increased activity of innate immunity system cells are involved in severe impairment of renal function. This has increased interest in identifying the molecular pathways regulating the receptor ligands of potential interest for therapeutic intervention. However, experimental studies are only available on the TLR2 blockade of chronic inflammation-induced fibrosis. A mouse model of sterile peritoneal fibrosis induced by continuous exposure to low-glucose (1.36% glucose) PDS was characterized by parietal peritoneal membrane thickening and increased peritoneal fluid levels of TNF-α, IL-1β, IL-6 and IFN-γ [111]. In such disease model co-administration of PDS + sTLR2 prevented peritoneal fibrosis, resulting in suppression of PDS-induced cytokines and fibrotic mediators. NKs mediated neutrophil infiltration, and subsequent renal damage was prevented in a murine kidney IRI by local infusion of anti-TLR2 mAbs [112] or i.v. OPN301, a mouse anti-human TLR2 antibody that cross-reacts with mouse TLR2 [113]. HMGB1 blockade inhibited the development of albuminuria, glomerular mesangial matrix expansion, hypercellularity, hypertrophy, and interstitial fibrosis in streptozotocin-induced DKD mice [114]. A large amount of evidence indicates the role of TLRs in the pathogenesis of human renal inflammaging diseases, and animal studies suggest that the blockade of such receptors could slow the progression to ESRD [115, 116]. The role of native immunity in modulating renal inflammaging could also be hypothesized after infusion of autologous MSCs, in the course of pANCA renal vasculitis [117]. Disease remission 12 months after autologous MSC infusion was associated with the reduction of IFN-γ, IL-6, and TNF serum levels (Table 3).
Conclusion
Recent evidence suggest that renal inflammaging could be diagnosed and treated as a clinical disorder. Blockade of PRRs and cytokines associated with native immunity (e.g. IL-6) appears to be an opportunity for treatment of renal inflammaging in animal models, opening a prospective translation for treating human aging-related kidney inflammatory disorders (Fig. 1).
Abbreviations
- ACE2 :
-
Angiotensin-converting enzyme 2
- aHUS :
-
Atypical hemolytic uremic syndrome (thrombotic microangiopathy)
- ANCA :
-
Anti-neutrophil cytoplasmic antibody
- AP :
-
Complement alternative pathway
- ASC :
-
Or PYCARD is an apoptosis-associated speck-like protein containing a CARD
- ATN :
-
Acute tubular necrosis
- AKI :
-
Acute kidney injury
- CARD :
-
Caspase recruitment domain
- CD :
-
Cluster of differentiation
- CKD :
-
Chronic kidney disease
- CMV :
-
Cytomegalovirus
- COVID-19 :
-
Corona virus disease—2019 novel coronavirus
- CXCL16 :
-
Chemokine (C-X-C motif) ligand 16
- CTLRs :
-
C-type lectin receptors
- CP :
-
Complement classical pathway
- CRP :
-
C-reactive protein
- CXCR6 :
-
C-X-C motif chemokine receptor 6
- DCs :
-
Dendritic cells
- DAMPs :
-
Dander-associated molecular patterns or alarmins
- DGF :
-
Delayed graft function
- DKD :
-
Diabetic kidney disease or diabetic nephropathy
- ds :
-
Double-stranded
- ECM :
-
Extracellular matrix
- eNOS :
-
Endothelial nitric oxide synthase
- ESRD :
-
End-stage renal disease
- FGF-2 :
-
Fibroblast growth factor 2
- GBM :
-
Glomerular basement membrane
- HCV :
-
Hepatitis C virus
- HD :
-
Hemodialysis
- HMGB1 :
-
High-mobility group box 1
- HUVECs :
-
Human umbilical vein endothelial cells
- hVSMCs :
-
Human vascular smooth muscle cells
- IDO1 :
-
Indoleamine 2,3-dioxygenase-1
- IFN :
-
Interferon
- IgAN :
-
IgA nephropathy
- IL :
-
Interleukin
- IRF :
-
Interferon regulatory factor family
- IRI :
-
Renal ischemia reperfusion injury
- IV :
-
Intravenous
- LN :
-
Lupus nephritis
- LP :
-
Complement lectin pathway
- LPS :
-
Lipopolysaccharides
- LRR :
-
Leucine-rich repeat receptor kinase
- M1 :
-
Classically activated macrophages
- M2 :
-
Alternatively activated macrophages
- Matrix Gla protein :
-
Small vitamin K dependent protein (VKDP) involved in bone calcification
- MDA5 :
-
Melanoma differentiation associated gene 5
- MICS :
-
Malnutrition-inflammation-cachexia syndrome
- miRNA :
-
MicroRNA
- MLKL :
-
Mixed lineage kinase like
- MN :
-
Membranous glomerulopathy
- MNP :
-
Mononuclear phagocytes
- mRNA :
-
Messenger RNA
- MasdSCs :
-
Mesenchymal stromal cells
- MyD88 :
-
Myeloid differentiation primary response protein 88
- NCAM :
-
Neural cell adhesion molecule
- NETs :
-
Neutrophil extracellular traps
- NFκB :
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NLRs :
-
NOD-like receptors
- NLRP3 :
-
NOD-, LRR- and pyrin domain-containing protein 3
- NO :
-
Nitric oxide
- NOD :
-
Nucleotide-binding oligomerization domain-like receptor
- NKs :
-
Natural killer cells
- NS :
-
Nephrotic syndrome
- PDS :
-
Peritoneal dialysis solution
- PMN :
-
Polymorphonuclear leukocytes
- PAMPs :
-
Pathogen associated molecular patterns
- PBMCs :
-
Peripheral blood mononuclear cells
- PD :
-
Peritoneal dialysis
- PDGF :
-
Platelet-derived growth factor
- PRRs :
-
Pattern recognition receptors
- PTH :
-
Parathyroid hormone
- RIG-I :
-
Retinoic acid-inducible gene-I
- RIPK3 :
-
Receptor interacting protein kinase-3
- RLRs :
-
RIG-I-like helicase receptors
- ROS :
-
Reactive oxygen species
- SARS-CoV-2 :
-
Severe acute respiratory syndrome coronavirus 2
- SASP :
-
Senescence-associated secretory phenotype
- sCD163 :
-
Soluble CD163
- sIL-6 :
-
Soluble IL-6
- IL-6R :
-
IL-6 receptor
- SRs :
-
Scavenger receptors
- ss :
-
Single-stranded
- sTLR2 :
-
Soluble TLR2
- TCR :
-
T-cell receptor
- TGF-β1 :
-
Transforming growth factor beta-1
- TIR :
-
Intracellular Toll/interleukin-1R
- TLRs :
-
Toll-like receptors
- TNF :
-
Tumor necrosis factor
- TRAF :
-
TNF receptor-associated factors
- Tregs :
-
Regulatory T cells
- TRIF :
-
TIR-domain-containing adapter-inducing interferon-β
References
Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A (2018) Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol 14(10):576–590. https://doi.org/10.1038/s41574-018-0059-4
Sepe V, Gregorini M, Rampino T, Esposito P, Coppo R, Galli F, Libetta C (2019) Vitamin E-loaded membrane dialyzers reduce hemodialysis inflammaging. BMC Nephrol 20(1):412. https://doi.org/10.1186/s12882-019-1585-6
Loeser RF, Collins JA, Diekman BO (2016) Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol 12(7):412–420. https://doi.org/10.1038/nrrheum.2016.65
Calabrese V, Santoro A, Monti D, Crupi R, Di Paola R, Latteri S, Cuzzocrea S, Zappia M, Giordano J, Calabrese EJ, Franceschi C (2018) Aging and Parkinson’s disease: inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic Biol Med 115:80–91. https://doi.org/10.1016/j.freeradbiomed.2017.10.379
Meftahi GH, Jangravi Z, Sahraei H, Bahari Z (2020) The possible pathophysiology mechanism of cytokine storm in elderly adults with COVID-19 infection: The contribution of “Inflame-Aging.” Inflamm Res. https://doi.org/10.1007/s00011-020-01372-8
Heath JJ, Grant MD (2020) The immune response against human cytomegalovirus links cellular to systemic senescence. Cells 9(3):766. https://doi.org/10.3390/cells9030766
Bottazzi B, Riboli E, Mantovani A (2018) Aging, inflammation and cancer. Semin Immunol 40:74–82. https://doi.org/10.1016/j.smim.2018.10.011
Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G (2000) Inflamm-aging. an evolutionary perspective on immunosenescence. Ann N Y Acad Sci 908:244–254. https://doi.org/10.1111/j.1749-6632.2000.tb06651.x
Santoro A, Bientinesi E, Monti D (2021) Immunosenescence and inflammaging in the aging process: age-related diseases or longevity? Ageing Res Rev. https://doi.org/10.1016/j.arr.2021.101422
Krishna C, Chowell D, Gönen M, Elhanati Y, Timothy AC (2020) Genetic and environmental determinants of human TCR repertoire diversity. Immun Ageing 17:26. https://doi.org/10.1186/s12979-020-00195-9
McDaniel MM, Meibers HE, Pasare C (2021) Innate control of adaptive immunity and adaptive instruction of innate immunity: bi-directional flow of information. Curr Opin Immunol 73:25–33. https://doi.org/10.1016/j.coi.2021.07.013
Yue Z, Nie L, Zhang P, Chen Q, Lv Q, Wang Q (2021) Tissue-resident macrophage inflammaging aggravates homeostasis dysregulation in age-related diseases. Cell Immunol 361:104278. https://doi.org/10.1016/j.cellimm.2020.104278
Shaw AC, Goldstein DR, Montgomery RR (2013) Age-dependent dysregulation of innate immunity. Nat Rev Immunol 13(12):875–887. https://doi.org/10.1038/nri3547
Akar-Ghibril N (2021) Defects of the innate immune system and related immune deficiencies. Clin Rev Allergy Immunol. https://doi.org/10.1007/s12016-021-08885-y
Takeda K (2005) Evolution and integration of innate immune recognition systems: the Toll-like receptors. J Endotoxin Res 11(1):51–55. https://doi.org/10.1179/096805105225006687
Olivieri F, Procopio AD, Montgomery RR (2014) Effect of aging on microRNAs and regulation of pathogen recognition receptors. Curr Opin Immunol 29:29–37. https://doi.org/10.1016/j.coi.2014.03.006
Duez H, Pourcet B (2021) Nuclear Receptors in the Control of the NLRP3 Inflammasome Pathway. Front Endocrinol (Lausanne) 12:630536. https://doi.org/10.3389/fendo.2021.630536
Latz E, Duewell P (2018) NLRP3 inflammasome activation in inflammaging. Semin Immunol 40:61–73. https://doi.org/10.1016/j.smim.2018.09.001
Kielar M, Gala-Błądzińska A, Dumnicka P, Ceranowicz P, Kapusta M, Naumnik B, Kubiak G, Kuźniewski M, Kuśnierz-Cabala B (2021) Complement components in the diagnosis and treatment after kidney transplantation-Is there a missing link? Biomolecules 11(6):773. https://doi.org/10.3390/biom11060773
Collins SE, Mossman KL (2014) Danger, diversity and priming in innate antiviral immunity. Cytokine Growth Factor Rev 25(5):525–531. https://doi.org/10.1016/j.cytogfr.2014.07.002
Jacquet A, Robinson C (2020) Proteolytic, lipidergic and polysaccharide molecular recognition shape innate responses to house dust mite allergens. Allergy 75(1):33–53. https://doi.org/10.1111/all.13940
Cui B, Lin H, Yu J, Yu J, Hu Z (2019) Autophagy and the immune response. Adv Exp Med Biol 1206:595–634. https://doi.org/10.1007/978-981-15-0602-4_27
Murao A, Aziz M, Wang H, Brenner M, Wang P (2021) Release mechanisms of major DAMPs. Apoptosis. https://doi.org/10.1007/s10495-021-01663-3
El-Naccache DW, Haskó G, Gause WC (2021) Early events triggering the initiation of a Type 2 immune response. Trends Immunol 42(2):151–164. https://doi.org/10.1016/j.it.2020.11.006
Degn SE, Thiel S (2013) Humoral pattern recognition and the complement system. Scand J Immunol 78(2):181–193. https://doi.org/10.1111/sji.12070
Tesch GH (2017) Diabetic nephropathy—Is this an immune disorder? Clin Sci (Lond) 131(16):2183–2199. https://doi.org/10.1042/CS20160636
Nie L, Cai SY, Shao JZ, Chen J (2018) Toll-Like Receptors, associated biological roles, and signaling networks in non-mammals. Front Immunol 9:1523. https://doi.org/10.3389/fimmu.2018.01523
Lorenz G, Lech M, Anders HJ (2017) Toll-like Receptor activation in the pathogenesis of lupus nephritis. Clin Immunol 185:86–94. https://doi.org/10.1016/j.clim.2016.07.015
Arnold-Schrauf C, Berod L, Sparwasser T (2015) Dendritic Cell Specific targeting of MyD88 signalling pathways in vivo. Eur J Immunol 45(1):32–39. https://doi.org/10.1002/eji.201444747
O’Reilly S (2018) Toll Like Receptors in systemic sclerosis: an emerging target. Immunol Lett 195:2–8. https://doi.org/10.1016/j.imlet.2017.09.001
Negishi H, Taniguchi T, Yanai H (2018) The interferon (IFN) class of cytokines and the IFN Regulatory Factor (IRF) Transcription Factor family. Cold Spring Harb Perspect Biol 10(11):a028423. https://doi.org/10.1101/cshperspect.a028423
Almuttaqi H, Udalova IA (2019) Advances and challenges in targeting IRF5, a key regulator of inflammation. FEBS J 286(9):1624–1637. https://doi.org/10.1111/febs.14654
Shimada M, Ishimoto T, Lee PY, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, Wymer DT, Yamabe H, Mathieson PW, Saleem MA, Garin EH, Johnson RJ (2012) Toll-like Receptor 3 ligands induce CD80 expression in human podocytes via an NF-κB-dependent pathway. Nephrol Dial Transpl 27(1):81–89. https://doi.org/10.1093/ndt/gfr271
Farhat K, Riekenberg S, Heine H, Debarry J, Lang R, Mages J, Buwitt-Beckmann U, Röschmann K, Jung G, Wiesmüller KH, Ulmer AJ (2004) Signaling Danger: Toll-Like receptors and their potential roles in Kidney Disease. J Am Soc Nephrol 15(4):854–867. https://doi.org/10.1097/01.asn.0000121781.89599.16
Upadhyay R, Ying WZ, Nasrin Z, Safah H, Jaimes EA, Feng W, Sanders PW, Batuman V (2020) Free light chains injure proximal tubule cells through STAT1-HMGB1-TLR axis. JCI Insight. https://doi.org/10.1172/jci.insight.137191
O’Sullivan KM, Ford SL, Longano A, Kitching AR, Holdsworth SR (2018) Intrarenal Toll-like Receptor 4 and Toll-like Receptor 2 expression correlates with injury in antineutrophil cytoplasmic antibody-associated vasculitis. Am J Physiol Renal Physiol 315(5):F1283–F1294. https://doi.org/10.1152/ajprenal.00040.2018
Wang H, Gou SJ, Zhao MH, Chen M (2014) The expression of Toll-like Receptors 2, 4 and 9 in kidneys of patients with anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Clin Exp Immunol 177(3):603–610. https://doi.org/10.1111/cei.12365
Sharbafi MH, Assadiasl S, Pour-Reza-Gholi F, Barzegari S, Mohammadi Torbati P, Samavat S, Nicknam MH, Amirzargar A (2019) TLR-2, TLR-4 and MyD88 genes expression in renal transplant acute and chronic rejections. Int J Immunogenet 46(6):427–436. https://doi.org/10.1111/iji.12446
Hosseinzadeh M, Ahmadpoor P, Yekaninejad MS, Pourrezagholi F, Foroughi F, Ghorbanpour M, Barabadi M, Shahbaz SK, Solgi G, Amirzargar A (2018) Expression patterns of Toll Like Receptor (TLR)-2, TLR-4 and myeloid differentiation primary response gene 88 (MYD88) in renal transplant patients developing allograft dysfunction; A cohort study. Transpl Immunol 48:26–31. https://doi.org/10.1016/j.trim.2018.02.005
Raby AC, Colmont CS, Kift-Morgan A, Köhl J, Eberl M, Fraser D, Topley N, Labéta MO (2017) Toll-Like Receptors 2 and 4 are potential therapeutic targets in peritoneal dialysis-associated fibrosis. J Am Soc Nephrol 28(2):461–478. https://doi.org/10.1681/ASN.2015080923
Elloumi N, Fakhfakh R, Ayadi L, Sellami K, Abida O, Ben Jmaa M, Sellami T, Kammoun K, Masmoudi H (2017) The increased expression of Toll-Like Receptor 4 in renal and skin lesions in lupus erythematosus. J Histochem Cytochem 65(7):389–398. https://doi.org/10.1369/0022155417709234
Imaizumi T, Hayakari R, Watanabe S, Aizawa T, Matsumiya T, Yoshida H, Tsuruga K, Kawaguchi S, Tanaka H (2017) Cylindromatosis (CYLD), a deubiquitinase, attenuates inflammatory signaling pathways by activating Toll-Like Receptor 3 in human mesangial cells. Kidney Blood Press Res 42(5):942–950. https://doi.org/10.1159/000485084
Mishra OP, Kumar R, Narayan G, Srivastava P, Abhinay A, Prasad R, Singh A, Batra VV (2017) Toll-like Receptor 3 (TLR-3), TLR-4 and CD80 expression in peripheral blood mononuclear cells and urinary CD80 levels in children with idiopathic nephrotic syndrome. Pediatr Nephrol 32(8):1355–1361. https://doi.org/10.1007/s00467-017-3613-8
Conti F, Spinelli FR, Truglia S, Miranda F, Alessandri C, Ceccarelli F, Bombardieri M, Giannakakis K, Valesini G (2016) Kidney expression of Toll Like Receptors in lupus nephritis: quantification and clinicopathological correlations. Mediators Inflamm 2016:7697592. https://doi.org/10.1155/2016/7697592
Jamin A, Dehoux L, Dossier C, Fila M, Heming N, Monteiro RC, Deschênes G (2015) Toll-like Receptor 3 expression and function in childhood idiopathic nephrotic syndrome. Clin Exp Immunol 182(3):332–345. https://doi.org/10.1111/cei.12659
Nakano D, Kitada K, Wan N, Zhang Y, Wiig H, Wararat K, Yanagita M, Lee S, Jia L, Titze JM, Nishiyama A (2020) Lipopolysaccharide induces filtrate leakage from renal tubular lumina into the interstitial space via proximal tubular Toll-like receptor 4-dependent pathway and limits sensitivity to fluid therapy in mice. Kidney Int 97(5):904–912. https://doi.org/10.1016/j.kint.2019.11.024
Wei Q (2020) Novel strategy for septic acute kidney injury rescue: maintenance of the tubular integrity. Kidney Int 97(5):847–849. https://doi.org/10.1016/j.kint.2019.12.024
Gollapudi P, Yoon JW, Gollapudi S, Pahl MV, Vaziri ND (2010) Leukocyte Toll-Like Receptor expression in end-stage kidney disease. Am J Nephrol 31(3):247–254. https://doi.org/10.1159/000276764
Frieri M, Samih MA, Dzhindzhikhashvili M, Liu H, Balsam L, Rubinstein S (2012) Toll-like Receptor 9 and vascular endothelial growth factor levels in human kidneys from lupus nephritis patients. J Nephrol Nov-Dec 25(6):1041–1046. https://doi.org/10.5301/jn.5000091
Elloumi N, Fakhfakh R, Abida O, Ayadi L, Marzouk S, Hachicha H, Fourati M, Bahloul Z, Mhiri MN, Kammoun K, Masmoudi H (2017) Relevant genetic polymorphisms and kidney expression of Toll-like Receptor (TLR)-5 and TLR-9 in lupus nephritis. Clin Exp Immunol 190(3):328–339. https://doi.org/10.1111/cei.13022
Lee SM, Kok KH, Jaume M, Cheung TK, Yip TF, Lai JC, Guan Y, Webster RG, Jin DY, Peiris JM (2014) Toll-like Receptor 10 is involved in induction of innate immune responses to influenza virus infection. Proc Natl Acad Sci USA 111(10):3793–3798. https://doi.org/10.1073/pnas.1324266111
Hari P, Millar FR, Tarrats N, Birch J, Quintanilla A, Rink CJ, Fernández-Duran I, Muir M, Finch AJ, Brunton VG, Passos JF, Morton JP, Boulter L, Acosta JC (2019) The innate immune sensor Toll-like Receptor 2 controls the senescence-associated secretory phenotype. Sci Adv 5(6):eaaw0254. https://doi.org/10.1126/sciadv.aaw0254
Pasparakis M, Vandenabeele P (2015) Necroptosis and its role in inflammation. Nature 517(7534):311–320. https://doi.org/10.1038/nature14191
Xi Y, Shao F, Bai XY, Cai G, Lv Y, Chen X (2014) Changes in the expression of the Toll-like receptor system in the aging rat kidneys. PLoS ONE 9(5):e96351. https://doi.org/10.1371/journal.pone.0096351
Drouin M, Saenz J, Chiffoleau E (2020) C-type lectin-like receptors: Head or tail in cell death immunity. Front Immunol 11:251. https://doi.org/10.3389/fimmu.2020.00251
Chiffoleau E (2018) C-type lectin-like receptors as emerging orchestrators of sterile inflammation represent potential therapeutic targets. Front Immunol 9:227. https://doi.org/10.3389/fimmu.2018.00227
Hadaya K, de Rham C, Bandelier C, Bandelier C, Ferrari-Lacraz S, Jendly S, Berney T, Buhler L, Kaiser L, Seebach JD, Tiercy JM, Martin PY, Villard J (2008) Natural killer cell receptor repertoire and their ligands, and the risk of CMV infection after kidney transplantation. Am J Transpl 8(12):2674–2683. https://doi.org/10.1111/j.1600-6143.2008.02431.x
Swanson KV, Deng M, Ting JP (2019) The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 19(8):477–489. https://doi.org/10.1038/s41577-019-0165-0
Mulay SR (2019) Multifactorial functions of the inflammasome component NLRP3 in pathogenesis of chronic kidney diseases. Kidney Int 96(1):58–66. https://doi.org/10.1016/j.kint.2019.01.014
Hutton HL, Ooi JD, Holdsworth SR, Kitching AR (2016) The NLRP3 inflammasome in kidney disease and autoimmunity. Nephrology (Carlton) 21(9):736–744. https://doi.org/10.1111/nep.12785
Anders HJ (2007) Innate pathogen recognition in the kidney: toll-like receptors, NOD-like receptors, and RIG-like helicases. Kidney Int 72(9):1051–1056. https://doi.org/10.1038/sj.ki.5002436
Suzuki K, Imaizumi T, Tsugawa K, Ito E, Tanaka H (2007) Expression of retinoic acid-inducible gene-I in lupus nephritis. Nephrol Dial Transpl 22(8):2407–2409. https://doi.org/10.1093/ndt/gfm175
PrabhuDas MR, Baldwin CL, Bollyky PL, Bowdish DME, Drickamer K, Febbraio M, Herz J, Kobzik L, Krieger M, Loike J, McVicker B, Means TK, Moestrup SK, Post SR, Sawamura T, Silverstein S, Speth RC, Telfer JC, Thiele GM, Wang XY, Wright SD, El Khoury J (2017) A consensus definitive classification of scavenger receptors and their roles in health and disease. J Immunol 198(10):3775–3789. https://doi.org/10.4049/jimmunol.1700373
Yang X, Okamura DM, Lu X, Chen Y, Moorhead J, Varghese Z, Ruan XZ (2017) CD36 in chronic kidney disease: novel insights and therapeutic opportunities. Nat Rev Nephrol 13(12):769–781. https://doi.org/10.1038/nrneph.2017.126
Zhang Y, Ma Y, Zhang C, Zhang Y, Zhuang R, Liu B, Yi J, Jin B (2015) Soluble scavenger receptor CD163 is associated with severe acute kidney injury in patients with hantaan virus infection. Viral Immunol 28(4):241–246. https://doi.org/10.1089/vim.2014.0112
O’Reilly VP, Wong L, Kennedy C, Elliot LA, O’Meachair S, Coughlan AM, O’Brien EC, Ryan MM, Sandoval D, Connolly E, Dekkema GJ, Lau J, Abdulahad WH, Sanders JS, Heeringa P, Buckley C, O’Brien C, Finn S, Cohen CD, Lindemeyer MT, Hickey FB, O’Hara PV, Feighery C, Moran SM, Mellotte G, Clarkson MR, Dorman AJ, Murray PT, Little MA (2016) Urinary soluble CD163 in active renal vasculitis. J Am Soc Nephrol 27(9):2906–2916. https://doi.org/10.1681/ASN.2015050511
Izquierdo MC, Martin-Cleary C, Fernandez-Fernandez B, Elewa U, Sanchez-Niño MD, Carrero JJ, Ortiz A (2014) CXCL16 in kidney and cardiovascular injury. Cytokine Growth Factor Rev 25(3):317–325. https://doi.org/10.1016/j.cytogfr.2014.04.002
Franzin R, Stasi A, Fiorentino M, Stallone G, Cantaluppi V, Gesualdo L, Castellano G (2020) Inflammaging and complement system: a link between acute kidney injury and chronic graft damage. Front Immunol 11:734. https://doi.org/10.3389/fimmu.2020.00734
Stewart BJ, Ferdinand JR, Young MD, Mitchell TJ, Loudon KW, Riding AM, Richoz N, Frazer GL, Staniforth JUL, Vieira Braga FA, Botting RA, Popescu DM, Vento-Tormo R, Stephenson E, Cagan A, Farndon SJ, Polanski K, Efremova M, Green K, Del Castillo V-H, Guzzo C, Collord G, Mamanova L, Aho T, Armitage JN, Riddick ACP, Mushtaq I, Farrell S, Rampling D, Nicholson J, Filby A, Burge J, Lisgo S, Lindsay S, Bajenoff M, Warren AY, Stewart GD, Sebire N, Coleman N, Haniffa M, Teichmann SA, Behjati S, Clatworthy MR (2019) Spatiotemporal immune zonation of the human kidney. Science 365(6460):1461–1466. https://doi.org/10.1126/science.aat5031
Rogers NM, Ferenbach DA, Isenberg JS, Thomson AW, Hughes J (2014) Dendritic cells and macrophages in the kidney: a spectrum of good and evil. Nat Rev Nephrol 10(11):625–643. https://doi.org/10.1038/nrneph.2014.170
Ricketts TD, Prieto-Dominguez N, Gowda PS, Ubil E (2021) Mechanisms of macrophage plasticity in the tumor environment: manipulating activation state to improve outcomes. Front Immunol 12:642285. https://doi.org/10.3389/fimmu.2021.642285
Vegting Y, Vogt L, Anders HJ, de Winther MPJ, Bemelman FJ, Hilhorst ML (2021) Monocytes and macrophages in ANCA-associated vasculitis. Autoimmun Rev 20(10):102911. https://doi.org/10.1016/j.autrev.2021.102911
Zhang H, Li Z, Li W (2021) M2 macrophages serve as critical executor of innate immunity in chronic allograft rejection. Front Immunol 12:648539. https://doi.org/10.1016/10.3389/fimmu.2021.648539
Bonavia A, Singbartl K (2018) A review of the role of immune cells in acute kidney injury. Pediatr Nephrol 33(10):1629–1639. https://doi.org/10.1007/s00467-017-3774-5
Cao Q, Harris DC, Wang Y (2015) Macrophages in kidney injury, inflammation, and fibrosis. Physiology (Bethesda) 30(3):183–194. https://doi.org/10.1152/physiol.00046.2014
Meng XM, Mak TS, Lan HY (2019) Macrophages in renal fibrosis. Adv Exp Med Biol 1165:285–303. https://doi.org/10.1007/978-981-13-8871-2_13
Rauen T, Floege J (2017) Inflammation in IgA nephropathy. Pediatr Nephrol 32(12):2215–2224. https://doi.org/10.1007/s00467-017-3628-1
Hu W, Lin J, Lian X, Yu F, Liu W, Wu Y, Fang X, Liang X, Hao W (2019) M2a and M2b macrophages predominate in kidney tissues and M2 subpopulations were associated with the severity of disease of IgAN patients. Clin Immunol 205:8–15. https://doi.org/10.1016/j.clim.2019.05.005
Maria NI, Davidson A (2017) Renal macrophages and dendritic cells in SLE nephritis. Curr Rheumatol Rep 19(12):81. https://doi.org/10.1007/s11926-017-0708-y
Ikezumi Y, Suzuki T, Yamada T, Hasegawa H, Kaneko U, Hara M, Yanagihara T, Nikolic-Paterson DJ, Saitoh A (2015) Alternatively activated macrophages in the pathogenesis of chronic kidney allograft injury. Pediatr Nephrol 30(6):1007–1017. https://doi.org/10.1007/s00467-014-3023-0
Meissner M, Viehmann SF, Kurts C (2019) DAMPening sterile inflammation of the kidney. Kidney Int 95(3):489–491. https://doi.org/10.1016/j.kint.2018.12.007
Nakazawa D, Marschner JA, Platen L, Anders HJ (2018) Extracellular traps in kidney disease. Kidney Int 94(6):1087–1098. https://doi.org/10.1016/j.kint.2018.08.035
Nakazawa D, Kumar SV, Marschner J, Desai J, Holderied A, Rath L, Kraft F, Lei Y, Fukasawa Y, Moeckel GW, Angelotti ML, Liapis H, Anders HJ (2017) Histones and neutrophil extracellular traps enhance tubular necrosis and remote organ injury in ischemic AKI. J Am Soc Nephrol 28(6):1753–1768. https://doi.org/10.1681/ASN.2016080925
Mulay SR, Kumar SV, Lech M, Desai J, Anders HJ (2016) How kidney kell death induces renal necroinflammation. Semin Nephrol 36(3):162–173. https://doi.org/10.1016/j.semnephrol.2016.03.004
Turner JE, Rickassel C, Healy H, Kassianos AJ (2019) Natural killer cells in kidney health and disease. Front Immunol 10:587. https://doi.org/10.3389/fimmu.2019.00587
Bahl D, Haddad Z, Datoo A, Qazi YA (2019) Delayed graft function in kidney transplantation. Curr Opin Organ Transpl 24(1):82–86. https://doi.org/10.1097/MOT.0000000000000604
Gómez-Massa E, Talayero P, Utrero-Rico A, Laguna-Goya R, Andrés A, Mancebo E, Leivas A, Polanco-Fernández N, Justo I, Jimenez-Romero C, Pleguezuelo D, Paz-Artal E (2020) Number and function of circulatory helper innate lymphoid cells are unaffected by immunosuppressive drugs used in solid organ recipients—a single centre cohort study. Transpl Int 33(4):402–413. https://doi.org/10.1111/tri.13567
Victorino F, Sojka DK, Brodsky KS, McNamee EN, Masterson JC, Homann D, Yokoyama WM, Eltzschig HK, Clambey ET (2015) Tissue-resident NK cells mediate ischemic kidney injury and are not depleted by anti-asialo-GM1 antibody. J Immunol 195(10):4973–4985. https://doi.org/10.4049/jimmunol.1500651
Turner JE, Becker M, Mittrücker HW, Panzer U (2018) Tissue-resident lymphocytes in the kidney. J Am Soc Nephrol 29(2):389–399. https://doi.org/10.1681/ASN.2017060599
Zhang Z-X, Wang S, Huang X, Min W-P, Sun H, Liu W, Garcia B, Jevnikar AM (2008) NK Cells induce apoptosis in tubular epithelial cells and contribute to renal ischemia-reperfusion injury. J Immunol 181(11):7489–7498. https://doi.org/10.4049/jimmunol.181.11.7489
Luo L, Lu J, Wei L, Long D, Guo JY, Shan J, Fu S, Li FS, Lu PY, Li PY, Feng L (2010) The role of HIF-1 in up-regulating MICA expression on human renal proximal tubular epithelial cells during hypoxia/reoxygenation. BMC Cell Biol 11:91. https://doi.org/10.1186/1471-2121-11-91
Libetta C, Sepe V, Esposito P, Galli F, Dal Canton A (2011) Oxidative stress and inflammation: Implications in uremia and hemodialysis. Clin Biochem 44(14–15):1189–1198. https://doi.org/10.1016/j.clinbiochem.2011.06.988
Losappio V, Franzin R, Infante B, Godeas G, Gesualdo L, Fersini A, Castellano G, Stallone G (2020) Molecular mechanisms of premature aging in hemodialysis: the complex interplay between innate and adaptive immune dysfunction. Int J Mol Sci 21(10):3422. https://doi.org/10.3390/ijms21103422
Sanchis P, Ho CY, Liu Y, Beltran LE, Ahmad S, Jacob AP, Furmanik M, Laycock J, Long DA, Shroff R, Shanahan CM (2019) Arterial “inflammaging” drives vascular calcification in children on dialysis. Kidney Int 95(4):958–972. https://doi.org/10.1016/j.kint.2018.12.014
Sepe V, Rampino T, Libetta C (2019) Arterial, “inflammaging” drives vascular calcification in children on dialysis. Kidney Int 96(2):522. https://doi.org/10.1016/j.kint.2019.05.021
Desai AA, Nissenson A, Chertow GM, Farid M, Singh I, Van Oijen MG, Esrailian E, Solomon MD, Spiegel BM (2009) The relationship between laboratory-based outcome measures and mortality in end-stage renal disease: a systematic review. Hemodial Int 13(3):347–359. https://doi.org/10.1111/j.1542-4758.2009.00377.x
Li PK, Ng JK, Mcintyre CW (2017) Inflammation and peritoneal dialysis. Semin Nephrol 37(1):54–65. https://doi.org/10.1016/j.semnephrol.2016.10.007
Elmarakby AA, Sullivan JC (2012) Relationship between oxidative stress and inflammatory cytokines in diabetic nephropathy. Cardiovasc Ther 30(1):49–59. https://doi.org/10.1111/j.1755-5922.2010.00218.x
Feroze U, Molnar MZ, Dukkipati R, Kovesdy CP, Kalantar-Zadeh K (2011) Insights into nutritional and inflammatory aspects of low parathyroid hormone in dialysis patients. J Ren Nutr 21(1):100–104. https://doi.org/10.1053/j.jrn.2010.10.006
Gandolfini I, Regolisti G, Bazzocchi A, Maggiore U, Palmisano A, Piotti G, Fiaccadori E, Sabatino A (2019) Frailty and sarcopenia in older patients receiving kidney transplantation. Front Nutr 6:169. https://doi.org/10.3389/fnut.2019.00169
Fuery MA, Liang L, Kaplan FS, Mohler ER 3rd (2018) Vascular ossification: pathology, mechanisms, and clinical implications. Bone 109:28–34. https://doi.org/10.1016/j.bone.2017.07.006
Düsing P, Zietzer A, Goody PR, Hosen MR, Kurts C, Nickenig G, Jansen F (2021) Vascular pathologies in chronic kidney disease: pathophysiological mechanisms and novel therapeutic approaches. J Mol Med (Berl) 99(3):335–348. https://doi.org/10.1007/s00109-021-02037-7
López-Mejías R, González-Gay MA (2019) IL-6: linking chronic inflammation and vascular calcification. Nat Rev Rheumatol 15(8):457–459. https://doi.org/10.1038/s41584-019-0259-x
Rampino T, Gregorini M, Perotti L, Ferrari F, Pattonieri EF, Grignano MA, Valente M, Garrone A, Islam T, Libetta C, Sepe V, Albertini R, Bruno R, Belliato M (2020) Hemoperfusion with CytoSorb as adjuvant therapy in critically ill patients with SARS-CoV2 pneumonia. Blood Purif. https://doi.org/10.1159/000511725
Cheng Y, Luo R, Wang K, Zhang M, Wang Z, Dong L, Li J, Yao Y, Ge S, Xu G (2020) Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int 97(5):829–838. https://doi.org/10.1016/j.kint.2020.03.005
Kudose S, Batal I, Santoriello D, Xu K, Barasch J, Peleg Y, Canetta P, Lloyd E, Ratner LE, Marasa M, Gharavi AG, Stokes MB, Markowitz GS, D’Agati VD (2020) Kidney biopsy findings in patients with COVID-19. J Am Soc Nephrol. https://doi.org/10.1681/ASN.2020060802
George JA, Khoza S (2021) SARS-CoV-2 infection and the kidneys: an evolving picture. Adv Exp Med Biol 1327:107–118. https://doi.org/10.1007/978-3-030-71697-4_8
D’Silva KM, Jorge A, Cohen A, McCormick N, Zhang Y, Wallace ZS, Choi HK (2021) COVID-19 outcomes in patients with systemic autoimmune rheumatic diseases compared to the general population: a US Multicenter, Comparative Cohort Study. Arthritis Rheumatol 73(6):914–920. https://doi.org/10.1002/art.41619
Yazdanpanah N, Rezaei N (2021) Autoimmune Complications of COVID-19. J Med Virol. https://doi.org/10.1002/jmv.27292
Bourgonje AR, Abdulle AE, Timens W, Hillebrands JL, Navis GJ, Gordijn SJ, Bolling MC, Dijkstra G, Voors AA, Osterhaus AD, van der Voort PH, Mulder DJ, van Goor H (2020) Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J Pathol 251(3):228–248. https://doi.org/10.1002/path.5471
Raby AC, González-Mateo GT, Williams A, Topley N, Fraser D, López-Cabrera M, Labéta MO (2018) Targeting Toll-like receptors with soluble Toll-like receptor 2 prevents peritoneal dialysis solution-induced fibrosis. Kidney Int 94(2):346–362. https://doi.org/10.1016/j.kint.2018.03.014
Kim HJ, Park SJ, Koo S, Cha HJ, Lee JS, Kwon B, Cho HR (2014) Inhibition of kidney ischemia-reperfusion injury through local infusion of a TLR2 blocker. J Immunol Methods 407:146–150. https://doi.org/10.1016/j.jim.2014.03.014
Farrar CA, Keogh B, McCormack W, O’Shaughnessy A, Parker A, Reilly M, Sacks SH (2012) Inhibition of TLR2 promotes graft function in a murine model of renal transplant ischemia-reperfusion injury. FASEB J 26(2):799–807. https://doi.org/10.1096/fj.11-195396
Chen X, Ma J, Kwan T, Stribos EGD, Messchendorp AL, Loh YW, Wang X, Paul M, Cunningham EC, Habib M, Alexander IE, Sharland AF, Chadban SJ, Wu H (2018) Blockade of HMGB1 attenuates diabetic nephropathy in mice. Sci Rep 8(1):8319. https://doi.org/10.1038/s41598-018-26637-5
Prattichizzo F, Giuliani A, Recchioni R, Bonafè M, Marcheselli F, De Carolis S, Campanati A, Giuliodori K, Rippo MR, Brugè F, Tiano L, Micucci C, Ceriello A, Offidani A, Procopio AD, Olivieri F (2016) Anti-TNF-α treatment modulates SASP and SASP-related microRNAs in endothelial cells and in circulating angiogenic cells. Oncotarget 7(11):1945–1958. https://doi.org/10.18632/oncotarget.7858
Xi T, Jin F, Zhu Y, Wang J, Tang L, Wang Y, Liebeskind DS, He Z (2017) MicroRNA-126-3p attenuates blood-brain barrier disruption, cerebral edema and neuronal injury following intracerebral hemorrhage by regulating PIK3R2 and Akt. Biochem Biophys Res Commun 494(1–2):144–151
Gregorini M, Maccario R, Avanzini MA, Corradetti V, Moretta A, Libetta C, Esposito P, Bosio F, Dal Canton A, Rampino T (2013) Antineutrophil cytoplasmic antibody-associated renal vasculitis treated with autologous mesenchymal stromal cells: Evaluation of the contribution of immune-mediated mechanisms. Mayo Clin Proc 88(10):1174–1179. https://doi.org/10.1016/j.mayocp.2013.06.021
Funding
None.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by CL, MG, TR and VS. The first draft of the manuscript was written by VS and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors have no conflict of interest to disclose.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sepe, V., Libetta, C., Gregorini, M. et al. The innate immune system in human kidney inflammaging. J Nephrol 35, 381–395 (2022). https://doi.org/10.1007/s40620-021-01153-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40620-021-01153-4