
Abstract
MicroRNAs are small, noncoding RNAs which regulate protein expression post-transcriptionally. They respond to changes in a cell's environment and can promote cell death or cell survival depending on the context. Recent studies have linked microRNAs to the unfolded protein response pathway. This pathway is activated in the endoplasmic reticulum by conditions which interfere with the normal functions of the endoplasmic reticulum. The cell fate outcomes consequent to the activation of the unfolded protein response are binary, either cell survival or cell death. MicroRNAs can regulate multiple components of this pathway to tip the cell towards either fate. Inositol-requiring enzyme 1 alpha, a canonical unfolded protein response sensor and mediator, has inherent endoribonuclease activity. Interestingly, recently, it has been demonstrated that it can target microRNAs in addition to its previously known targets. This review highlights key papers in this rapidly emerging field.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The endoplasmic reticulum (ER) is a vital organelle with multiple cellular functions. Predominant among the functions is the folding of newly synthesized proteins into their native configuration [1]. The ER also stores most of the intracellular calcium in a cell. Furthermore, lipid metabolism is closely linked to the ER with cholesterol biosynthesis, lipid droplet formation, very low density lipoprotein (VLDL) particle formation occurring either in the ER or juxtaposed to the ER membrane [2]. This diversity of function makes the ER vulnerable to extracellular stimuli and intracellular perturbations which interfere with these functions. Such stimuli are said to have caused ER stress, a condition best recognized by a signature response, the unfolded protein response (UPR), which canonically is activated by the accumulation of misfolded proteins within the ER lumen. In the absence of a direct measure for misfolded proteins, activation of the UPR remains the best method to monitor ER stress.
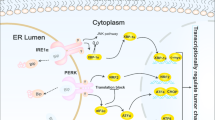
The three canonical UPR sensors are activating transcription factor 6 alpha (ATF6α), inositol-requiring enzyme 1 alpha (IRE1α), and protein kinase-RNA-like ER kinase (PERK). The UPR pathways collectively mediate global attenuation of mRNA translation and increase both the abundance and the folding capacity of the ER. Thus, the ER attempts to handle the load of accumulated misfolded proteins and thus ameliorate ER stress. Cellular homeostasis is restored if the ER stress can be successfully ameliorated. The restoration of cellular homeostasis ensures survival of the stressed cell. However, if a cell is unable to resolve ER stress, it activates cell death pathways, termed ER stress-induced cell death. MicroRNAs are small, noncoding RNAs that regulate protein expression at the post-transcriptional level and regulate cell death pathways in response to environmental changes in a cell [3, 4]. Recent studies have uncovered a close link between ER stress responses and microRNAs such that microRNAs fine-tune the outcome of a cell to ER stress, promoting either cell survival or cell death (Fig. 1). The UPR sensors, and how microRNAs contribute to cell fate decisions under ER stress conditions, are discussed herein.

MicroRNAs involved in the unfolded protein response pathway. Several microRNAs have recently been described as having a role in the cell fate outcome, survival or apoptosis, under conditions of endoplasmic reticulum (ER) stress. The three canonical unfolded protein response sensors, activating transcription factor 6 alpha (ATF6α), inositol requiring enzyme 1 alpha (IRE1α), and protein kinase-like ER kinase (PERK), are activated under ER stress. ATF6α inhibits miR-455, which inhibits calreticulin, and mir-702 is an inhibitor of ATF6α. Mir-30c-2-3p antagonizes the IRE1α target mRNA X-box-binding protein-1 (XBP-1). IRE1α directly cleaves several microRNAs, including miR-17, miR-34a, miR-96, and miR-125b. In itself, IRE1α is repressed by the microRNA, miR-199a-5p. IRE1α-dependent degradation of these microRNAs derepresses caspase 2 and thioredoxin interacting protein (TXNIP), thus promoting apoptosis. PERK induces the XBP-1 inhibitory miR-30c-2-3p. It also induces miR-211, which inhibits Chop transcription via hypermethylation of its promoter. CHOP-dependent microRNA miR-708 promotes cell survival by inhibiting the ER folded protein, Rhodopsin
The Unfolded Protein Response Sensors
Under ER stress conditions ATF6α translocates to the Golgi where it is cleaved by regulated intramembrane proteolysis to release its N-terminal domain which is an active transcription factor. The N-terminal domain of ATF6α translocates to the nucleus and regulates target genes which encode ER chaperones that facilitate protein folding [5]. Its target genes are also involved in the degradation of terminally misfolded proteins by the proteasome, a process termed ER-associated degradation (ERAD) [6]. IRE1α is a kinase and endoribonuclease [7]. It autotransphosphorylates upon dimerization, and one of its defined phosphorylation targets is the stress-activated kinase c-jun N-terminal kinase (JNK) [8, 9]. X-box-binding protein 1 (XBP-1) mRNA is the canonical IRE1α endoribonuclease target [10, 11]. IRE1α removes 26 nucleotides from the XBP-1 mRNA to generate a spliced mRNA which encodes a potent transcription factor spliced XBP-1 (XBP-1s). XBP-1s target genes facilitate adaptation to ER stress by leading to ER expansion and increased folding capacity of the ER [12]. Recently, other endoribonuclease targets have been defined. It has been shown that active IRE1α can degrade mRNAs in a process termed regulated IRE1α-dependent decay (RIDD) [13]. The targeting of pro-survival mRNAs facilitates cell death under ER stress conditions, whereas the degradation of targeted mRNAs promotes cell survival by reducing the mRNAs available for translation [13, 14].
PERK phosphorylates eukaryotic translation initiation factor 2-alpha (eIF2α) leading to a global halting of mRNA translation, while simultaneously promoting selective translation of a few transcripts, including activating transcription factor 4 (ATF4) [15]. ATF4-regulated genes include ER chaperones and ERAD components. It also regulates amino acid transporters, and redox stress response genes, and also C/EBP homologous protein (CHOP) [16–18]. CHOP is a stress-induced transcription factor known to mediate ER stress-induced cell death [19]. ATF4 and CHOP also plays an important role in limiting translation repression and promoting new protein synthesis [20]. The activation of nuclear factor κB (NFκB) occurs under ER stress conditions due to impaired synthesis of its inhibitory regulator, inhibitor of κB (IκB) [21].
Thus, output from the three UPR sensors orchestrates multiple adaptive responses, the sum total of which is to deal with the ER stress-inducing agent, refold or degrade the accumulated misfolded proteins and return the ER and the cell to a state of homeostasis. And in the event of unrelenting ER stress, apoptosis occurs.
MicroRNAs that Promote Adaptation
One of the first studies to establish a link between microRNAs and ER stress utilized a human airway epithelial cell line [22•]. They treated this cell line with either tunicamycin or the proteasome inhibitor ALLN. They identified 47 tunicamycin-induced microRNAs and 39 ALLN-induced microRNAs. Only two microRNAs were common to the two data sets, these were miR-346 and miR-885-3p. MiR-346 was induced to a greater extent than miR-885; therefore, it was verified in several cell types. MiR-346 is encoded within intron 2 of the GRID1 gene; however, GRID1 levels did not change with ER stress, suggesting that miR-346 was induced independently of GRID1. They identified a putative XBP-1 binding site in the promoter region of miR-346, and then induced it by the enforced expression of exogenous XBP-1. Using wild-type and XBP-1 knockout mouse embryonic fibroblasts (MEFs), they confirm the lack of ER stress-induced increase in miR-346 in XBP-1 knockout MEFs.
To identify targets of miR-346, they expressed a mimic of miR-346 and a control microRNA and expression profiled the mRNAs to identify those most significantly repressed by miR-346. Then, by comparative analyses with ER stress-induced mRNAs expression profiling, they identified 21 common mRNAs that were downregulated by ER stress in Calu-3 cells and the miR-346 mimic in HeLa cells. These 21 genes encoded products with functional roles in the immune response: MHC-associated genes and interferon-inducible genes. They focused on one of these, antigen peptide transporter (TAP1), as it has an miR-346 binding site in its 3′UTR. Using a mimic or inhibitor, they show that the combination of ER stress and an miR-346 mimic virtually eliminates mRNA expression of TAP1 and significantly reduced protein expression of TAP1, whereas the presence of the antagomir of miR-346 prevented an ER stress-induced decrease in TAP1 protein levels. These studies confirmed that the putative miR-346 binding site in the TAP1 mRNA was functional in cultured cells. Thus, they identified an ER stress-induced microRNA, miR-346 and its direct target, TAP1, both of which are reciprocally regulated under ER stress conditions. The ER stress-induced repression of TAP1 protein would presumably promote cell survival by limiting new polypeptides entering the ER to undergo folding. How this particular microRNA and other candidate microRNAs might regulate the ER stress-induced immune response and cell fate downstream of that will need additional studies.
The PERK axis of UPR signaling has been linked to microRNAs that favor cell survival, thus perhaps antagonizing the output from the proapoptotic signaling of the ATF4-CHOP axis. Behrman and Walter performed expression profiling for microRNAs in wild-type and CHOP deficient MEFs, and identified miR-708 as a CHOP-dependent ER stress-induced microRNA [23•]. In addition to miR-708, they identified seven additional microRNAs (miR-689, miR-711, miR-1897-3p, miR-2137, miR-762, miR-712*, miR-2132) that were induced by ER stress, and three (miR-322, miR-351, miR-503) that were inhibited. MiR-708 targets included several proteins expressed in the eye. They further show that rhodopsin, an ER folded protein, is down-regulated by mir-708. Thus, CHOP appears to be cytoprotective, by inducing the expression of miR-708, which is located in intron 1 of a known CHOP target gene, Odz4. Under ER stress conditions, the induction of CHOP and mir-708 would decrease the synthesis of rhodopsin, thus decreasing the protein folding load of the ER and promoting adaptation. This is one of the first pro-survival functions attributed to CHOP. In this study, CHOP wild-type and knockout MEFs were treated with either tunicamycin or thapsigargin, conditions that are quite well-defined for inducing the UPR, though the authors do not show activation of either PERK or ATF4, or even CHOP induction. MiR-708 is also highly expressed in the brain, skeletal muscle and small intestine. The authors have shown a cytoprotective role for CHOP-induced miR-708 in the eye; it will be interesting to see if this is true in the other tissues where miR-708 is highly expressed.
MiR-211 was shown to be a direct PERK-induced microRNA by Chitnis et al. [24]. They profiled microRNAs induced early in the onset of ER stress, treating PERK knockout and wild-type MEFs with tunicamycin for 2 h. They identified seven microRNAs that were increased and ten that were decreased under these conditions. Of these, miR-211 was most induced. MiR-211 is encoded by intron 2 of the Trpm1 gene. The primary transcript of miR-211 (pri-211) was upregulated along with Trpm1 mRNA under ER stress conditions in a PERK- and ATF4-dependent manner. They identified binding sites in the promoter region of the Chop (ddit3) gene, and using antagomir based loss of function demonstrated increased Chop mRNA accumulation; and with miR-211 mimic based gain of function decreased Chop mRNA accumulation under ER stress conditions. Interestingly, there were no binding sites for miR-211 in the 3′UTR of the Chop transcript. However, there were two potential binding sites in the proximal promoter region of the Chop gene. They go on to identify histone 3 lysine 27 trimethylation of the Chop promoter as the mechanism for mir-211-mediated suppression of Chop transcription. Furthermore, the kinetics of miR-211 are inverse to the kinetics of CHOP expression. MiR-211 levels peak early (5 h), and decline to basal levels by about 8 h under ER stress. Correspondingly, a significant increase in CHOP protein levels occurs from 5 to 8 h of treatment. Antagonism of miR-211 increased CHOP protein expression at early timepoints and sensitized cells to ER stress-induced apoptosis. To expand further on a biological role for this process, the authors demonstrate inverse expression of miR-211 and Chop mRNA in experimental mouse mammary cancer tissues, and much reduced miR-211 in PERK negative cancer tissues.
ATF6α has been best studied in the context of the heart with both ATF6α-regulated microRNAs and a microRNA that regulates ATF6α described under ER stress conditions [25, 26]. Both sets of microRNAs favor cell survival under ER stress conditions. In an interesting approach to identify ATF6α microRNA targets, the authors of this study used heart-specific ATF6α transgenic mice [25]. Upon induction of the ATF6α transgene, 13 microRNAs were regulated. Of these, they identified eight microRNAs that were downregulated and five that were upregulated. Of the downregulated microRNAs, they identified miR-455 as one which could potentially regulate several of the mRNAs reciprocally upregulated by the enforced expression of active ATF6α. Furthermore, they verified that tunicamycin-induced ER stress and adenovirally expressed active ATF6α decreased miR-455 and increased calreticulin (Calr) mRNA. They then characterized, with loss-of-function and gain-of-function approaches, miR-455′s target, calreticulin protein, as being upregulated and downregulated, respectively. The cardioprotective function of ATF6α under ischemic conditions in the heart has previously been shown by the same group, and calreticulin is known to promote ischemic preconditioning in the heart [27, 28]. The identification of miR-455 and its target, the cardioprotective protein, calreticulin provides a mechanistic explanation for the salutary effects of ATF6 signaling under conditions of cardiac ischemia.
In an alternative approach, miR-702 was identified as a microRNA that regulates ATF6α [26]. The authors utilized isoproterenol injection in mice to show that the apoptotic stress of isoproterenol was associated with ER stress and an upregulation of miR-702. They computationally identified four potential miR-702 binding sites in the 3′ UTR of Atf6α, and two sites in the 3′ UTR of Chop. Using luciferase reporter constructs, they identified one of the four sites in the 3′ UTR of Atf6α, as actually regulated by miR-702 in cultured cells. Then using mimic of miR-702 they show that apoptosis is reduced in isoproterenol treated cultured cardiomyocytes. They hypothesize that ATF6α is proapoptotic in their model, and its inhibition by miR-702 is the potential mechanism for the anti-apoptotic activity of miR-702.
In one of the few liver-related studies on ER stress and microRNAs, the authors generated a human hepatocyte cell line genetically deficient in DICER (by RNA interference), and demonstrated enhanced sensitivity to ER stress-induced apoptosis, utilizing deoxycholic acid as a model of cholestatic liver disease or thapsigargin, to induce ER stress [29]. They then used computational tools and published transcription profiles to identify miR-199a/b-5p cluster as a potential candidate which could regulate GRP78, ATF6α and IRE1α. Kinetic studies to validate this miRNA and its potential targets, show that upon deoxycholic acid treatment, the levels of GRP78, ATF6α, and IRE1α mRNA rise and peak at 12 h after the induction of stress, followed by progressive decline, whereas miR-199a-5p levels peak at 36 h. The upregulation of miR-199a-5p was transcriptional as both the primary and precursor transcripts were increased. Next, using reporter constructs, they demonstrate miR-199a-5p binding sites in the 3′UTRs of GRP78, ATF6α and IRE1α mRNA for exogenous and endogenous miR-199a-5p.
Having demonstrated that deoxycholic acid can induce miR-199a-5p, they next confirmed its functional significance by using an inhibitor of miR-199a-5p. In the presence of the inhibitor cells miR-199a-5p is sensitized to ER stress-induced apoptosis, which can be partially abrogated by silencing IRE1α suggesting that IRE1α signaling under these conditions is proapoptotic. On the other hand, silencing either GRP78 or ATF6α potentiated ER stress-induced apoptosis, suggesting that these two molecules promote adaptation. They define the mechanism for the transcriptional upregulation of miR-199a-5p under ER stress conditions, via the transcription factor AP1. Lastly, they show that pharmacologic JNK inhibition promotes cell death under ER stress condition, and exogenous restoration of miR-199a-5p activity rescues cells from cell death. Thus, in their model, ER stress may itself activate JNK and the transcription factor AP1, induce miR-199a-5p, thus inhibiting prolonged IRE1α activity and cell death. whereas inhibition of miR-199a-5p would result in prolonged IRE1α activity and cell death.
MicroRNAs that Promote Apoptosis
Conversely, microRNAs have also been implicated in promoting ER stress-induced apoptosis. In one of the earlier studies in this context, the authors computationally searched and identified miR-30c-2-3p as a potential regulator of XBP-1 on the basis of conserved binding sites in the 3′ UTR of the human, mouse, and rat XBP-1 mRNA [30•]. They go on to validate that, by overexpressing miR-30c-2-3p, it does repress XBP-1 protein levels, is induced by ER stress, and is PERK-dependent. The activation of NFκB via lack of synthesis of its inhibitor IκB following PERK activation and eIF2α phosphorylation has been previously described [21]. The authors of the present study show that miR-30c-2p is a transcriptional target of NFκB. Furthermore, antagonism of miR-30c-2p in HeLa cells attenuated tunicamycin-induced apoptosis in these cells, suggesting that a consequent increase in XBP-1 protein levels enhanced the cells' capacity to resolve ER stress. Thus, by extension, one could extrapolate that miR-30c-2p may exert a proapoptotic effect by reducing XBP-1 translation under ER stress conditions. Another possible role, as suggested by the authors, is that it may keep exuberant XBP-1 expression in check. This study is also an example of regulatory cross-talk between UPR sensors, as the PERK-dependent expression of miR-30c-2p represses XBP-1.
In an effort to find mediators of cell fate outcomes under ER stress conditions the authors of this study approached the question by performing an unbiased screen for mRNAs that are preferentially translated early under ER stress conditions that are ultimately apoptotic [31]. They identified that thioredoxin-interacting protein (TXNIP) mRNA and protein are induced rapidly under ER stress conditions, and that this occurs downstream of PERK and IRE1α activation. Looking for a mechanism, they computationally identified two conserved miR-17 binding sites in the 3′UTR of TXNIP, validate these binding sites with reporter constructs, and show that the ER stress-induced decrease in miR-17 is dependent on IRE1α and JNK. Cultured β cells and pancreatic islets in mice lacking Txnip were protected from ER stress-induced apoptosis. Furthermore, in the human macrophage cell line THP1, the secretion of interleukin 1β via activation of the NLRP3 inflammasome under ER stress conditions occurred downstream of TXNIP and was prevented by pharmacologic inhibition of IRE1α. The authors of this study show an IRE1α–dependent decrease in miR-17; however, a mechanism for the decrease is not elucidated by them. It is likely that precursor-miR-17 is degraded by IRE1α as subsequently shown by Upton et al.
In a landmark study Upton et al. [32•] demonstrate that microRNAs are targeted and cleaved by the endoribonuclease activity of IRE1α. The authors had previously shown that ER stress-induced apoptosis is mediated by caspase 2 [33]. In the present study they expand on that to show that caspase 2 expression requires IRE1α activity, and further define that the endoribonuclease activity of IRE1α is needed for caspase 2 expression, in a manner that does not require XBP-1. Therefore, they search for microRNAs that have binding sites in the 3′UTR of caspase 2 mRNA, and indeed show that miR-17, miR-34a, miR-96, and miR-125b levels are reduced by IRE1α. The authors then show that miR-17 and pre-miR-17 are both reduced due to direct cleavage of pre-miR-17 at sites that are distinct from the DICER cleavage sites; however, have some similarity to the XBP-1 cleavage sites. Though the generalizability of caspase 2-mediated apoptosis is questionable, the identification of the cleavage of microRNAs by IRE1α is not surprising [34]. It remains to be seen what additional pathways and cell fate decisions this newly described property of IRE1α may regulate.
Conclusion
Changes in microRNA levels under ER stress conditions have been described by many research groups, with several microRNAs induced or repressed by the respective UPR sensors, and their downstream effectors. microRNAs which regulate the UPR sensors themselves, have been described. As the two cell fates possible under ER stress conditions are either cell survival or cell death, these microRNAs have been shown to fine-tune a cell's response to ER stress. The signaling output from the PERK branch of the UPR was known to be dichotomous with the salutary transcriptional profile activated by ATF4 and the cell death susceptibility imparted by CHOP. Similarly, for IRE1α, its initially identified conserved target, XBP-1 promotes adaptation to ER stress conditions. However, the degradation of mRNAs which favor cell survival, and now the degradation of microRNAs which repress caspase 2, are two signaling pathways by which IRE1α can activate the apoptotic machinery in a cell. Future studies may identify additional RNA targets of IRE1α endoribonuclease activity. IRE1α, though predominantly located on the ER membrane, can also be detected on the nuclear membrane, thus its presence in two locations may impart distinct RNA targets to IRE1α. Similarly, ATF6α can transcriptionally increase expression of many microRNAs. The mechanism for ATF6α–induced microRNA repression remains to be elucidated. Thus, microRNAs are clearly pieces of the UPR pathway. The identification of additional microRNAs and how they target critical components of the UPR will help define if they have broader roles under conditions of ER stress, in addition to the recently described role as regulators of cell survival.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Malhi H, Kaufman RJ (2011) Endoplasmic reticulum stress in liver disease. J Hepatol 54(4):795–809
Wang S, Kaufman RJ (2014) How does protein misfolding in the endoplasmic reticulum affect lipid metabolism in the liver? Curr Opin Lipidol 25(2):125–132
Bartel DP (2009) microRNAs: target recognition and regulatory functions. Cell 136(2):215–233
Guicciardi ME et al (2013) Apoptosis and necrosis in the liver. Compr Phys 3(2):977–1010
Okada T et al (2002) Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem J 366(Pt 2):585–594
Yamamoto K et al (2007) Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell 13(3):365–376
Cox JS, Shamu CE, Walter P (1993) Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73(6):1197–1206
Nishitoh H et al (2002) ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev 16(11):1345–1355
Urano F et al (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287(5453):664–666
Calfon M et al (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415(6867):92–96
Lee K et al (2002) IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev 16(4):452–466
Lee AH, Iwakoshi NN, Glimcher LH (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23(21):7448–7459
Hollien J, Weissman JS (2006) Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313(5783):104–107
Han D et al (2009) IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138(3):562–575
Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397(6716):271–274
Harding HP et al (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6(5):1099–1108
Lange PS et al (2008) ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J Exp Med 205(5):1227–1242
Harding HP et al (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11(3):619–633
Zinszner H et al (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 12(7):982–995
Han J et al (2013) ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15(5):481–490
Jiang HY et al (2003) Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol Cell Biol 23(16):5651–5663
• Bartoszewski, R et al (2011) The unfolded protein response (UPR)-activated transcription factor X-box-binding protein 1 (XBP1) induces microRNA-346 expression that targets the human antigen peptide transporter 1 (TAP1) mRNA and governs immune regulatory genes. J Biol Chem 286(48):41862–41870. This is one of the first papers demonstrating a link between microRNAs and the UPR pathway. They show that microRNA-346 is a transcriptional target of XBP-1
• Behrman, S, Acosta-Alvear D, Walter P (2011) A CHOP-regulated microRNA controls rhodopsin expression. J Cell Biol 192(6):919–927. This study demonstrates that microRNA-708 is a direct transcriptional target of CHOP, and shows a prosurvival role for CHOP under ER stress conditions by limiting the syntehsis of the ER folded protein rhodopsin
Chitnis NS et al (2012) miR-211 is a prosurvival microRNA that regulates chop expression in a PERK-dependent manner. Mol Cell 48(3):353–364
Belmont PJ et al (2012) Regulation of microRNA expression in the heart by the ATF6 branch of the ER stress response. J Mol Cell Cardiol 52(5):1176–1182
Zhang WG et al (2013) Mmu-miR-702 functions as an anti-apoptotic mirtron by mediating ATF6 inhibition in mice. Gene 531(2):235–242
Doroudgar S et al (2009) Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J Biol Chem 284(43):29735–29745
Wu X et al (2007) Hypoxic preconditioning induces delayed cardioprotection through p38 MAPK-mediated calreticulin upregulation. Shock 27(5):572–577
Dai BH et al (2013) microRNA-199a-5p protects hepatocytes from bile acid-induced sustained endoplasmic reticulum stress. Cell Death Dis 4:e604
• Byrd AE, Aragon IV, Brewer JW (2012) microRNA-30c-2* limits expression of proadaptive factor XBP1 in the unfolded protein response. J Cell Biol 196(6):689–698. In one of the earlier papers in the field the authors show that the PERK branch of the UPR induces microRNA-30c-2p, which represses XBP-1 expression, thus regulating the IRE1α branch of the UPR
Lerner AG et al (2012) IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab 16(2):250–264
• Upton JP et al (2012) IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 338(6108):818–822. This landmark paper describes that the endoribonuclease activity of IRE1α can directly cleave microRNAs
Upton JP et al (2008) Caspase-2 cleavage of BID is a critical apoptotic signal downstream of endoplasmic reticulum stress. Mol Cell Biol 28(12):3943–3951
Sandow JJ et al (2014) ER stress does not cause upregulation and activation of caspase-2 to initiate apoptosis. Cell Death Differ 21(3):475–480
Acknowledgments
This work was supported by NIH Grant DK97178 (to HM). The author is grateful to Ms. Courtney Hoover for her excellent secretarial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Malhi, H. MicroRNAs in ER Stress: Divergent Roles in Cell Fate Decisions. Curr Pathobiol Rep 2, 117–122 (2014). https://doi.org/10.1007/s40139-014-0046-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40139-014-0046-y