
Abstract
Background
STUB1 has been first associated with autosomal recessive (SCAR16, MIM# 615768) and later with dominant forms of ataxia (SCA48, MIM# 618093). Pathogenic variations in STUB1 are now considered a frequent cause of cerebellar ataxia.
Objective
We aimed to improve the clinical, radiological, and molecular delineation of SCAR16 and SCA48.
Methods
Retrospective collection of patients with SCAR16 or SCA48 diagnosed in three French genetic centers (Montpellier, Strasbourg and Nancy).
Results
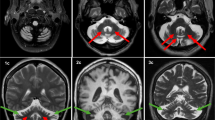
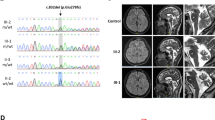
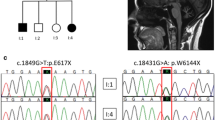
Here, we report four SCAR16 and nine SCA48 patients from two SCAR16 and five SCA48 unrelated French families. All presented with slowly progressive cerebellar ataxia. Additional findings included cognitive decline, dystonia, parkinsonism and swallowing difficulties. The age at onset was highly variable, ranging from 14 to 76 years. Brain MRI showed marked cerebellar atrophy in all patients. Phenotypic findings associated with STUB1 pathogenic variations cover a broad spectrum, ranging from isolated slowly progressive ataxia to severe encephalopathy, and include extrapyramidal features. We described five new pathogenic variations, two previously reported pathogenic variations, and two rare variants of unknown significance in association with STUB1-related disorders. We also report the first pathogenic variation associated with both dominant and recessive forms of inheritance (SCAR16 and SCA48).
Conclusion
Even though differences are observed between the recessive and dominant forms, it appears that a continuum exists between these two entities. While adding new symptoms associated with STUB1 pathogenic variations, we insist on the difficulty of genetic counselling in STUB1-related pathologies. Finally, we underscore the usefulness of DAT-scan as an additional clue for diagnosis.



Similar content being viewed by others
References
Marelli C et al (2016) Mini-exome coupled to read-depth based copy number variation analysis in patients with inherited ataxias. Hum Mutat 37(12):1340–1353
Renaud M et al (2018) Clinical, biomarker, and molecular delineations and genotype-phenotype correlations of ataxia with oculomotor apraxia Type 1. JAMA Neurol 75(4):495–502
Renaud M et al (2014) Autosomal recessive cerebellar ataxia type 3 due to ANO10 mutations: delineation and genotype-phenotype correlation study. JAMA Neurol 71(10):1305–1310
Anheim M, Tranchant C, Koenig M (2012) The autosomal recessive cerebellar ataxias. N Engl J Med 366(7):636–646
Renaud M et al (2017) A recessive ataxia diagnosis algorithm for the next generation sequencing era. Ann Neurol 82(6):892–899
Gebus O et al (2017) Deciphering the causes of sporadic late-onset cerebellar ataxias: a prospective study with implications for diagnostic work. J Neurol 264(6):1118–1126
Mallaret M et al (2016) Validation of a clinical practice-based algorithm for the diagnosis of autosomal recessive cerebellar ataxias based on NGS identified cases. J Neurol 263(7):1314–1322
Coutelier M et al (2017) A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelopathies. Brain 140(6):1579–1594
Shi Y et al (2013) Identification of CHIP as a novel causative gene for autosomal recessive cerebellar ataxia. PLoS ONE 8(12):e81884
Min JN et al (2008) CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol Cell Biol 28(12):4018–4025
Hayer SN et al (2017) STUB1/CHIP mutations cause Gordon Holmes syndrome as part of a widespread multisystemic neurodegeneration: evidence from four novel mutations. Orphanet J Rare Dis 12(1):31
Genis D et al (2018) Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48). Neurology 91(21):e1988–e1998
De Michele G et al (2019) Spinocerebellar ataxia 48 presenting with ataxia associated with cognitive, psychiatric, and extrapyramidal features: a report of two Italian families. Parkinsonism Relat Disord 65:91–96
Palvadeau R et al (2020) Cerebellar cognitive-affective syndrome preceding ataxia associated with complex extrapyramidal features in a Turkish SCA48 family. Neurogenetics 21(1):51–58
Mol MO et al (2020) Clinical and pathologic phenotype of a large family with heterozygous STUB1 mutation. Neurol Genet 6(3):e417
Roux T et al (2020) Clinical, neuropathological, and genetic characterization of STUB1 variants in cerebellar ataxias: a frequent cause of predominant cognitive impairment. Genet Med
De Michele G et al (2020) Spinocerebellar ataxia type 48: last but not least. Neurol Sci 41(9):2423–2432
Anheim M et al (2010) Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: implications for clinical management. Neurogenetics 11(1):1–12
Schmitz-Hübsch T et al (2006) Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 66(11):1717–1720
Montaut S et al (2018) Assessment of a targeted gene panel for identification of genes associated with movement disorders. JAMA Neurol 75(10):1234–1245
Oussalah A et al (2020) Population and evolutionary genetics of the PAH locus to uncover overdominance and adaptive mechanisms in phenylketonuria: results from a multiethnic study. EBioMedicine 51:102623
Wiedemann A et al (2020) Mutations in MTHFR and POLG impaired activity of the mitochondrial respiratory chain in 46-year-old twins with spastic paraparesis. J Hum Genet 65(2):91–98
Renard E et al (2019) Exome sequencing of cases with neural tube defects identifies candidate genes involved in one-carbon/vitamin B12 metabolisms and Sonic Hedgehog pathway. Hum Genet 138(7):703–713
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16):e164
Chiu HH et al (2020) Clinical and genetic characterization of autosomal recessive spinocerebellar ataxia type 16 (SCAR16) in Taiwan. Cerebellum 19(4):544–549
Synofzik M et al (2014) Phenotype and frequency of STUB1 mutations: next-generation screenings in Caucasian ataxia and spastic paraplegia cohorts. Orphanet J Rare Dis 9:57
Depondt C et al (2014) Autosomal recessive cerebellar ataxia of adult onset due to STUB1 mutations 82(19):1749–1750
Heimdal K et al (2014) STUB1 mutations in autosomal recessive ataxias—evidence for mutation-specific clinical heterogeneity. Orphanet J Rare Dis 9:146
Bettencourt C et al (2015) Clinical and neuropathological features of spastic ataxia in a spanish family with novel compound heterozygous mutations in STUB1. Cerebellum 14(3):378–381
Gazulla J et al (2018) Inaugural cognitive decline, late disease onset and novel STUB1 variants in SCAR16. Neurol Sci 39(12):2231–2233
Kawarai T et al (2016) Choreoathetosis, dystonia, and myoclonus in 3 siblings with autosomal recessive spinocerebellar ataxia Type 16. JAMA Neurol 73(7):888–890
Madrigal SC et al (2019) Changes in protein function underlie the disease spectrum in patients with CHIP mutations. J Biol Chem 294(50):19236–19245
Lieto M et al (2020) The complex phenotype of spinocerebellar ataxia type 48 in eight unrelated Italian families. Eur J Neurol 27(3):498–505
Cocozza S, Santorelli FM, De Michele G (2020) STUB1-related ataxias: a challenging diagnosis. Mov Disord Clin Pract 7(6):733–734
Chen DH et al (2020) Heterozygous STUB1 missense variants cause ataxia, cognitive decline, and STUB1 mislocalization. Neurol Genet 6(2):1–13
Nicita F et al (2019) Heterozygous missense variants of SPTBN2 are a frequent cause of congenital cerebellar ataxia. Clin Genet 96(2):169–175
Nemani T et al (2020) KIF1A-related disorders in children: a wide spectrum of central and peripheral nervous system involvement. J Peripher Nerv Syst 25(2):117–124
Coarelli G et al (2019) Loss of paraplegin drives spasticity rather than ataxia in a cohort of 241 patients with SPG7. Neurology 92(23):e2679–e2690
Acknowledgements
We wish to thank David Baux and Olivier Ardouin for bioinformatics support, Karine Choquet for careful reading and Véronique Braun and Antoine Verger for fruitful discussion.
Funding
This work was in part supported by a grant from the French “Connaitre les Syndromes Cerebelleux” (CSC) association.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical approval
The study was approved by the local ethics committee.
Consent to participate
Written informed consent was obtained from all individuals contributing a blood sample for molecular investigations and the local ethics committees approved the study. A distinct consent form was also signed if videos were recorded.
Consent for publication
The final manuscript has been read and approved by all authors who accepted full responsibility for the design and undertaking of the original article. They had access to the data and controlled the decision to publish.
Supplementary Information
Below is the link to the electronic supplementary material.
415_2020_10348_MOESM1_ESM.png
Figure S1. Electro-encephalogram anomalies. (A) Patient 1. (1) EEE during wakefulness. Slow background activity around 6Hz, bilaterally. (2) Sharp waves with sometimes triphasic aspect over bilateral suprasylvian areas, mostly on the right side. (3) Periods of several minutes of rhythmic theta activity (4hz) in bilateral frontotemporal areas (4) At the photic stimulation, short EEG activation with appearance of a rhythmic theta background activity (6hz) in the posterior electrodes followed by a delta range slow activity over frontotemporal areas. (B) Patient 2. Right posterior temporo-parieto-occipital fast epileptiform discharge, involving in a second step the entire right hemisphere, then followed by contralateral involvement with fast epileptiform discharge in the left frontocentral and posterior temporoparietal occipital areas. At the end of the ictal discharge, presence of diffuse bilateral slowing (PNG 2330 kb)
415_2020_10348_MOESM2_ESM.png
Figure S2. DAT-scan showing dopaminergic impairment. (A) Patient 4. Moderate impairment of predominantly presynaptic dopaminergic pathways (B) Patient 5. Diffuse impairment of presynaptic dopaminergic pathways. (C) Patient 10. Marked dopaminergic alteration. (PNG 3071 kb)
Video 1 to 3. SCAR16 patient (Patient 1). Video 1: Moderate, but still intelligible, cerebellar dysarthria. Moderate head tremor. (MP4 4012 kb)
Video 2: Cerebellar oculomotor abnormalities: saccadic pursuit and horizontal nystagmus. (MP4 6129 kb)
Video 3: Dystonia of the upper limbs (mainly hands and fingers). Dysmetria during the nose-finger test. (MP4 10483 kb)
Video 4. SCA48 patient (Patient 6). Patient confined to wheelchair, unable to walk, even with support. Extrapyramidal signs: rigidity, akinesia. Rest and action tremor on upper limbs. (MP4 39560 kb)
Video 5. SCA48 patient (Patient 6). Patient confined to wheelchair, unable to walk, even with support. Extrapyramidal signs: rigidity, akinesia. Rest and action tremor on upper limbs. (MP4 10007 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Ravel, JM., Benkirane, M., Calmels, N. et al. Expanding the clinical spectrum of STIP1 homology and U-box containing protein 1-associated ataxia. J Neurol 268, 1927–1937 (2021). https://doi.org/10.1007/s00415-020-10348-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-020-10348-x