
Abstract
Rheumatoid arthritis (RA) is a chronic autoimmune disease affecting 0.5–1% of the population worldwide. The disease has a heterogeneous character, including clinical subsets of anti-citrullinated protein antibody (ACPA)-positive and APCA-negative disease. Although the pathogenesis of RA is poorly understood, progress has been made in identifying genetic factors that contribute to the disease. The most important genetic risk factor for RA is found in the human leukocyte antigen (HLA) locus. In particular, the HLA molecules carrying the amino acid sequence QKRAA, QRRAA, or RRRAA at positions 70–74 of the DRβ1 chain are associated with the disease. The HLA molecules carrying these “shared epitope” sequences only predispose for ACPA-positive disease. More than two decades after the discovery of HLA-DRB1 as a genetic risk factor, the second genetic risk factor for RA was identified in 2003. The introduction of new techniques, such as methods to perform genome-wide association has led to the identification of more than 20 additional genetic risk factors within the last 4 years, with most of these factors being located near genes implicated in immunological pathways. These findings underscore the role of the immune system in RA pathogenesis and may provide valuable insight into the specific pathways that cause RA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis
Rheumatoid arthritis (RA) is characterized by the chronic inflammation of synovial joints, and affects 0.5–1% of the adult population worldwide (Silman and Pearson 2002). This inflammation can affect any synovial-lined diarthrodial joint (Imboden 2009), although, small joints of the hands and feet are most commonly affected. Larger joints like the shoulder and the knee can also be involved, but this varies according to the individual. Radiographic joint damage accumulates at a constant rate over time when left untreated (Wolfe and Sharp 1998), and erosion of the joint surface eventually causes joint deformity and loss of function, which is associated with significant disability and premature mortality (Majithia and Geraci 2007; Silman and Pearson 2002). The definition of RA is phenotypic and was developed on the basis of a consensus procedure by clinical experts, resulting in the 1987 and 2010 Rheumatoid Arthritis Classification Criteria (Aletaha et al. 2010; Arnett et al. 1988).
Clinical subsets of ACPA-positive and ACPA-negative RA patients
The clinical course of RA is extremely variable, showing a wide spectrum of clinical manifestations ranging from mild and self-limiting disease to rapidly progressive inflammation, joint destruction, and severe physical disability (Lee and Weinblatt 2001). The heterogeneous character of the disease results in differential responses to a range of therapies, including anti-tumor necrosis factor (anti-TNF) therapy (Plenge and Criswell 2008). A sub-classification of RA could be helpful to predict clinical outcomes of therapies. One way to sub-classify the phenotype RA could be achieved on the basis of serologic factors, such as RA-associated autoantibodies rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPA). RF is not unique to RA, whereas ACPA are highly specific for RA. ACPA are antibodies directed against citrullinated proteins, while citrullination is a physiologic process occurring under different conditions including inflammation. Citrullination is established by the conversion of arginine into citrullin by peptidylarginine deiminase (PAD) (Makrygiannakis et al. 2006). ACPA are predictive for RA; 75% of the patients with ACPA who presented with undifferentiated arthritis will progress to RA within 3 years of follow-up (van Gaalen et al. 2004a). Furthermore, patients with ACPA-positive RA have a more aggressive clinical course as compared to patients without ACPA (van Gaalen et al. 2004b). ACPA can also induce and aggravate arthritis in mice, suggesting that ACPA is involved in disease pathogenesis in humans (Kuhn et al. 2006; Uysal et al. 2009).
Genetics in RA
The prevalence of RA in the general population is <1%; however, among siblings, the prevalence increases to 2–4% (Seldin et al. 1999). In addition, in cross-selectional twin studies, concordance rates for RA were found between 12.3% and 15.4% for monozygotic twins, compared to 3.5% for dizygotic twins (MacGregor et al. 2000). These sibling and twin pair studies demonstrate that genetic factors have a substantial impact on susceptibility to RA resulting in an estimated genetic contribution to RA ranging from 50% to 60% (MacGregor et al. 2000; Seldin et al. 1999).
HLA and the shared epitope hypothesis
The most important genetic risk factor for RA is the HLA locus, which accounts for 30% to 50% of overall genetic susceptibility to RA (Bowes and Barton 2008; Imboden 2009). In 1969, it was demonstrated that in mixed lymphocyte cultures, peripheral blood lymphocytes from patients with RA were non-reactive to peripheral blood lymphocytes from other RA patients (Astorga and Williams 1969). Seven years later, in 1976, it was discovered that this non-reactivity was due to the sharing of genes in the HLA region (Stastny 1976), indicating a contribution of the HLA region to RA. Stastny noted in 1978 that 78% of Caucasian RA patients were HLA-DRw4 positive compared to 28% of healthy controls (Stastny 1978). A decade later, it was discovered that multiple RA risk alleles within the HLA-DRB1 gene share a conserved amino acid sequence (Gregersen et al. 1986a, b). This led to a new theory: the “shared epitope” (SE) hypothesis (Gregersen et al. 1987). Positions 70–74 in the third hypervariable region of the DRβ1 chain of the RA-associated HLA-DRB1 molecules all contain the conserved amino acids QKRAA, QRRAA, or RRRAA. This sequence of amino acids is called the SE, and the risk alleles carrying this sequence are widely known as SE alleles. These specific amino acid sequences were not only shared between HLA-DRB1*04 alleles but also between the RA-associated HLA-DRB1*01 alleles (Gregersen et al. 1987). The OR for one SE allele is 4.37, whereas two copies of HLA-DRB1 SE alleles have an OR of 11.79 (Huizinga et al. 2005). Surprisingly, the association between SE-encoding HLA-DRB1 alleles and RA was only observed for ACPA-positive disease (Huizinga et al. 2005). Apparently, ACPA-positive and ACPA-negative RA have different genetic risk profiles (De Rycke et al. 2004; Imboden 2009; van der Helm-van Mil and Huizinga 2008), indicating that not only from clinical and pathogenic perspective but also from genetic perspective, ACPA-positive versus ACPA-negative RA represents two forms of RA.
Putative mechanisms behind the HLA SE in ACPA-positive RA
Although it has been known for more than 30 years that the HLA locus carries the most prominent genetic association for RA, the underlying mechanism by which particular HLA-DRB1 alleles predispose to the development of ACPA-positive RA is still not understood, despite progress in understanding the structure and function of HLA-DRB1 molecules. The HLA-DR molecule is a heterodimer consisting of an alpha (DRA) and a beta chain (DRB), both anchored in the membrane of antigen-presenting cells. The function of HLA-DR molecules is to present antigenic peptides to T lymphocytes. For efficient antigen presentation to T cells, the T cell receptor recognizes residues from both the peptide as well as the HLA-DR molecule itself. The part of HLA-DR that binds to the peptide, the peptide-binding groove, is composed of two α-helical walls and a floor of β-pleated sheet (Brown et al. 1993). The SE is situated in the α-helix wall of the peptide-binding groove (Gregersen et al. 1987). In this position, the SE influences peptide binding to the HLA molecule as well as T cell presentation (Fig. 1). Therefore, it has been postulated that the SE motif itself is directly involved in the pathogenesis of RA by allowing the presentation of an arthritogenic peptide to T cells (Gregersen et al. 1987; van der Helm-van Mil et al. 2007). Unfortunately, this hypothesis has not yet been confirmed by the identification of specific arthritogenic peptides that bind to SE HLA-DR molecules in RA, although it has been reported that citrullinated peptides bind to HLA-SE molecules for presentation to T cells (Feitsma et al. 2010; Hill et al. 2003). T cells that recognize citrullinated antigen presented by HLA could subsequently help B cells to secrete ACPA. However, several alternative hypotheses regarding SE have been proposed that could also explain the contribution of the HLA locus to RA. These hypotheses include the idea that the HLA-SE molecules shape the T cell repertoire to permit escape from tolerance or survival of autoreactive clones, or serve as a target for autoreactive T cells owing to molecular mimicry with a pathogen, and even fail to bind an arthritogenic peptide, leading to an inadequate tolerant immune response (Firestein 2003). In ACPA-positive RA patients, SE alleles have been shown to influence ACPA specificity by predisposing to the development of antibodies against citrullinated vimentin but not to the development of antibodies against citrullinated fibrinogen. This indicates that SE alleles act as classic immune response genes in the ACPA response because they influence both the magnitude and the specificity of this RA-specific antibody response (Verpoort et al. 2007). Because of the heterogeneous nature of RA, it is suggested that in addition to genetic factors, environmental factors might be of importance as well for the development of RA (Firestein 2003). Smoking has been established as a relevant environmental risk factor for ACPA-positive disease, especially in subjects carrying one or two copies of the HLA-DRB1 SE (Kallberg et al. 2011; Linn-Rasker et al. 2006). The association of ACPA-positive RA and smoking could be explained by the observation of citrullinated proteins in the lungs of smokers, which are possibly well presented by HLA with SE (Makrygiannakis et al. 2008).

The HLA-DR4 molecule with the position of the SE sequence. The crystal structure of HLA-DRB1*04:01/DRA1*01:01 complexed with a human collagen II-derived peptide (dark grey circles) shows the peptide-binding groove created by the helical structures of the HLA-DR alpha and beta chain with (indicated in red) the SE at peptide-binding pocket 4 (Polyview-3D)
Genetic risk factors in addition to HLA
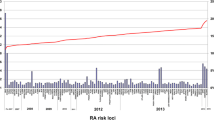
Almost 30 years after the identification of HLA alleles as a risk factor for RA, an RA-associated gene outside the HLA region was recognized. It was not until 2003, however, that the gene peptidylarginine deiminase type 4 (PADI4) was identified in a Japanese population as a second risk factor for RA (Suzuki et al. 2003). Interestingly, PADI genes encode the enzymes to change arginine into citrulline, the target of ACPA. The discovery of PADI4 as a risk factor was followed by the identification of protein tyrosine phosphatase non-receptor type 22 (PTPN22) in 2004 (Begovich et al. 2004; Carlton et al. 2005; Gregersen 2005). After 2004, the identification of new genetic risk factors underwent an unexpected acceleration. One year later, in 2005, CTLA4 was found during a candidate gene study (Plenge et al. 2005). In 2007, by means of a candidate gene approach (Kurreeman et al. 2007), a novel genetic risk factor was identified in the 9q33 region of the genome containing TRAF1/C5; it was also detected concurrently in a genome-wide study (Plenge et al. 2007b). Two more additional risk loci were discovered in 2007, the signal transducer and activator of transcription (STAT4) gene region on chromosome 2q, following a mapping of genes under a linkage peak (Remmers et al. 2007), as well as single-nucleotide polymorphisms (SNPs) near the gene TNF-α induced protein 3 (TNFAIP3), following genome-wide association studies (Plenge et al. 2007a; Thomson et al. 2007). The application of this latter technique allowed the identification of new risk alleles for RA to take enormous steps forward (Plenge 2009). In 2008, another seven RA risk loci genes, of which six were identified in a meta-analysis of three genome-wide association study (GWAS) (Barton et al. 2008; Raychaudhuri et al. 2008) were detected, with the most significant finding localized in the CD40 gene region. At the beginning of 2011, more than 30 established RA risk loci were identified (Chen et al. 2011; Gregersen et al. 2009; Raychaudhuri et al. 2009; Stahl et al. 2010; Zhernakova et al. 2011) (Fig. 2).

Susceptibility genes for RA (Barton et al. 2008; Begovich et al. 2004; Chen et al. 2011; Gregersen et al. 1987, 2009; Kurreeman et al. 2007; Plenge et al. 2005, 2007a, b; Raychaudhuri et al. 2008, 2009; Remmers et al. 2007; Stahl et al. 2010; Stastny 1978; Suzuki et al. 2003; Thomson et al. 2007; Zhernakova et al. 2011)
GWAS studies rapidly expanded the number of SNPs associated with ACPA-positive RA; however, this gave rise to the question as to what could we learn from these associations and whether we could use these SNPs to better understand RA pathogenesis. Most of the identified SNPs are located near genes that are linked to immunological pathways, implicating—not surprisingly—immune-related events as an important component of RA. The associated genes are often linked to T or B cell activation or to differentiation and cytokine signaling (Zhernakova et al. 2009). Some of the associated genes—for instance, PADI4—are only found to be associated with RA. This directly implicates differences in citrullination as a mechanism important for RA development. Other genetic associations are shared between different autoimmune diseases (Zhernakova et al. 2009). PTPN22, for example, is linked to RA, systemic lupus erythematosus, type 1 diabetes, and other autoimmune diseases (Bottini et al. 2004; Jawaheer et al. 2003; Kyogoku et al. 2004). PTPN22 encodes a tyrosine phosphatase and is hypothesized to affect the threshold for B and T cell receptor signaling (Gregersen 2005). For many of the associated genes, the immunological mechanism explaining the genetic association is difficult to demonstrate. For most of the associated loci, the functional SNP is still unknown. Identifying the functional SNP is important for determining the effect of the functional variation on gene expression and function, while another complicating factor is the fact that most of the proteins expressed by the associated genes play multiple important roles in the immune system and affect different immunological pathways. For example, genetic variants near CD40 have been shown to be associated with RA (Raychaudhuri et al. 2008). CD40 is a member of the TNF-receptor family and is expressed on many different immune cells including B cells, macrophages, and dendritic cells. It is important not only for the priming of T cells and activation of B cells but also for the activation of macophages and dendritic cells. Furthermore, the expression of CD40 on RA synovial cells was hypothesized to contribute to joint destruction (Peters et al. 2009). Dissecting these different pathways in a human system to study which pathways are important for RA pathogenesis is a challenge, as it will be difficult to show which CD40-expressing cell associated with certain CD40 genotypes is primarily responsible for the enhanced risk to develop RA. Like CD40, most of the identified genes play complex roles in the immune system, indicating that even if it is elucidated as to how a certain genetic variant influences gene expression or function, it is still not known how it contributes to disease. Thus, although the number of SNPs associated with RA is rapidly expanding, the challenge for the future will be to unravel the mechanisms by which the proteins expressed by these genes contribute to RA pathogenesis.
Genetic risk factors for ACPA-negative RA
Many HLA-DRB1 alleles are described as risk factors for ACPA-positive RA. For ACPA-negative disease, the situation is clearly different, as only HLA-DR3 predisposes to ACPA-negative RA (Irigoyen et al. 2005; Verpoort et al. 2005). Another risk factor for ACPA-negative RA—interferon regulatory factor 5 (Sigurdsson et al. 2007)—has been described, although this association could not be replicated in other study populations (Garnier et al. 2007; Rueda et al. 2006). Recently, it has been suggested that neuropeptide S receptor gene polymorphisms might be implicated in ACPA-negative RA susceptibility and its clinical manifestations (D’Amato et al. 2010). Although many more genetic risk factors have thus far been described for ACPA-positive RA compared to ACPA-negative RA, this does not imply that genetic factors contribute more to ACPA-positive RA. A recent study showed that the heritability of RA among twin pairs for ACPA-positive RA is 68% and for ACPA-negative RA it is 66% (van der Woude et al. 2009). This suggests that the heritability of ACPA-positive and ACPA-negative disease is comparable, although for ACPA-negative disease, the genetic risk factors remain to be identified since cohorts used for genetic risk factors mainly consist of ACPA-positive RA patients.
Protection against the development of RA
Protection against RA is predominantly associated with HLA-DRB1*13:01
In addition to HLA-DRB1 alleles that contribute to RA susceptibility, other HLA-DRB1 alleles confer protection against the disease. These protective HLA-DRB1 alleles are more often present in healthy controls compared to RA patients and have been categorized according to several models. One of the classifications put forward in the literature postulates that protection against RA is conferred by the DERAA sequence at position 70–74 of the HLA-DRB1 allele, which is at the same position on the HLA-DRB1 alleles as the SE sequence. Likewise, other models suggest that protection is conferred by isoleucine (I) at position 67 (I67) (de Vries et al. 2002) or propose an association mainly with aspartic acid (D) at position 70 (D70) (del Ricon and Escalante 1999; Mattey et al. 2001; Reviron et al. 2001). Data from a recent study did not support the hypothesis that I67 confers protection against RA; in this study, only the presence of D70 appeared to confer some degree of protection (Morgan et al. 2008). Due to conflicting results, a meta-analysis was performed involving four European populations with >2,800 patients and >3,000 control subjects to investigate thoroughly which HLA-DRB1 alleles were associated with protection against ACPA-positive RA and ACPA-negative RA. To correct for skewing due to this association, the analysis for ACPA-positive RA was stratified for the SE alleles. Interestingly, this study showed that the only association that conveyed protection against ACPA-positive RA after stratification of SE involved HLA-DRB1*13 alleles. The protective effect of other classifications, including DERAA and D70, was no longer present after the exclusion of HLA-DRB1*13. This indicates that HLA-DRB1*13 rather than DERAA, D70, or I67 is associated with protection. Among the DRB1*13 alleles, only DRB1*13:01 appeared to be associated with protection. For ACPA-negative RA, no associations with protective HLA-DRB1 alleles exist (van der Woude et al. 2010).
Non-inherited maternal antigen
In 1954, a biological effect of non-inherited maternal antigen (NIMA) was reported for the first time. It was demonstrated that rhesus D (RhD)-negative children were tolerant to the RhD antigen if their mother was RhD positive (Owen et al. 1954). In addition to the RhD antigen, HLA is also mentioned as NIMA in a study showing that haploidentical NIMA-mismatched sibling transplants have a graft survival rate similar to that of HLA identical siblings, whereas non-inherited paternal antigen (NIPA)-mismatched sibling transplants did as poorly as the recipients of maternal and paternal grafts (Burlingham et al. 1998). Recently, it was shown that HLA-DRB1 molecules that contain the amino acid sequence DERAA when present as NIMA also have a protective effect on the development of RA (Feitsma et al. 2007). By using a cohort of Dutch RA patients together with their parents, it was shown that the mothers of patients with RA had a significantly lower frequency (16.1%) of DERAA-containing HLA-DRB1 alleles than did the Dutch control population (29.3%). In contrast, the frequencies of DERAA-containing HLA-DRB1 alleles in fathers of the patients with RA (26.2%) were comparable to the frequencies of DERAA-containing HLA-DRB1 alleles in fathers of healthy controls. These findings were replicated in an English cohort (Feitsma et al. 2007). Due to the dominance of HLA-DRB1*13:01 in DERAA-positive HLA alleles, it is not known whether the NIMA effect derives from DERAA-containing HLA-DRB1 alleles or only from HLA-DRB1*13:01. If replicated further, the finding that certain HLA-alleles—when seen as NIMA—can protect offspring is highly interesting, as it suggests directions in which to manipulate the immune system in such a way that it will not be able to precipitate RA.
Conclusions: genetics and RA
The first genetic risk factor for RA was identified in 1978. After the discovery of this association between HLA and RA, it took 25 years until PADI4, the second genetic risk factor for RA, was identified. On the basis of new techniques, the discovery of new genes associated with a high risk of developing RA improved. Since 2004, more than 20 genes have been found to be implicated, mainly with ACPA-positive disease. The identification of disease-associated genes could provide valuable insight into the genetic variations prior to disease onset in order to identify the pathways important for RA pathogenesis. Future challenges will be, among others, the translation of genetic associations into biological pathways that are responsible for RA, as this knowledge may prove to be exceedingly useful for the invention of curative therapies for RA.
Abbreviations
- ACPA:
-
Anti-citrullinated protein antibody
- NIMA:
-
Non-inherited maternal antigen
- NIPA:
-
Non-inherited paternal antigen
- PAD:
-
Peptidylarginine deiminase
- PADI4:
-
Peptidylarginine deiminase type 4
- PTPN22:
-
Protein tyrosine phosphatase non-receptor type 22
- RA:
-
Rheumatoid arthritis
- RF:
-
Rheumatoid factor
- RhD:
-
Rhesus D
- SNP:
-
Single-nucleotide polymorphism
- STAT4:
-
Signal transducer and activator of transcription 4
- SE:
-
Shared epitope
- TNF:
-
Tumor necrosis factor
- TNFAIP3:
-
Tumor necrosis factor, alpha-induced protein 3
References
Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO III, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, Combe B, Costenbader KH, Dougados M, Emery P, Ferraccioli G, Hazes JM, Hobbs K, Huizinga TW, Kavanaugh A, Kay J, Kvien TK, Laing T, Mease P, Menard HA, Moreland LW, Naden RL, Pincus T, Smolen JS, Stanislawska-Biernat E, Symmons D, Tak PP, Upchurch KS, Vencovsky J, Wolfe F, Hawker G (2010) 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. Arthritis Rheum 62:2569–2581
Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS (1988) The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 31:315–324
Astorga GP, Williams RC Jr (1969) Altered reactivity in mixed lymphocyte culture of lymphocytes from patients with rheumatoid arthritis. Arthritis Rheum 12:547–554
Barton A, Thomson W, Ke X, Eyre S, Hinks A, Bowes J, Plant D, Gibbons LJ, Wilson AG, Bax DE, Morgan AW, Emery P, Steer S, Hocking L, Reid DM, Wordsworth P, Harrison P, Worthington J (2008) Rheumatoid arthritis susceptibility loci at chromosomes 10p15, 12q13 and 22q13. Nat Genet 40:1156–1159
Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang Q, Smith AM, Spoerke JM, Conn MT, Chang M, Chang SY, Saiki RK, Catanese JJ, Leong DU, Garcia VE, McAllister LB, Jeffery DA, Lee AT, Batliwalla F, Remmers E, Criswell LA, Seldin MF, Kastner DL, Amos CI, Sninsky JJ, Gregersen PK (2004) A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet 75:330–337
Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, MacMurray J, Meloni GF, Lucarelli P, Pellecchia M, Eisenbarth GS, Comings D, Mustelin T (2004) A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet 36:337–338
Bowes J, Barton A (2008) Recent advances in the genetics of RA susceptibility. Rheumatology (Oxford) 47:399–402
Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL, Wiley DC (1993) Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364:33–39
Burlingham WJ, Grailer AP, Heisey DM, Claas FH, Norman D, Mohanakumar T, Brennan DC, de Fijter H, van Gelder T, Pirsch JD, Sollinger HW, Bean MA (1998) The effect of tolerance to noninherited maternal HLA antigens on the survival of renal transplants from sibling donors. N Engl J Med 339:1657–1664
Carlton VE, Hu X, Chokkalingam AP, Schrodi SJ, Brandon R, Alexander HC, Chang M, Catanese JJ, Leong DU, Ardlie KG, Kastner DL, Seldin MF, Criswell LA, Gregersen PK, Beasley E, Thomson G, Amos CI, Begovich AB (2005) PTPN22 genetic variation: evidence for multiple variants associated with rheumatoid arthritis. Am J Hum Genet 77:567–581
Chen R, Stahl EA, Kurreeman FA, Gregersen PK, Siminovitch KA, Worthington J, Padyukov L, Raychaudhuri S, Plenge RM (2011) Fine mapping the TAGAP risk locus in rheumatoid arthritis. Genes Immun doi:10.1038/gene.2011.8
D’Amato M, Zucchelli M, Seddighzadeh M, Anedda F, Lindblad S, Kere J, Alfredsson L, Klareskog L, Padyukov L (2010) Analysis of neuropeptide S receptor gene (NPSR1) polymorphism in rheumatoid arthritis. PLoS ONE 5:e9315
De Rycke L, Peene I, Hoffman IE, Kruithof E, Union A, Meheus L, Lebeer K, Wyns B, Vincent C, Mielants H, Boullart L, Serre G, Veys EM, De Keyser F (2004) Rheumatoid factor and anticitrullinated protein antibodies in rheumatoid arthritis: diagnostic value, associations with radiological progression rate, and extra-articular manifestations. Ann Rheum Dis 63:1587–1593
De Vries N, Tijssen H, van Riel PL, van de Putte LB (2002) Reshaping the shared epitope hypothesis: HLA-associated risk for rheumatoid arthritis is encoded by amino acid substitutions at positions 67–74 of the HLA-DRB1 molecule. Arthritis Rheum 46:921–928
del Ricon I, Escalante A (1999) HLA-DRB1 alleles associated with susceptibility or resistance to rheumatoid arthritis, articular deformities, and disability in Mexican Americans. Arthritis Rheum 42:1329–1338
Feitsma AL, Worthington J, van der Helm-van Mil AH, Plant D, Thomson W, Ursum J, van Schaardenburg D, van der Horst-Bruinsma IE, van Rood JJ, Huizinga TW, Toes RE, de Vries RR (2007) Protective effect of noninherited maternal HLA-DR antigens on rheumatoid arthritis development. Proc Natl Acad Sci U S A 104:19966–19970
Feitsma AL, van der Voort EI, Franken KL, el Bannoudi H, Elferink BG, Drijfhout JW, Huizinga TW, de Vries RR, Toes RE, Ioan-Facsinay A (2010) Identification of citrullinated vimentin peptides as T cell epitopes in HLA-DR4-positive patients with rheumatoid arthritis. Arthritis Rheum 62:117–125
Firestein GS (2003) Evolving concepts of rheumatoid arthritis. Nature 423:356–361
Garnier S, Dieude P, Michou L, Barbet S, Tan A, Lasbleiz S, Bardin T, Prum B, Cornelis F (2007) IRF5 rs2004640-T allele, the new genetic factor for systemic lupus erythematosus, is not associated with rheumatoid arthritis. Ann Rheum Dis 66:828–831
Gregersen PK (2005) Gaining insight into PTPN22 and autoimmunity. Nat Genet 37:1300–1302
Gregersen PK, Moriuchi T, Karr RW, Obata F, Moriuchi J, Maccari J, Goldberg D, Winchester RJ, Silver J (1986a) Polymorphism of HLA-DR beta chains in DR4, −7, and −9 haplotypes: implications for the mechanisms of allelic variation. Proc Natl Acad Sci U S A 83:9149–9153
Gregersen PK, Shen M, Song QL, Merryman P, Degar S, Seki T, Maccari J, Goldberg D, Murphy H, Schwenzer J (1986b) Molecular diversity of HLA-DR4 haplotypes. Proc Natl Acad Sci U S A 83:2642–2646
Gregersen PK, Silver J, Winchester RJ (1987) The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum 30:1205–1213
Gregersen PK, Amos CI, Lee AT, Lu Y, Remmers EF, Kastner DL, Seldin MF, Criswell LA, Plenge RM, Holers VM, Mikuls TR, Sokka T, Moreland LW, Bridges SL Jr, Xie G, Begovich AB, Siminovitch KA (2009) REL, encoding a member of the NF-kappaB family of transcription factors, is a newly defined risk locus for rheumatoid arthritis. Nat Genet 41:820–823
Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E (2003) Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J Immunol 171:538–541
Huizinga TW, Amos CI, van der Helm-van Mil AH, Chen W, van Gaalen FA, Jawaheer D, Schreuder GM, Wener M, Breedveld FC, Ahmad N, Lum RF, de Vries RR, Gregersen PK, Toes RE, Criswell LA (2005) Refining the complex rheumatoid arthritis phenotype based on specificity of the HLA-DRB1 shared epitope for antibodies to citrullinated proteins. Arthritis Rheum 52:3433–3438
Imboden JB (2009) The immunopathogenesis of rheumatoid arthritis. Annu Rev Pathol 4:417–434
Irigoyen P, Lee AT, Wener MH, Li W, Kern M, Batliwalla F, Lum RF, Massarotti E, Weisman M, Bombardier C, Remmers EF, Kastner DL, Seldin MF, Criswell LA, Gregersen PK (2005) Regulation of anti-cyclic citrullinated peptide antibodies in rheumatoid arthritis: contrasting effects of HLA-DR3 and the shared epitope alleles. Arthritis Rheum 52:3813–3818
Jawaheer D, Seldin MF, Amos CI, Chen WV, Shigeta R, Etzel C, Damle A, Xiao X, Chen D, Lum RF, Monteiro J, Kern M, Criswell LA, Albani S, Nelson JL, Clegg DO, Pope R, Schroeder HW Jr, Bridges SL Jr, Pisetsky DS, Ward R, Kastner DL, Wilder RL, Pincus T, Callahan LF, Flemming D, Wener MH, Gregersen PK (2003) Screening the genome for rheumatoid arthritis susceptibility genes: a replication study and combined analysis of 512 multicase families. Arthritis Rheum 48:906–916
Kallberg H, Ding B, Padyukov L, Bengtsson C, Ronnelid J, Klareskog L, Alfredsson L (2011) Smoking is a major preventable risk factor for rheumatoid arthritis: estimations of risks after various exposures to cigarette smoke. Ann Rheum Dis 70:508–511
Kuhn KA, Kulik L, Tomooka B, Braschler KJ, Arend WP, Robinson WH, Holers VM (2006) Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J Clin Invest 116:961–973
Kurreeman FA, Padyukov L, Marques RB, Schrodi SJ, Seddighzadeh M, Stoeken-Rijsbergen G, van der Helm-van Mil AH, Allaart CF, Verduyn W, Houwing-Duistermaat J, Alfredsson L, Begovich AB, Klareskog L, Huizinga TW, Toes RE (2007) A candidate gene approach identifies the TRAF1/C5 region as a risk factor for rheumatoid arthritis. PLoS Med 4:e278
Kyogoku C, Langefeld CD, Ortmann WA, Lee A, Selby S, Carlton VE, Chang M, Ramos P, Baechler EC, Batliwalla FM, Novitzke J, Williams AH, Gillett C, Rodine P, Graham RR, Ardlie KG, Gaffney PM, Moser KL, Petri M, Begovich AB, Gregersen PK, Behrens TW (2004) Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am J Hum Genet 75:504–507
Lee DM, Weinblatt ME (2001) Rheumatoid arthritis. Lancet 358:903–911
Linn-Rasker SP, van der Helm-van Mil AH, van Gaalen FA, Kloppenburg M, de Vries RR, le Cessie S, Breedveld FC, Toes RE, Huizinga TW (2006) Smoking is a risk factor for anti-CCP antibodies only in rheumatoid arthritis patients who carry HLA-DRB1 shared epitope alleles. Ann Rheum Dis 65:366–371
MacGregor AJ, Snieder H, Rigby AS, Koskenvuo M, Kaprio J, Aho K, Silman AJ (2000) Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum 43:30–37
Majithia V, Geraci SA (2007) Rheumatoid arthritis: diagnosis and management. Am J Med 120:936–939
Makrygiannakis D, af Klint E, Lundberg IE, Lofberg R, Ulfgren AK, Klareskog L, Catrina AI (2006) Citrullination is an inflammation-dependent process. Ann Rheum Dis 65:1219–1222
Makrygiannakis D, Hermansson M, Ulfgren AK, Nicholas AP, Zendman AJ, Eklund A, Grunewald J, Skold CM, Klareskog L, Catrina AI (2008) Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann Rheum Dis 67:1488–1492
Mattey DL, Dawes PT, Gonzalez-Gay MA, Garcia-Porrua C, Thomson W, Hajeer AH, Ollier WE (2001) HLA-DRB1 alleles encoding an aspartic acid at position 70 protect against development of rheumatoid arthritis. J Rheumatol 28:232–239
Morgan AW, Haroon-Rashid L, Martin SG, Gooi HC, Worthington J, Thomson W, Barrett JH, Emery P (2008) The shared epitope hypothesis in rheumatoid arthritis: evaluation of alternative classification criteria in a large UK Caucasian cohort. Arthritis Rheum 58:1275–1283
Owen RD, Wood HR, Foord AG, Sturgeon P, Baldwin LG (1954) Evidence for actively acquired tolerance to Rh antigens. Proc Natl Acad Sci U S A 40:420–424
Peters AL, Stunz LL, Bishop GA (2009) CD40 and autoimmunity: the dark side of a great activator. Semin Immunol 21:293–300
Plenge RM (2009) Recent progress in rheumatoid arthritis genetics: one step towards improved patient care. Curr Opin Rheumatol 21:262–271
Plenge RM, Criswell LA (2008) Genetic variants that predict response to anti-tumor necrosis factor therapy in rheumatoid arthritis: current challenges and future directions. Curr Opin Rheumatol 20:145–152
Plenge RM, Padyukov L, Remmers EF, Purcell S, Lee AT, Karlson EW, Wolfe F, Kastner DL, Alfredsson L, Altshuler D, Gregersen PK, Klareskog L, Rioux JD (2005) Replication of putative candidate-gene associations with rheumatoid arthritis in >4,000 samples from North America and Sweden: association of susceptibility with PTPN22, CTLA4, and PADI4. Am J Hum Genet 77:1044–1060
Plenge RM, Cotsapas C, Davies L, Price AL, de Bakker PI, Maller J, Pe’er I, Burtt NP, Blumenstiel B, DeFelice M, Parkin M, Barry R, Winslow W, Healy C, Graham RR, Neale BM, Izmailova E, Roubenoff R, Parker AN, Glass R, Karlson EW, Maher N, Hafler DA, Lee DM, Seldin MF, Remmers EF, Lee AT, Padyukov L, Alfredsson L, Coblyn J, Weinblatt ME, Gabriel SB, Purcell S, Klareskog L, Gregersen PK, Shadick NA, Daly MJ, Altshuler D (2007a) Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet 39:1477–1482
Plenge RM, Seielstad M, Padyukov L, Lee AT, Remmers EF, Ding B, Liew A, Khalili H, Chandrasekaran A, Davies LR, Li W, Tan AK, Bonnard C, Ong RT, Thalamuthu A, Pettersson S, Liu C, Tian C, Chen WV, Carulli JP, Beckman EM, Altshuler D, Alfredsson L, Criswell LA, Amos CI, Seldin MF, Kastner DL, Klareskog L, Gregersen PK (2007b) TRAF1–C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med 357:1199–1209
Raychaudhuri S, Remmers EF, Lee AT, Hackett R, Guiducci C, Burtt NP, Gianniny L, Korman BD, Padyukov L, Kurreeman FA, Chang M, Catanese JJ, Ding B, Wong S, van der Helm-van Mil AH, Neale BM, Coblyn J, Cui J, Tak PP, Wolbink GJ, Crusius JB, van der Horst-Bruinsma IE, Criswell LA, Amos CI, Seldin MF, Kastner DL, Ardlie KG, Alfredsson L, Costenbader KH, Altshuler D, Huizinga TW, Shadick NA, Weinblatt ME, de Vries N, Worthington J, Seielstad M, Toes RE, Karlson EW, Begovich AB, Klareskog L, Gregersen PK, Daly MJ, Plenge RM (2008) Common variants at CD40 and other loci confer risk of rheumatoid arthritis. Nat Genet 40:1216–1223
Raychaudhuri S, Thomson BP, Remmers EF, Eyre S, Hinks A, Guiducci C, Catanese JJ, Xie G, Stahl EA, Chen R, Alfredsson L, Amos CI, Ardlie KG, Barton A, Bowes J, Burtt NP, Chang M, Coblyn J, Costenbader KH, Criswell LA, Crusius JB, Cui J, De Jager PL, Ding B, Emery P, Flynn E, Harrison P, Hocking LJ, Huizinga TW, Kastner DL, Ke X, Kurreeman FA, Lee AT, Liu X, Li Y, Martin P, Morgan AW, Padyukov L, Reid DM, Seielstad M, Seldin MF, Shadick NA, Steer S, Tak PP, Thomson W, van der Helm-van Mil AH, van der Horst-Bruinsma IE, Weinblatt ME, Wilson AG, Wolbink GJ, Wordsworth P, Altshuler D, Karlson EW, Toes RE, de Vries N, Begovich AB, Siminovitch KA, Worthington J, Klareskog L, Gregersen PK, Daly MJ, Plenge RM (2009) Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat Genet 41:1313–1318
Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, de Bakker PI, Le JM, Lee HS, Batliwalla F, Li W, Masters SL, Booty MG, Carulli JP, Padyukov L, Alfredsson L, Klareskog L, Chen WV, Amos CI, Criswell LA, Seldin MF, Kastner DL, Gregersen PK (2007) STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med 357:977–986
Reviron D, Perdriger A, Toussirot E, Wendling D, Balandraud N, Guis S, Semana G, Tiberghien P, Mercier P, Roudier J (2001) Influence of shared epitope-negative HLA-DRB1 alleles on genetic susceptibility to rheumatoid arthritis. Arthritis Rheum 44:535–540
Rueda B, Reddy MV, Gonzalez-Gay MA, Balsa A, Pascual-Salcedo D, Petersson IF, Eimon A, Paira S, Scherbarth HR, Pons-Estel BA, Gonzalez-Escribano MF, Alarcon-Riquelme ME, Martin J (2006) Analysis of IRF5 gene functional polymorphisms in rheumatoid arthritis. Arthritis Rheum 54:3815–3819
Seldin MF, Amos CI, Ward R, Gregersen PK (1999) The genetics revolution and the assault on rheumatoid arthritis. Arthritis Rheum 42:1071–1079
Sigurdsson S, Padyukov L, Kurreeman FA, Liljedahl U, Wiman AC, Alfredsson L, Toes R, Ronnelid J, Klareskog L, Huizinga TW, Alm G, Syvanen AC, Ronnblom L (2007) Association of a haplotype in the promoter region of the interferon regulatory factor 5 gene with rheumatoid arthritis. Arthritis Rheum 56:2202–2210
Silman AJ, Pearson JE (2002) Epidemiology and genetics of rheumatoid arthritis. Arthritis Res 4(Suppl 3):S265–S272
Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, Li Y, Kurreeman FA, Zhernakova A, Hinks A, Guiducci C, Chen R, Alfredsson L, Amos CI, Ardlie KG, Barton A, Bowes J, Brouwer E, Burtt NP, Catanese JJ, Coblyn J, Coenen MJ, Costenbader KH, Criswell LA, Crusius JB, Cui J, de Bakker PI, De Jager PL, Ding B, Emery P, Flynn E, Harrison P, Hocking LJ, Huizinga TW, Kastner DL, Ke X, Lee AT, Liu X, Martin P, Morgan AW, Padyukov L, Posthumus MD, Radstake TR, Reid DM, Seielstad M, Seldin MF, Shadick NA, Steer S, Tak PP, Thomson W, van der Helm-van Mil AH, van der Horst-Bruinsma IE, van der Schoot CE, van Riel PL, Weinblatt ME, Wilson AG, Wolbink GJ, Wordsworth BP, Wijmenga C, Karlson EW, Toes RE, de Vries N, Begovich AB, Worthington J, Siminovitch KA, Gregersen PK, Klareskog L, Plenge RM (2010) Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet 42:508–514
Stastny P (1976) Mixed lymphocyte cultures in rheumatoid arthritis. J Clin Invest 57:1148–1157
Stastny P (1978) Association of the B-cell alloantigen DRw4 with rheumatoid arthritis. N Engl J Med 298:869–871
Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, Nagasaki M, Nakayama-Hamada M, Kawaida R, Ono M, Ohtsuki M, Furukawa H, Yoshino S, Yukioka M, Tohma S, Matsubara T, Wakitani S, Teshima R, Nishioka Y, Sekine A, Iida A, Takahashi A, Tsunoda T, Nakamura Y, Yamamoto K (2003) Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet 34:395–402
Thomson W, Barton A, Ke X, Eyre S, Hinks A, Bowes J, Donn R, Symmons D, Hider S, Bruce IN, Wilson AG, Marinou I, Morgan A, Emery P, Carter A, Steer S, Hocking L, Reid DM, Wordsworth P, Harrison P, Strachan D, Worthington J (2007) Rheumatoid arthritis association at 6q23. Nat Genet 39:1431–1433
Uysal H, Bockermann R, Nandakumar KS, Sehnert B, Bajtner E, Engstrom A, Serre G, Burkhardt H, Thunnissen MM, Holmdahl R (2009) Structure and pathogenicity of antibodies specific for citrullinated collagen type II in experimental arthritis. J Exp Med 206:449–462
van der Helm-van Mil AH, Huizinga TW (2008) Advances in the genetics of rheumatoid arthritis point to subclassification into distinct disease subsets. Arthritis Res Ther 10:205
van der Helm-van Mil AH, Huizinga TW, de Vries RR, Toes RE (2007) Emerging patterns of risk factor make-up enable subclassification of rheumatoid arthritis. Arthritis Rheum 56:1728–1735
van der Woude D, Houwing-Duistermaat JJ, Toes RE, Huizinga TW, Thomson W, Worthington J, van der Helm-van Mil AH, de Vries RR (2009) Quantitative heritability of anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis. Arthritis Rheum 60:916–923
van der Woude D, Lie BA, Lundstrom E, Balsa A, Feitsma AL, Houwing-Duistermaat JJ, Verduijn W, Nordang GB, Alfredsson L, Klareskog L, Pascual-Salcedo D, Gonzalez-Gay MA, Lopez-Nevot MA, Valero F, Roep BO, Huizinga TW, Kvien TK, Martin J, Padyukov L, de Vries RR, Toes RE (2010) Protection against anti-citrullinated protein antibody-positive rheumatoid arthritis is predominantly associated with HLA-DRB1*1301: a meta-analysis of HLA-DRB1 associations with anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis in four European populations. Arthritis Rheum 62:1236–1245
van Gaalen FA, Linn-Rasker SP, van Venrooij WJ, de Jong BA, Breedveld FC, Verweij CL, Toes RE, Huizinga TW (2004a) Autoantibodies to cyclic citrullinated peptides predict progression to rheumatoid arthritis in patients with undifferentiated arthritis: a prospective cohort study. Arthritis Rheum 50:709–715
van Gaalen FA, van Aken J, Huizinga TW, Schreuder GM, Breedveld FC, Zanelli E, van Venrooij WJ, Verweij CL, Toes RE, de Vries RR (2004b) Association between HLA class II genes and autoantibodies to cyclic citrullinated peptides (CCPs) influences the severity of rheumatoid arthritis. Arthritis Rheum 50:2113–2121
Verpoort KN, van Gaalen FA, van der Helm-van Mil AH, Schreuder GM, Breedveld FC, Huizinga TW, de Vries RR, Toes RE (2005) Association of HLA-DR3 with anti-cyclic citrullinated peptide antibody-negative rheumatoid arthritis. Arthritis Rheum 52:3058–3062
Verpoort KN, Cheung K, Ioan-Facsinay A, van der Helm-van Mil AH, de Vries-Bouwstra JK, Allaart CF, Drijfhout JW, de Vries RR, Breedveld FC, Huizinga TW, Pruijn GJ, Toes RE (2007) Fine specificity of the anti-citrullinated protein antibody response is influenced by the shared epitope alleles. Arthritis Rheum 56:3949–3952
Wolfe F, Sharp JT (1998) Radiographic outcome of recent-onset rheumatoid arthritis: a 19-year study of radiographic progression. Arthritis Rheum 41:1571–1582
Zhernakova A, van Diemen CC, Wijmenga C (2009) Detecting shared pathogenesis from the shared genetics of immune-related diseases. Nat Rev Genet 10:43–55
Zhernakova A, Stahl EA, Trynka G, Raychaudhuri S, Festen EA, Franke L, Westra HJ, Fehrmann RS, Kurreeman FA, Thomson B, Gupta N, Romanos J, McManus R, Ryan AW, Turner G, Brouwer E, Posthumus MD, Remmers EF, Tucci F, Toes R, Grandone E, Mazzilli MC, Rybak A, Cukrowska B, Coenen MJ, Radstake TR, van Riel PL, Li Y, de Bakker PI, Gregersen PK, Worthington J, Siminovitch KA, Klareskog L, Huizinga TW, Wijmenga C, Plenge RM (2011) Meta-analysis of genome-wide association studies in celiac disease and rheumatoid arthritis identifies fourteen non-HLA shared loci. PLoS Genet 7:e1002004
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bax, M., van Heemst, J., Huizinga, T.W.J. et al. Genetics of rheumatoid arthritis: what have we learned?. Immunogenetics 63, 459–466 (2011). https://doi.org/10.1007/s00251-011-0528-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-011-0528-6