
Abstract
Pathophysiology of sporadic Alzheimer’s disease (SAD) and familial Alzheimer’s disease (FAD) remains poorly known, including the exact role of neuroglia and specifically astroglia, in part because studies of astrocytes in human Alzheimer’s disease (AD) brain samples are scarce. As far as we know, this is the first study of a 3-D immunohistochemical and microstructural analysis of glial fibrillary acidic protein (GFAP)- and glutamine synthetase (GS)-positive astrocytes performed in the entorhinal cortex (EC) of human SAD and FAD samples. In this study, we report prominent atrophic changes in GFAP and GS astrocytes in the EC of both SAD and FAD characterised by a decrease in area and volume when compared with non-demented control samples (ND). Furthermore, we did not find neither astrocytic loss nor astrocyte proliferation or hypertrophy (gliosis). In contrast with the astrogliosis classically accepted hypothesis, our results show a highly marked astrocyte atrophy that could have a major relevance in AD pathological processes being fundamental and key for AD mnesic and cognitive alterations equivalent in both SAD and FAD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The pathophysiology of Alzheimer’s disease (AD) in both the sporadic (SAD) and familial (FAD) forms still remain ill-known. Indeed, genetics and a variety of environmental influences play a role in AD aetiology and cerebral amyloid angiopathy (Braak and Braak 1991; Rodríguez et al. 2016). FAD (5% AD) is characterised by mutations in the amyloid precursor protein (APP), presenilin 1 (PSN1) and presenilin 2 (PSN2) (Barber 2012), whereas SAD (95% of AD) is associated to mutations in thousands of genes (Barber 2012). On the other hand, astrocyte and microglial activation are also related with the pathogenesis of AD. In fact, astrogliosis is classically considered as the astrocytic reaction to AD pathology in human brain and animal models (Rodríguez et al. 2016; Nagele et al. 2003).
The distinctive cellular events in SAD and FAD are amyloid beta peptide (Aβ) accumulation, Tau neurofibrillary tangles and their hyperphosphorylation, which triggers the so-called neurotoxic cascade (Karran et al. 2011) neuroinflammation and neuronal death (Selkoe and Hardy 2016). On the other hand, extracellular Aβ induces astrogliosis, and microstructural analysis shows that astrocytes are found in the core of the amyloid plaque and its surroundings, affecting the physiology of astrocytes and impairing cognition (Rodríguez et al. 2016).
However, our group has shown that, in the triple transgenic mouse model of AD (3xTg-AD), a generalised astroglia atrophy exists with just a restricted astrogliosis in the vicinity of Aβ plaques, in addition to glutamate glial metabolism and trophic factors alterations in the hippocampus (HPC), but not in the entorhinal cortex (EC) and prefrontal cortex (PFC) showing a differential region behaviour that also implies the involvement of Glutamate transporter and receptors (Rodríguez et al. 2016).
In this context, the glutamate balance via the glutamate–glutamine (Glu–Gln) shuttle is critical for cognitive functions and excitotoxicity in AD being glutamine synthetase (GS) a key element in the Glu–Gln cycle without forgetting the (Glu–Gln-GABA metabolism cycle) fundamental for both excitatory and inhibitory transmission (Eid et al. 2012). Our group has reported a reduction of GS-immunoreactive astrocytes in two major cognitive areas of the HPC (DG and CA1) in 3xTg-AD. This reduction of GS-immunoreactive astrocytes was paralleled by a decrease in global GS suggesting alteration of glutamate balance (Olabarría et al. 2011). On the contrary, we did not find changes in GS-immunoreactive astrocytes or double-labelled GS/GFAP in the same animal model in the EC (Yeh et al. 2013).
Despite the 3xTg-AD we used in these previous studies shows similar distribution of Aβ and Tau alterations present in human AD and demonstrates synaptic dysfunction with impaired long-term potentiation (LTP; Oddo et al. 2003), the interpretations of these studies are limited since these are not performed in human pathological samples. In this work, we carried out a 3-D anatomical, immunohistochemical and microstructural analysis of glial fibrillary acidic protein (GFAP)- and glutamine synthetase (GS)-positive astrocytes performed in the entorhinal cortex (EC) study using post-mortem human brain samples of patients who suffered from SAD and FAD compared with ND. Our results show the relevance of astrocyte atrophy in human AD which are equivalent to the observed in animal models, suggesting that in the human pathology, astrocyte atrophy is fundamental to the cognitive and mnesic alterations in early and late onset of AD contributing to neuronal damage and CNS dysfunction shown by cytoskeletal alterations—GFAP—and metabolic dysfunctions—GS—(Eid et al. 2012; Rodríguez et al. 2016, 2023). Therefore, we consider that development of therapeutic strategies treating both astrocytes and neurons would improve their altered connectivity, function together with synaptic functionality and interaction, including abnormal accumulation of Gln driving to potential astroglial toxicity through hyperammonemia (Eid et al. 2012). Thus, this innovative approach and potential treatment would hopefully reduce the deleterious effects of this devastating disease.
Material and methods
Human material
Post-mortem human AD brain samples ranging from 40 years of age till over 80 of both SAD (n = 8) and their equivalent non-demented controls (ND, n = 5) were obtained from the Netherlands Brain Bank, whereas FAD (n = 9) brain samples were obtained from the University of Antioquia, in Colombia (kindly provided by Profs. Francisco Lopera and Diego Sepulveda). All human samples were obtained from the Netherlands Brain Bank (NBB) and from the University of Antioquia, Colombia (UA), according to their guidelines and ethical committees’ approval. Briefly, all donors singed the Informed Consent form, gave permissions for post-mortem brain autopsies and for the use of their brain material and medical records for research purposes. The informed consent form of the NBB and UA meets all current legal and ethical requirements for brain autopsy, tissue storage and use of tissue and clinical data for scientific research worldwide (https://www.brainbank.nl/).
All samples were fixed by immersion in a solution of 4% paraformaldehyde (Sigma, Germany) and 0.1 M phosphate buffer (PB) pH 7.4. Then, they were cut into 40–50-μm-thick coronal sections using a vibrating microtome (MICROM HM 650 V, Thermo Scientific, USA). Free-floating brain sections in 0.1 mM PB, pH 7.4 were collected and stored in a cryoprotectant solution containing 25% sucrose and 3.5% glycerol in 0.05 M PB at pH 7.4.
Double immunofluorescence for human paraffin-embedded sections
Sections were incubated in 30% methanol and 3% hydrogen peroxide in 0.1 M PB at pH 7.4 for endogenous peroxidase inactivation, following by incubation in 0.3 M glycine in 0.1 M PB for autofluorescence elimination. Once rinsed with 0.1 M PB, tissue slides were treated with 1% sodium borohydride in 0.1 mM PB to remove the excess of aldehyde groups and subsequently, blocked in 0.5% bovine serum albumin in 0.1 M Tris Saline (TS) with 1% Triton-X pH 7.6. Then, they were incubated during 48 h at room temperature (RT) in the primary single and/or antibodies cocktail (Rabbit anti-GFAP, 1:10,000, Sigma and Mouse anti-GS, 1:2000, Millipore). After that, sections were incubated during 2 h at RT in the corresponding secondary antibodies (Goat anti-Rabbit AlexaFluor 594 and Goat anti-Mouse AlexaFluor 48, 1:400, Invitrogen). Finally, sections were rinsed with 0.1 mM PB and cover slipped using Vectashield.
Tri-dimensional reconstruction of astrocytic morphology and population count
Single GFAP-, single GS- and GFAP/GS-immunopositive astrocytes of the entorhinal cortex were imaged using a confocal microscope (Leica TCS STED CW SP8 microscope), at least 35 per sample. Parallel confocal planes were superimposed and morphological analysis was carried out by Cell Analyst (Chvátal et al. 2007) using digital filters (average 3 × 3, convolution, gauss 5 × 5) to determine the surface area (S) and volume (V) of the GFAP, GS and GFAP/GS-stained astrocytes.
To determine whether the changes in the PFC GFAP-positive astrocytes, GS and GFAP/GS cytoskeletal surface area and volume are linked with changes in the number of astrocytes expressing these markers, we determined the numerical density (Nv, Number of cells/mm3) of astrocyte population in at least three representative non-consecutive sections, analysing an area of 600,000 μm2 in coronal sections of 40 μm thickness, thus representing a total volume of 24,000,000 μm3 per section (Olabarría et al. 2011; Yeh et al. 2013). GFAP-, GS- and GFAP-positive astrocytes were intensively labelled against a dark background that made them easy to identify with an equal chance of being counted. The number of GFAP-, GS- and GFAP-positive astrocytes was determined and quantified blindly on fluorescent microscope images by a single observer to reduce counting bias to a minimum.
Statistical analysis
One-way Analysis of Variance (ANOVA) with the post hoc Tukey test was performed to examine the significant alterations in the morphometric parameters of the different astroglial populations between groups. Data are expressed as mean ± SEM. Significance was accepted at p < 0.05. Initially, we also compared the potential statistical differences in the age difference onset of SAD patients 65–75 years and 75 years onwards). As there was no difference, we amalgamate all cases as a unique group.
Results
Changes in GFAP-positive astrocytes morphology in SAD and FAD compared to ND human brain samples
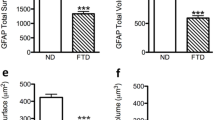
GFAP-positive astrocytes analysis of FAD and SAD human entorhinal cortex revealed differences of total surface area and volume, processes surface area and volume as well as somata surface and volume compared to control ND human EC slides [F3,386 = 14.23, 16.17, 27.51, 26.64, 11.55, 12.84, respectively; p < 0.001] (Fig. 1, Table 1). When compared ND to SAD, we found a decrease of 38.29% of the total GFAP surface area (1956.0 ± 111.5 µm2 vs. 1207 ± 63.43 µm2; p < 0.001; Fig. 1d). Similar decrease of surface area was observed when comparing ND with FAD samples (26.43%; 1956.0 ± 111.5 µm2 vs. 1439 ± 80.63 µm2; p < 0.001; Fig. 1d). Accordingly, the total volume of GFAP-positive astrocytes was significantly reduced when comparing ND to SAD with a 45% decrease (950.9 ± 62.94 µm3 vs. 523.0 ± 29.69 µm3; p = 0.001; Fig. 1e) or when comparing ND to FAD (29.53%; 950.9 ± 62.94 µm3 vs. 670.1 ± 45.01 µm3, p < 0.001; Fig. 1e). The somata surface also showed a significant 40.65% decrease when comparing ND to SAD (422.9 ± 18.48 µm2 vs. 251 ± 10.42 µm2; p < 0.001; Fig. 1f) or a non-significant change when comparing ND to FAD (422. 9 ± 18.48 µm2 vs. 379.1 ± 18,20 µm2; p > 0,05; Fig. 1f); despite the evident tendency to decrease as shown by a 10–15% reduction. On the other hand, somata volume showed a significant reduction of 51.63% when comparing ND to SAD (254.7 ± 16.56 µm3 vs. 123.2 ± 6,37 µm3; p < 0.001; Fig. 1g) and non-significant changes when comparing ND to FAD (254.7 ± 16.56 µm3 vs. 212,1 ± 13.18 µm3; p > 0.05; Fig. 1g) notwithstanding the evident tendency to decrease as shown by a 10–15% reduction. We also find an extremely interesting difference between SAD and FAD, since both somata surface and volume were also reduced in SAD compared to FAD (251 ± 10.42 µm2 vs 379.1 ± 18,20 µm2; p < 0,001; 123.2 ± 6,37 µm3 vs 212,1 ± 13.18 µm3; p < 0.001, respectively).

Confocal micrographs illustrating GFAP immunoreactive astrocytes cytoskeleton in the human entorhinal cortex of ND subjects (a), SAD (b) and FAD (c). Bar graphs showing the astrocytic cytoskeleton total surface (d), total volume (e), somata surface (f), somata volume (g), processes surface (h) and processes volume (i). Bars represent means ± SEM (***p ≤ 0.001, comparing SAD or FAD with ND and p ≤ 0.001, comparing SAD with FAD)
A detailed analysis of the GFAP-positive astrocytic processes demonstrated a reduction of both surface and volume of these processes. When compared ND to SAD, we found a decrease of 37.63% of processes’ surface area (1533 ± 98.07 µm2 vs. 956.2 ± 56.10 µm2; p < 0.001; Fig. 1h). Similarly, a reduction was found in processes’ surface area when comparing NAD to FAD (30.86%; 1533 ± 98.07 µm2 vs. 1060.0 ± 67.60 µm2, p < 0.001; Fig. 1h). The volume of GFAP-positive processes was also reduced. When compared ND to SAD, we found a decrease of 42.56% of the total processes’ volume (696.2 ± 49.17 µm3 vs. 399.8 ± 24.90 µm;3 p < 0.001; Fig. 1i). Similar decrease of processes’ volume was also observed when comparing ND with FAD samples (34.2%; 696.2 ± 49.17 µm3 vs. 458.1 ± 33.88 µm3; p < 0.001; Fig. 1 i), whilst the changes were equivalent when compared both SAD and FAD (Fig. 1h and i).
Changes in GS-positive astrocytes morphology in SAD and FAD compared to ND human brain samples
GS-positive astrocytes analysis of FAD and SAD human EC samples revealed a loss of total surface area and volume as well as processes surface area and volume compared to control ND human EC slides [F3,364 = 28.22, 38.20, 29.05, 47.91, 26.01, 33.55, respectively; p < 0.001] (Fig. 2, Table 2). When compared ND to SAD, we found a decrease of 39.0% of the total surface area (2077.0 ± 132.1 µm2 vs. 1267.0 ± 66.97 µm2; p < 0.001; Fig. 2d). Similar decrease of surface area was observed when comparing ND with FAD samples (43.62%; 2077.0 ± 132.1 µm2 vs. 1171.0 ± 97.31 µm2 p < 0.001; Fig. 2d). Accordingly, the total volume of GS-positive astrocytes was significantly reduced when comparing ND to SAD with a 41.22% decrease (1078.0 ± 81.70 µm3 vs. 633.7 ± 36.51 µm3; p < 0.001; Fig. 2e) or when comparing ND to FAD (58.87%; 1078.0 ± 81.70 µm3 vs. 443.4 ± 36.61 µm3; p < 0.001; Fig. 2e). We also found differences between SAD and FAD total volume (633.7 ± 36.51 µm3 vs 443.4 ± 36.61 µm3; p < 0.05; Fig. 2e). The somata surface also showed a significant 29.15% decrease when comparing ND to SAD (452.5 ± 17.82 vs. 320.6 ± 11.66; p < 0.001; Fig. 2f) and a decrease of 38.92% when comparing ND to FAD (452.5 ± 17.82 µm2 vs. 276.4 ± 14 µm2; p < 0.001; Fig. 2f). On the other hand, somata volume showed a significant reduction of 39.08% when comparing ND to SAD (299.1 ± 15.94 µm3 vs. 182.2 ± 8.98 µm3; p < 0.001; Fig. 2g) and a decrease of a 58.48% when comparing ND to FAD (299.1 ± 15.94 µm3 vs. 124.2 ± 7.89 µm3; p < 0.001; Fig. 2g). When compared the total volume of GS astrocytes and their somata, we observe a further reduction in SAD when compared to FAD (182.2 ± 8.98 µm3 vs 124.2 ± 7.89 µm3; p < 0,001; Fig. 2 g). A detailed analysis of the GS-positive astrocytic processes demonstrated a reduction of both surface and volume of these processes. When compared ND to SAD, we found a decrease of 41.75% of processes’ surface area (1625.0 ± 119.6 µm2 vs. 946.6 ± 59.94 µm2; p < 0.001; Fig. 2h). Equally, we found a reduction in processes surface area when comparing SAD to FAD (44.94%; 1625.0 ± 119.6 µm2 vs. 894.7 ± 88.0 µm2; p < 0.001; Fig. 2h). As indicated, the volume of GS-positive processes was also reduced. When compared ND to SAD, we found a decrease of 42.06% of the total processes volume (779.2 ± 68.36 µm2 vs. 451.5 ± 29.85 µm2; p < 0.001; Figure 2i). Similar decrease of processes volume was observed when comparing ND with FAD samples (59.04%; 779.2 ± 68.36 µm2 vs. 319.2 ± 31.68 µm2; p = 0.001; Fig. 2i).

Confocal micrographs illustrating GS-immunoreactive astrocytes morphology in the human entorhinal cortex of ND subjects (a), SAD (b) and FAD (c). Bar graphs showing the astrocytic total surface. (d), total volume (e), somata surface (f), somata volume (g) processes surface (h) and processes volume (i) Bars represent means ± SEM (***p ≤ 0.001, comparing SAD or FAD with ND and ⋅p ≤ 0.05, p ≤ 0.001, comparing SAD with FAD)
Changes in double GFAP/GS-positive astrocytes morphology in SAD and FAD compared to ND human brain samples
Furthermore, we analysed the changes in the co-localised astrocytic populations of GFAP/ GS within the EC of the different AD subtypes and ND patients (Fig. 3, Table 3). When compared ND to SAD, we found no changes in the total GFAP/GS surface area (1704.0 ± 180.6 µm2 vs. 1267.0 ± 125.8 µm2; p > 0.05; Fig. 3j) nor when compared ND with FAD (1704.0 ± 180.6 µm2 vs. 1337.0 ± 117.1 µm2; Fig. 3j). On the other hand, the total volume of GFAP/GS-positive astrocytes was significantly reduced when comparing ND to SAD with a 37.88% decrease (850.1 ± 99.78 µm3 vs. 528.1 ± 59.23 µm3; p < 0.05; Fig. 3k) but non-significant changes were observed when comparing ND to FAD (850.1 ± 99.78 µm3 vs. 627.5 ± 61.18 µm3, p > 0.05; Fig. 3k) independently of the reduction. When compared the total volume of GFAP/GS, we observe a reduction in SAD when compared to ND (528.1 ± 59.23 µm3 vs 850.1 ± 99.78 µm3; p < 0.05; Fig. 3k). The somata surface of GFAP/GS-positive astrocytes showed a significant 40.35% decrease when comparing ND to SAD (441.1 ± 31.13 µm2 vs. 263.1 ± 27.29 µm2; p < 0.001; Fig. 3l) or non-significant differences when comparing ND to FAD (441.1 ± 31.13 µm2 vs. 382.7 ± 23.18 µm2; p > 0.05; Fig. 3l). On the other hand, somata volume showed a significant reduction of 56.41% when comparing ND to SAD (273.2 ± 29.49 µm3 vs. 119.1 ± 14.94 µm3; p < 0.001; Fig. 3m) and non-significant differences when comparing ND to FAD (273.2 ± 29.49 µm3 vs. 221.1 ± 19.63 µm3; p > 0.05; Fig. 3m). When compared the somata surface and volume of GFAP/GS, we observed a reduction in SAD when compared to FAD (263.1 ± 27.29 µm2 vs 382.7 ± 23.18 µm2; 119.1 ± 14.94 µm3 vs 221.1 ± 19.63 µm3; p < 0,05; Fig. 3l and m).

Dual confocal images (GFAP in red, GS in green and co-localisation of GFAP/GS in yellow) within the human entorhinal cortex of ND subjects (a, d and g), SAD (b, e and h) and FAD (c, f and i). Bar graphs showing the astrocytic total surface (j), total volume (k), somata surface (l), somata volume (m) processes surface (n) and processes volume (o). Bars represent means ± SEM (*p ≤ 0.05, ***p ≤ 0.001, comparing SAD or FAD with ND and ⋅p ≤ 0.05, comparing SAD with FAD)
GFAP/GS-positive astrocytic processes surface and volume demonstrated non-significant differences between ND, SAD and FAD samples. When compared ND to SAD, we found no statistical differences in the surface area (1263.0 ± 156.4 µm2 vs. 1004.0 ± 107.1 µm2; p > 0.05; Fig. 3n). Similarly, non-significant changes were observed in processes surface area when comparing ND to FAD (1263.0 ± 156.4 µm2 vs. 954.1 ± 100.7 µm2, p > 0.05; Fig. 3n). The volume of GFAP/GS-positive processes also showed non-significant differences. When compared ND to SAD, we found non-significant changes in the total processes volume (576.9 ± 75.59 µm3 vs. 409.0 ± 48.15 µm3; p > 0.05; Fig. 3o). Similarly, non-significant differences in the processes volume were observed when comparing ND with FAD samples (576.9 ± 75.59 µm3 vs. 406.3 ± 45.71 µm3; p > 0.05; Fig. 3o).
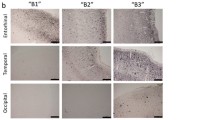
After performing and in-depth analysis and determination of the numerical density, it was evident that astrocytic population was equivalent in the different layers of the EC and did not show percentage changes of all three populations GFA, GS and dual-labelled GFAP/GS neither in ND nor in SAD and FAD (Fig. 4).

Confocal micrographs showing the different distribution of astrocytes labelled with GFAP (a, b, c), GS (d, e, f) and the co-localising cells (g, h, i) in the EC of ND subjects, SAD, FAD patient samples
Discussion
In this study, we show a prominent atrophy of SAD and FAD astrocytes in the EC demonstrated by the decrease in size and volume of GFAP- and GS-positive astrocytes and in co-expressing GFAP/GS astrocytes.
In AD, the EC is the first affected region before it spreads to other areas and is essential for memory and learning (Bevilaqua et al. 2008). Studies in animals have tried to elucidate the astrocyte modifications related with normal ageing and AD, since astrocytes are key for brain physiology and pathology. Studies in 3xTg-AD revealed morphological changes in astrocytes associated with age-dependent cognitive decline and AD pathogenesis showing both gliosis and atrophy in the HPC (Olabarría et al. 2011), whereas in EC and PFC, gliosis is minimal or inexistent (Kulijewicz-Nawrot et al. 2013). Moreover, astroglia atrophy is also accompanied by a glutamate homeostatic alteration by the glutamine (Glu-Gln) which also in consequence affects the Glutamine-Glutamate-GABA metabolic cycle as well as the shuttle (Eid et al. 2012; Andersen et al. 2022) fundamental to sustain neurotransmission and neurotransmitter recycling directly linked to astrocyte energy metabolism. Astrocytes in AD suffer important metabolic changes as we have clearly shown GS-immunoreactive astrocytes reduction in the HPC and PFC but not in the EC (Olabarría et al. 2011; Yeh et al. 2013; Kulijewicz-Nawrot et al. 2013). All these findings were found in the 3xTg-AD animal model (Olabarría et al. 2011; Yeh et al. 2013; Kulijewicz-Nawrot et al. 2013; Rodríguez et al. 2023). This atrophy of GS-immunoreactive astrocytes leads to glutamate and GABA maintenance impairing since both glutamate and GABA are generated via GS, compromising transmission in the EC circuitry and projections towards the HPC and PFC (Rodríguez et al. 2014). This is clearly evident with reduced activity, synaptic transmission and of course the regional homeostasis (Eid et al. 2012; Rodríguez et al. 2014, 2016; Andersen et al. 2022) and mitochondrial dysfunction (Albrecht et al. 2007). However, to our knowledge, it is unknown if the above described GFAP- and GS-positive atrophic alterations described in the 3xTg-AD are also present in human SAD or FAD. Thus, the purpose of this study was to show evidence of these rarely measured changes. We showed that the GFAP and GS atrophy appeared in all layers of the EC in either SAD and FAD but was never present in ND independent of the layer or age, on the contrary to what occurs in animal models (Yeh et al. 2013). GFAP atrophy appears as an important reduction of primary processes and a massive reduction of secondary and distal processes in the EC layers. This atrophy shown in both SAD and FAD in our knowledge is a novel observation. Since always has been considered the neuroinflammation processes associated with AD Pathology (Singh 2022). We consider that this has always been somehow biased since few in-depth studies have been performed till the present manuscript and even less comparing both SAD and FAD.
On the contrary to our observation, a variety of post-mortem studies have shown astrogliosis in the EC of AD human samples (Muramori et al. 1998; Porchet et al. 2003; Vanzani et al. 2005). Furthermore, microarray analysis also showed a decrease in gene transcription of astrocytic cytoskeleton proteins in AD, implying a down-regulation of astrocytic cytoskeleton (Simpson et al. 2011) consistent with atrophy. The spatiotemporal incidence of astroglia atrophy is consistent with the pathological hallmarks of AD. The changes in EC astrocytes are observed mainly within layers II, III, and IV at early onset; layer V was affected at middle onset, whilst at a very late onset, they are restricted layer VI as also described previously in patients by MRI in EC, neocortex and hippocampus (Pini et al. 2016). Nevertheless, it should be noted that there is neither recovery of atrophic astrocytes at any stage nor cell loss as indicated by the Nv (Fig. 4). As described previously in animal models, astroglial atrophy reduced synaptic coverage and decreased metabolic support to neurons (Heneka et al. 2010; Verkhratsky et al. 2010). EC, therefore, is important for cognitive functions and is homologous to the dorsolateral PFC in primates and humans (Ongür et al. 2000; Hoover et al. 2007; Milà-Alomà et al. 2020).
Astroglia releases trophic factors, sustains metabolic support, and extracellular ion buffering supporting neuronal connectivity (Rodríguez et al. 2023; Verkhratsky et al. 2010; Nedergaard et al. 2003; Lalo et al. 2011). Therefore, the SAD and FAD astroglial atrophy observed in EC may result in impaired synaptic connectivity and EC homeostasis (Verkhratsky et al. 2010). It is likely that astrocyte atrophy in EC might compromise its output to other areas, especially the HPC, resulting in loss of synapses and/or dendritic spines causing long-term potentiation (Oddo et al. 2003; Bertoni-Freddari et al. 2008; Noristani et al. 2011). We do not have to forget that this atrophy could be directly related with the oxidative stress via NADPH oxidase 4 (NOX4) impairing mitochondrial function (Park et al. 2021), affecting the morphology, functionality and metabolism, as well as glutamate and GABA production together with their dysregulation in neurodegenerative diseases (Verkhratsky and Nedergaard 2014; Milà-Alomà 2020; Sood et al. 2021). In addition, there is and evident morphological difference (size and shape) and functionality together with gene expression changes that, as mentioned above, could play a role in mitochondrial impairment (Park et al. 2021; Arranz et al. 2019). All the above points out the differences on GS affection in the EC of 3xTg-AD compared to the human EC suggesting different astrocyte functionality between the mice animal model EC and the human AD (Rodríguez et al. 2014; Arranz et al. 2019).
The severe atrophy observed in both SAD and FAD would also imply, in addition to astrocytic atrophy, an important Neurovascular Unit (NVU) alteration via the release of astrocyte extravascular vesicles (A-EVs) (González-Molina et al. 2021). In a recent study, SAD was modelled with human induced pluripotent stem cell (hiPSC)-derived organoids and treated with serum to replicate the blood brain breakdown in AD recapitulating Aβ and p-Tau accumulation and a reduced function of astrocytes (Chen et al. 2021). In a similar study, carried out with iPSC-derived astrocytes from FAD and SAD patients, astrocytes exhibited pathological and morphological changes (Jones et al. 2017) with no significant difference in the relative proportions of each cell type between the FAD and SAD astrocyte groups.
Surprisingly our study has demonstrated that despite the astrocyte atrophy, we do not have any reduction in the number of cells. In this regard, S100β is a trophic factor that marks the largest population of astrocytes (Ogata and Kosaka 2002; Araque et al. 1999) and occupy the entire extent of the EC (Rodríguez et al. 2023). S100β was overexpressed in brain tissue of patients with AD and Down syndrome (Griffin et al. 1989; Jørgensen et al. 1990; Sheng et al. 1994). All these findings are in agreement with our recent observations showing an increase in S100β expression in 3xTg-AD (Rodriguez et al. 2023) responding to the fact that there is not astrocytic death in the EC of AD patients, as shown in the present study.
Finally, the anatomical differences observed in this study amongst SAD and FAD can be explained by the fact that the onset of SAD is decades latter than the onset of FAD (Hunter et al. 2013), and therefore, the atrophy in FAD should be less prominent.
Considering all these new results in SAD and FAD, together with our previous results in the 3xTg-AD mouse model, we could state that AD is not only a neuropathological process, but also an early and chronic gliopathology affecting brain areas prior to neuronal alterations. Thus, therapeutic approaches targeting simultaneously glial and neural impairments might be of relevance for AD treatment.
Data availability
Enquiries about data availability should be directed to the corresponding author.
References
Albrecht J, Sonnewald U, Waagepetersen HS, Schousboe A (2007) Glutamine in the central nervous system: function and dysfunction. Front Biosci 12:332–343. https://doi.org/10.2741/2067
Andersen JV, Schousboe A, Verkhratsky A (2022) Astrocyte energy and neurotransmitter metabolism in Alzheimer’s disease: integration of the glutamate/GABA-glutamine cycle. Prog Neurobiol. https://doi.org/10.1016/j.pneurobio.2022.102331
Araque A, Parpura V, Sanzgiri RP, Haydon PG (1999) Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 22:208–215. https://doi.org/10.1016/s01662236(98)01349-6
Arranz AM, De Strooper B (2019) The role of astroglia in Alzheimer’s disease: pathophysiology and clinical implications. Lancet Neurol 18:406–414. https://doi.org/10.1016/S1474-4422(18)30490-3
Barber RC (2012) The Genetics of Alzheimer’s Disease. Scientifica. https://doi.org/10.6064/2012/246210
Bertoni-Freddari C, Sensi SL, Giorgetti B, Balietti M, Di Stefano G, Canzoniero LM, Casoli T, Fattoretti P (2008) Decreased presence of perforated synapses in a triple-transgenic mouse model of Alzheimer’s disease. Rejuvenation Res 11:309–313. https://doi.org/10.1089/rej.2008.0660
Bevilaqua LR, Rossato JI, Bonini JS, Myskiw JC, Clarke JR, Monteiro S, Lima RH, Medina JH, Cammarota M, Izquierdo I (2008) The role of the entorhinal cortex in extinction: Influences of aging. Neural Plast. https://doi.org/10.1155/2008/595282
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259. https://doi.org/10.1007/BF00308809
Chen X, Sun G, Tian E, Zhang M, Davtyan H, Beach TG, Reiman EM, Blurton-Jones M, Holtzman DM, Shi Y (2021) Modeling Sporadic Alzheimer’s Disease in Human Brain Organoids under Serum Exposure. Adv Sci (weinh). https://doi.org/10.1002/advs.202101462
Chvátal A, Anderová M, Hock M, Prajerová I, Neprasová H, Chvátal V, Kirchhoff F, Syková E (2007) Three-dimensional confocal morphometry reveals structural changes in astrocyte morphology in situ. J Neurosci Res 85:260–271. https://doi.org/10.1002/jnr.21113
Eid T, Behar K, Dhaher R, Bumanglag AV, Lee T-SW (2012) Roles of glutamine Synthetase inhibition in Epilepsy. Neurochem Res 37:20339–20350. https://doi.org/10.1007/s11064-012-0766-5
González-Molina LA, Villar-Vesga J, Henao-Restrepo J, Villegas A, Lopera F, CardonaGómez GP, Posada-Duque R (2021) Extracellular Vesicles From 3xTg-AD Mouse and Alzheimer’s Disease Patient Astrocytes Impair Neuroglial and Vascular Components. Front Aging Neurosci. https://doi.org/10.3389/fnagi.2021.593927
Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL 3rd, Araoz C (1989) Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA 86:7611–7615. https://doi.org/10.1073/pnas.86.19.7611
Heneka MT, Rodríguez JJ, Verkhratsky A (2010) Neuroglia in neurodegeneration. Brain Res Rev 63:189–211. https://doi.org/10.1016/j.brainresrev.2009.11.004
Hoover WB, Vertes RP (2007) Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct Funct 212:149–179. https://doi.org/10.1007/s00429-0070150-4
Hunter S, Arendt T, Brayne C (2013) The senencence hypothesis of sisease progression in Alzheimer disease: an integrated matrix of disease pathways for FAD and SAD. Mol Neurobiol 48:556–570. https://doi.org/10.1007/s12035-013-8445-3
Jones VC, Atkinson-Dell R, Verkhratsky A, Mohamet L (2017) Aberrant iPSC-derived human astrocytes in Alzheimer’s disease. Cell Death Dis. https://doi.org/10.1038/cddis.2017.89
Jørgensen OS, Brooksbank BW, Balázs R (1990) Neuronal plasticity and astrocytic reaction in Down syndrome and Alzheimer disease. J Neurol Sci 98:63–79. https://doi.org/10.1016/0022-510x(90)90182-m
Karran E, Mercken M, De Strooper B (2011) The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat Rev Drug Discov 10:698–712. https://doi.org/10.1038/nrd3505
Kulijewicz-Nawrot M, Syková E, Chvátal A, Verkhratsky A, Rodríguez JJ (2013) Astrocytes and glutamate homoeostasis in Alzheimer’s disease: a decrease in glutamine synthetase, but not in glutamate transporter-1, in the prefrontal cortex. ASN Neuro 5:273–282
Lalo U, Pankratov Y, Parpura V, Verkhratsky A (2011) Ionotropic receptors in neuronalastroglial signalling: what is the role of “excitable” molecules in non-excitable cells. Biochim Biophys Acta 1813:992–1002. https://doi.org/10.1016/j.bbamcr.2010.09.007
Milà-Alomà M, Salvadó G, Gispert JM, Vilor-Tejedor N, Grau-Rivera O, Sala-Vila A et al. (2020) Amyloid beta, tau, synaptic, neurodegeneration, and glial biomarkers in the preclinical stage of the Alzheimer’s continuum. 16: 1358–1371. https://doi.org/10.1002/alz12131
Muramori F, Kobayasi K, Nakamura I (1998) A quantitative study of neurofibrillary tangles, senile plaques and astrocytes in the hippocampal subdivisions and entorhinal cortex in Alzheimer’s disease, normal controls and non-Alzheimer neuropsychiatric diseases. Psychyatric Clin Neurosci 52:593–599. https://doi.org/10.1111/j.1440-1819.1998.tb02706.x
Nagele RG, D’Andrea MR, Lee H, Venkataraman V, Wang HY (2003) Astrocytes accumulate Aβ42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res 971:197–209. https://doi.org/10.1016/s0006-8993(03)02361-8
Nedergaard M, Ransom B, Goldman SA (2003) New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci 26:523–530. https://doi.org/10.1016/j.tins.2003.08.008
Noristani HN, Meadows RS, Olabarria M, Verkhratsky A, Rodríguez JJ (2011) Increased hippocampal CA1 density of serotonergic terminals in a triple transgenic mouse model of Alzheimer’s disease: an ultrastructural study. Cell Death Dis. https://doi.org/10.1038/cddis.2011.79
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer’s Disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron 39:409–421. https://doi.org/10.1016/s0896-6273(03)00434-3
Ogata K, Kosaka T (2002) Structural and quantitative analysis of astrocytes in the mouse hippocampus. Neuroscience 113:221–233. https://doi.org/10.1016/s0306-4522(02)00041-6
Olabarría M, Noristani HN, Verkhratsky A, Rodríguez JJ (2011) Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer’s disease mouse model: Mechanism for deficient glutamatergic transmission? Mol Neurodegener 6:1–9. https://doi.org/10.1186/1750-1326-6-55
Ongür D, Price JL (2000) The organization of networks within the orbital and medial prefrontal cortex of rats, monkeys and humans. Cereb Cortex 10:206–219. https://doi.org/10.1093/cercor/10.3.206
Park MW, Cha HW, Kim J, Kim JH, Yang H, Yoon S, Boonpraman N, Yi SS, Yoo ID, Moon J-S (2021) NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer’s diseases. Redox Biol. https://doi.org/10.1016/j.redox.2021.101947
Pini L, Pievani M, Bocchetta M, Altomare D, Bosco P, Cavedo E et al (2016) Brain atrophy in Alzheimer’s disease and aging. Ageing Res Rev 30:25–48. https://doi.org/10.1016/j.arr.2016.01.002
Porchet R, Probst A, Bouras C, Draberova E, Drabe P, Riederer PM (2003) Analysis of glial acidic fibrillary protein in the human entorhinal cortex during aging and in Alzheimer’s disease. Proteomics 3:1476–1485. https://doi.org/10.1002/pmic.200300456
Rodríguez JJ, Yeh CY, Terzieva S, Olabarria M, Kulijewicz-Nawrot M, Verkhratsky A (2014) Complex and region-specific changes in astroglial markers in the aging brain. Neurobiol Aging 35:15–23. https://doi.org/10.1016/j.neurobiolaging.2013.07.002
Rodríguez JJ, Butt AM, Gardenal E, Parpura V, Verkhratsky A (2016) Complex and differential glial responses in Alzheimer’s disease and ageing. Curr Alzheimer Res 13:343–358. https://doi.org/10.2174/1567205013666160229112911
Rodríguez JJ, Terzieva S, Yeh CY, Gardenal E, Zallo F, Verkhratsky A, Busquets X (2023) Neuroanatomical and morphometric study of S100β positive astrocytes in the entorhinal cortex during ageing in the 3xTg-Alzehimer’s disease mouse model. Neurosci Lett. https://doi.org/10.1016/j.neulet.2023.137167
Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8:595–608
Sheng JG, Mrak RE, Griffin WS (1994) S100 beta protein expression in Alzheimer disease: potential role in the pathogenesis of neuritic plaques. J Neurosci Res 39:398–404. https://doi.org/10.1002/jnr.490390406
Simpson JE, Ince PG, Shaw PJ, Heath PR, Raman R, Garwood CJ, Gelsthorpe C, Baxter L, Forster G, Matthews FE, Brayne C, Wharton SB (2011) Microarray analysis of the astrocyte transcriptome in the aging brain: relationship to Alzheimer’s pathology and APOE genotype. Neurobiol Aging 32:1795–1807. https://doi.org/10.1016/j.neurobiolaging.2011.04.013
Singh D (2022) Astrocytic and microglial cells as the modulators of neuroinflammation in Alzheimer’s disease. J Neuroinflammation 19:206. https://doi.org/10.1186/s12974-022-02565-0
Sood A, Preeti K, Fernandes V, Khatri DK, Singh SB (2021) Glia: a major player in glutamate-GABA dysregulation-mediated neurodegeneration. J Neurosci Res 99:3148–3189. https://doi.org/10.1002/jnr.2497
Vanzani MC, Iacono RF, Caccuri RL, Berria MI (2005) Immunochemical and morphometric features of astrocyte reactivity vs. plaque location in Alzheimer’s disease. Medicina 65:213–218
Verkhratsky A, Nedergaard M (2014) Astroglial cradle in the life of the synapse. Philos Trans R Soc Lond B Biol Sci 369:20130595. https://doi.org/10.1098/rstb.2013.0595
Verkhratsky A, Olabarria M, Noristani HN, Yeh CY, Rodriguez JJ (2010) Astrocytes in Alzheimer’s Disease. Neurotherapeutics 7:399–412. https://doi.org/10.1016/j.nurt.2010.05.017
Yeh CY, Verkhratsky A, Terzieva S, Rodríguez JJ (2013) Glutamine synthetase in astrocytes from entorhinal cortex of the triple transgenic animal model of Alzheimer’s disease is not affected by pathological progression. Biogerontology 14:777–787. https://doi.org/10.1007/s10522-013-9456-1
Acknowledgements
The authors acknowledge The Netherlands Brain Bank (NBB) for kindly providing the samples of sporadic Alzheimer’s disease (SAD) and the professors Francisco Lopera and Diego Sepulveda (University of Antioquia, Colombia) for kindly providing the samples of familial Alzheimer’s disease (FAD). This work was supported by the Grant Agency of the Czech Republic [GACR 309/09/1696 (to José J. Rodríguez)]; Instituto de Salud Carlos III (ISCIII) Subdirección General de Evaluación y Fomento de la investigación co-financed by FEDER [PI10/02738 (to José J. Rodríguez)]. The Government of the Basque Country [AE2010-1-28; AEGV10/16, GV2011111020 (to JJ Rodríguez)]; as well as by the Spanish Ministerio de Economía y Competitividad, RETOS Colaboración [RTC-2015-3542-1 co-financed by FEDER (to José J. Rodríguez and Xavier Busquets)].
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. The research was supported by the Grant Agency of the Czech Republic [GACR 309/09/1696 (to JJR)]; as well as by the Spanish Government Plan Nacional de I + D + I 2008–2011, and ISCIII Subdirección General de Evaluación y Fomento de la investigación co-financed by FEDER [PI10/02738 (to JJR)]. The Government of the Basque Country [AE-2010-1-28; AEGV10/16, GV2011111020 (to JJR)]; as well as by the Spanish Ministerio de Economía y Competitividad, RETOS Colaboración [RTC-2015–3542-1 co-financed by FEDER (to JJR and XB)] and by the Ministry of Italian University and Research (MIUR) to EG.
Author information
Authors and Affiliations
Contributions
J.J.R contributed to the study conception and design. Material preparation and data collection were performed by J.J.R, F.Z and E.G. Data analysis was performed by J.J.R, F.Z, E.G, J.C and X.B. The first draft of the manuscript was written by J.J.R and X.B and all the authors commented on previous versions of the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interests
All the authors declare no actual or potential conflicts of interest including any financial, personal or other relationship with other people or organisations that could inappropriately influence the present work. All human samples were obtained from the donor institutions according to the guidelines their ethical committees.
Ethics approval
All human samples were obtained from the Netherlands Brain Bank according to their guidelines and ethical committees approval. Briefly, all donors singed the Informed Consent form, gave permission for post-mortem brain autopsy and use of their brain material and medical records for research purposes. The informed consent form of the NBB meets all current legal and ethical requirements for brain autopsy, tissue storage and use of tissue and clinical data for scientific research worldwide. https://www.brainbank.nl/.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rodríguez, J.J., Zallo, F., Gardenal, E. et al. Prominent and conspicuous astrocyte atrophy in human sporadic and familial Alzheimer’s disease. Brain Struct Funct 228, 2103–2113 (2023). https://doi.org/10.1007/s00429-023-02707-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00429-023-02707-x