
Abstract
Regeneration of mature cells that produce functional insulin represents a major focus and a challenge of current diabetes research aimed at restoring beta cell mass in patients with most forms of diabetes, as well as in ageing. The capacity to adapt to diverse physiological states during life and the consequent ability to cope with increased metabolic demands in the normal regulation of glucose homeostasis is a distinctive feature of the endocrine pancreas in mammals. Both beta and alpha cells, and presumably other islet cells, are dynamically regulated via nutrient, neural and/or hormonal activation of growth factor signalling and the post-transcriptional modification of a variety of genes or via the microbiome to continually maintain a balance between regeneration (e.g. proliferation, neogenesis) and apoptosis. Here we review key regulators that determine islet cell mass at different ages in mammals. Understanding the chronobiology and the dynamics and age-dependent processes that regulate the relationship between the different cell types in the overall maintenance of an optimally functional islet cell mass could provide important insights into planning therapeutic approaches to counter and/or prevent the development of diabetes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
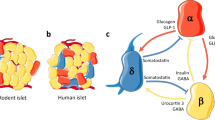
Approximately one million islets are distributed throughout the pancreas of a healthy adult human, and the combined mass of the islets is ∼2 g, which constitutes between 1% and 2% of the mass of the pancreas [1]. The islets, also termed ‘endocrine micro-organs’, comprise alpha, beta, delta, epsilon and pancreatic polypeptide hormone-producing cells. Among these, the beta cells are necessary for insulin production and the maintenance of glucose homeostasis. Current therapeutic approaches for both type 1 and type 2 diabetes are primarily focused on enhancing insulin secretion and/or insulin supplementation. Among the strategies that are continually evolving to preserve and/or replenish the functional beta cell mass, differentiation of embryonic and adult induced pluripotent stem cells in vitro [2] is appealing but is currently faced with limited efficiency in generating mature glucose-responsive beta cells. Other approaches include enhancing in vivo regeneration, either by improving resistance against destructive signals and apoptotic pathways or by careful and selective proliferation of existing beta cells, and differentiation and de-differentiation processes have recently been suggested to revive beta cells [3]. In favour of the former, several factors have been described as protecting beta cells in animal models, such as hepatocyte growth factor, gastric inhibitory polypeptide (GIP), insulin-like growth factor 1 (IGF-1), and prolactin. In terms of evidence for the latter, glucagon-like peptide 1 (GLP-1), liver-derived factors [4] and betatrophin (also termed angiopoietin-like 8, RIFL or lipasin) [5] have been reported to increase beta cell proliferation. While expansion of existing beta cells in vivo has mostly been explored in rodents [6, 7], limited data from obese or insulin-resistant humans also indicate a potential for islet mass expansion. Nevertheless, it continues to be a challenge to translate current approaches used to expand beta cell mass in rodents to human islet cells even in vitro [8]. Possible explanations for the resistance of human islet cells to proliferation include loss of expression of a key transcription factor(s) and increased expression of cell cycle inhibitors during senescence. It is notable that a renewed interest in the mechanisms that underlie defects in glucagon secretion in both type 1 and type 2 diabetes [9, 10] has prompted research into understanding the regulation of alpha cell mass and function [11]. Indeed, several reports suggest that appropriate in vitro and in vivo factors/signals can promote the transdifferentiation of alpha cells into beta cells. The notion that alpha cells represent an in vivo source of new beta cells, is supported by (1) data from Collombat et al [12], who demonstrated in mice that ectopic expression of Pax4 converts progenitor cells into insulin-producing beta-like cells; and (2) lineage-tracing evidence by Thorel and colleagues [13] for beta cell regeneration from glucagon-producing alpha cells in a transgenic mouse model of diphtheria-toxin-induced acute selective near-total beta cell ablation. Other cell types that have been suggested as sources of beta cells in rodents include ductal or acinar cells, intra-islet precursor cells and replication of pre-existing beta cells [14]. Studies in rodents clearly indicate that beta cell mass exhibits a remarkable plasticity to adapt, beginning in embryogenesis and continuing over a lifetime, largely in response to altered insulin demand [15]. In this review we will focus on the chronobiology of islet cell plasticity, particularly in the context of growth factor regulation of proliferation and cell death.
Prenatal and neonatal period
In rodents, the endocrine cell population undergoes a dramatic expansion beginning at embryonic day 14.5 followed by remodelling, during which there is regulated alteration in apoptosis and neogenesis and a progressive decrease in beta cell replication [16].
Insights into the normal development of the pancreas have emerged from observations made from late gestation throughout the neonatal period in a non-human primate model (see text box: ‘Age-dependent changes in islet morphology in non-human primates’). Pancreases from fetal/newborn baboons reveal that the islet architecture becomes more organised as gestational age advances. Thus, in early life, the total per cent of endocrine cell area, expressed as a percentage of total pancreas area, is greater than in adults, with no prevalence of any endocrine cell type. However, endocrine cells, as well as cells with mixed endocrine–exocrine lineage, were also detected in exocrine ductal and acinar cells in fetuses [17].
Age-dependent changes in islet morphology in non-human primates | |
Age | Changes in islet architecture |
Fetal period | • Islet architecture: insulin-positive cells surrounded by glucagon-positive cells • Margin between the endocrine and the exocrine tissue not always clearly defined • Islet architecture becomes organised as gestational age advances • A subpopulation of cells show both an endocrine and acinar phenotype • 2% of fetal cells have both glucagon immunoreactive alpha-type secretory granules and trypsin-positive zymogen granules in the cytoplasm • 1% of fetal cells contain both insulin-immunoreactive beta-type secretory granules and trypsin-positive zymogen granules in the cytoplasm • Co-localisation of glucagon and pancreatic polypeptide has been noted in islets of the vervet monkey (Chlorocebus pygerythrus) |
Newborn | • Islet architecture: glucagon-positive cells show peripheral localisation; insulin-positive cells are mainly in the islet core • ↑ Pancreas weight (greater than twofold) at 5 days after birth in baboons • ↑ Percentage of beta, alpha, and delta area: 15-, 5- and 94-fold higher, respectively, than in adult baboons |
Adulthood | • Islet architecture: glucagon-positive cells show prevalent peripheral localisation, while insulin-positive cells are mainly detected in the islet core • Modest overweight- and obesity-induced effects on relative beta cell volume • Being overweight induces an increase in alpha cell mass, which increases with the duration and severity of being overweight • IL-6 and TNF-α soluble receptors are positively correlated with body weight, suggesting that the induction of an abnormal activation of IL-6 and TNF-α signalling systems is associated with increased alpha cell mass in baboons. Evidence of islet amyloidosis in diabetic macaques, vervet monkeys and baboons, while type 2 diabetic monkeys show variable insulin staining, depending on the stage of disease development • No change in alpha cell mass in diabetic vervet monkeys • In adult non-human primates a decrement in fractional beta cell area of <50% or more leads to loss of glycaemic control • The islets of streptozotocin-induced diabetic rhesus monkeys (Macaca mulatta) have decreased numbers of beta cells and a relative increase in the proportion of alpha and delta cells |
Old age | • Islet amyloidosis plays a role in beta cell death in the Celebes crested macaque (Macaca nigra) and rhesus monkeys • Progressive islet amyloidosis is associated not only with increased beta cell apoptosis (reduced relative beta cell volume), but also with increased alpha cell replication and hypertrophy (increased relative alpha cell volume) in baboons |
The Notch pathway has been considered a major regulator during pancreas development in both humans and rodents [18]; cells in which the pathway is activated maintain their proliferative capacity, while those that do not express neurogenin 3 exit the cell cycle and differentiate into endocrine cells. Consistently, a mouse model with impaired Notch signalling exhibits increased endocrine cell differentiation at the expense of the pancreatic progenitor population [19]. In humans, a high rate of proliferation characterises late embryogenesis [20] and begins to decline postnatally [16, 21]. Specifically, proliferation of insulin-expressing cells occurs at relatively high rates during early development and decreases to low levels by 24 weeks [22, 23]. Analysing human pancreatic samples using Ki67+ insulin-expressing cells, Gregg et al [21] detected a high level of beta cell proliferation in neonates and in children of up to about 2 years of age that has been considered responsible for doubling human beta cells by 5 years of age to establish an organism’s beta cell mass.
Studies in genetic mouse models indicate the different regulators determine embryonic vs postnatal beta cell proliferation [24]. For example, D and E cyclin–cyclin-dependent kinase (CDK) complexes regulated by CDK inhibitors, including p27kip1 and p16cip1, play a critical role in progression of the cell cycle from G1 to S phase in the postnatal period [7]. Mouse models cyclin D2 or CDK4 knockout or overexpression of p27kip1 exhibit relatively normal islet structure, composition and mass in the neonatal stages, but manifest adult-onset diabetic phenotypes, suggesting that cell cycle regulators contribute to adaptation of islet function in the post-neonatal stage. Forkhead box protein (FOX)M1 has been investigated using a pancreas-specific knockout mouse. While whole pancreas deletion of FOXM1, a key cell cycle regulator of both G1/S progression and G2/M transition, does not affect embryonic pancreas development, it leads to impaired postnatal beta cell mass expansion via reduced proliferation. These studies suggest that cell cycle regulators are unlikely to be critical for endocrine progenitors and/or beta cell proliferation during embryogenesis, and that the mechanisms necessary for embryonic beta cell replication are likely to differ from those in adults [25].
In addition to regulating insulin secretion, the alpha cell hormone, glucagon, has been suggested to affect the development of beta cells [26] and regulate islet microcirculation [27]. Altered alpha cell function in mice, secondary to alpha cell-specific knockout of insulin receptors (alpha-IRKO), has been reported to either directly or indirectly influence beta cell proliferation in the mutants [28]. Lack of glucagon itself is reported to induce alpha cell hyperplasia [29]. Mice with disruption of prohormone convertase 2, which is essential for processing proglucagon into the mature hormone, glucagon, display increased proliferation of proglucagon cells in the perinatal period and a dramatic postnatal alpha cell hyperplasia and mild hypoglycaemia [30]. Notably, mice with alpha cell-specific mutated menin exhibit an increased frequency of glucagonomas, insulinomas and mixed islet tumours, arising from cells expressing the proglucagon gene; pointing to menin as an in vivo regulator of alpha cell plasticity, growth and transdifferentiation [31]. Glucagon-expressing cells can indeed transdifferentiate into insulin-expressing cells upon menin inactivation, suggesting that alpha cells are more plastic than previously appreciated. However, Wilcox et al [32] reported that ablation of the Arx gene in neonatal alpha cells results in loss of glucagon expression and conversion of this cell population to adopt an insulin-producing beta cell fate. The transcription factor encoded by the Arx gene, which is expressed in a subset of endocrine progenitors and restricted to alpha cells [33, 12], is involved in the specification and maintenance of alpha cell fate.
It is logical that adequate nutrition during embryogenesis is necessary to ensure appropriate development of beta cell mass. Nutritional insults that occur during early life may result in decreased beta cell mass and function that persist in adulthood to promote increased risk of glucose intolerance and type 2 diabetes [34]. Maternal restricted energy intake during parturition and the consequent increased levels of glucocorticoids [35] have been reported to decrease fetal beta cell mass as a result of reduced differentiation and decreased beta cell expression of the key transcription factors pancreatic and duodenal homeobox 1 (PDX1), paired box protein 4 (PAX4) and NK6 homeobox 1 (NKX6.1). A maternal diet low in protein content causes intrauterine growth retardation, and the newborn rodents exhibit underdeveloped beta cell mass owing to reduced proliferation and increased apoptosis associated with reduced levels of growth factors [36]. Protein restriction during pregnancy has also been associated with impaired beta cell replacement after streptozotocin treatment [37]. Maternal malnutrition can also alter the development of tissue vasculogenesis in the offspring, which may limit beta cell plasticity by disrupting the inductive signals from the microvasculature and an altered presence of endothelial progenitors [34]. Offspring of pregnant rats fed a low-protein diet during gestation exhibit a reduced capillary density in the islets [38]. In summary, nutritional insults can directly impact beta cell plasticity by effects on progenitors and the function of mature beta cells, and may also have indirect effects resulting in impaired trophic signalling between the vascular endothelium and beta cells [39].
Puberty and adolescence
The incidence of both type 1 and type 2 diabetes has increased over the past decade, and the increase in the latter has been attributed to obesity, insulin resistance and deficient beta cell function [40]. A pubertal increase in insulin resistance and an inability to mount an adequate beta cell insulin response results in hyperglycaemia. Intensive glycaemic regulation may allow for beta cell recovery in children [41] and is suggestive of plasticity of islets during this developmental stage.
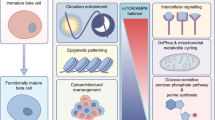
Hormonal changes related to puberty, along with race and genetic factors, has been recognised as a non-modifiable risk factor underlying the increased incidence of type 2 diabetes among adolescents [42, 43]. The transient insulin resistance of puberty [44] that is associated with a compensatory increase in insulin secretion [45] might accelerate progression of beta cell failure because of the additional stress on insulin-producing cells during crucial periods of growth and development, but the mechanism(s) contributing to the resistance has not been clearly determined. The hypothesis that reduced insulin sensitivity is driven by transient changes in growth hormone levels during puberty [46] is attractive, since growth hormone and IGF-1 levels are transiently higher in mid-puberty and mirror the changes in insulin sensitivity [47]. In addition, growth hormone-deficient children exhibit increased insulin sensitivity [48], and growth hormone is known to significantly affect beta cells [49]. Difficulty in designing approaches to easily and accurately measure beta cell mass in vivo has precluded a clear understanding of the precise changes that occur in the endocrine pancreas during the transition from childhood through to adulthood. Available data are derived from autopsy studies, including those by Butler and colleagues [50, 51], who measured pancreas volumes for 46 individuals aged from 2 weeks to 21 years using abdominal computerised tomography. These data indicate that beta cell mass expands by several fold from birth to adulthood, with the largest increment occurring prior to 2 years of age [21, 52]. This increase in islet mass is secondary to a greater number of beta cells per islet and increased islet size rather than an increase in the number of islets or alterations in apoptosis during the pancreatic remodelling phase. Thus, early childhood has been identified as the period with the most rapid expansion of beta cell numbers, while the rate of growth and the replication of existing beta cells decline from youth through to adulthood. A study using histological approaches reported beta cell neogenesis and a regenerative response in children and adolescents with type 1 diabetes [53]. In the same study, proliferation was detected in the remnant beta cell mass in those with new-onset type 1 diabetes but not in those with type 2 diabetes (Fig. 1).

Schematic illustration of age-dependent changes and regulators of islet cell morphology and function in humans. Because we do not have access to images of human islets, representative images of rodent islets at different ages are shown here. Insulin (red), Ki67 (green), DAPI (blue) immunoreactivity are shown in each picture. Arrow indicates Ki67+ beta cell. Magnification ×200
Adulthood
The presence of a functional islet mass that is optimal for the different stages of the lifespan of an organism depends on multiple regulatory processes. Under normal circumstances, islet mass is relatively stable, with the cells undergoing a slow renewal because of low levels of proliferation and apoptosis. Diverse regulatory factors include hormones, metabolites and growth factors which have all been proposed to modulate islet cell mass by direct or indirect effects. For example, growth factors including IGF-1 and IGF-2, insulin, platelet-derived growth factor (PDGF), epidermal growth factor (EGF) and others [54] exert a direct stimulatory effect on beta cell replication in vivo or in vitro [55].
Cell cycle regulation
As discussed in the ‘Prenatal and neonatal period’ section, several genetic mouse models of cell cycle dysregulation manifest an adult-onset diabetes phenotype, characterised by decreased beta cell mass via reduced proliferation. This suggests that mature beta cells are sensitive to perturbations in cell cycle control, and this is probably due to differential involvement of cell cycle regulators compared with most other cell types as exemplified by models lacking CDK6 [56] and/or redundant expression of cyclin D2 [57, 58]. Furthermore, overexpression of the inhibitor p27kip1 [59] in beta cells impairs proliferation, resulting in reduced beta cell mass. In contrast, global deletion of p27kip1 enhances beta cell proliferation, as also observed in genetic mouse models of insulin resistance (e.g. IRS2 KO, db/db) [59]. Beta cell hyperplasia is also observed in humans with a focal loss of heterozygosity of p57kip2, who suffer from hyperinsulinism of infancy [60].
Considering that mice with pancreas-specific knockout of FOXM1 leads to impaired alpha and beta cell proliferation and impaired beta cell mass regeneration after partial pancreatectomy, it is possible that FOXM1 is required for beta cell proliferation and hyperplasia of pre-existing beta cells following injury [61]. Recent studies point to FOXM1 being involved in the adaptive response of the beta cells to alterations in nutrients [62]. Thus, FOXM1 is critical for maintaining beta cell mass during adulthood by coordinating cell cycle progression. Another regulator of cell cycle genes is menin [63]; beta cell-specific deletion of menin leads to beta cell hyperplasia and insulinomas owing to increased proliferation secondary to reduced levels of p18 and p27kip1 [64].
Al-Hasani et al [65] recently reported that upon Pax4 misexpression, adult alpha cells are converted into insulin-producing cells, independent of their age, mutation state or microenvironment. They also report a duct cell to beta-like cell regeneration pathway that could be repeatedly activated by streptozotocin injury in an age-dependent manner. The newly formed beta cells are functional and able to repopulate and increase the size of islets (see text box: ‘Contributors to maintenance of islet cell mass in adult rodents’).
Contributors to maintenance of islet cell mass in adult rodents | ||
Regulators | Beta cell | Alpha cell |
Cell cycle regulation | ↑ in CDK6 KO, redundant expression of cyclin D2, and global deletion of p27kip1 ↓ in overexpression of p27kip1, pancreas-specific knockout mice for FOXM1, deletion of MEN1 Neogenesis from alpha cells and ducts in Pax4 misexpression | ↓ in pancreas-specific knockout mice for FOXM1 |
Insulin/IGF-1 and glucagon signalling | ↑ in alpha-IRKO ↓ in beta-IRKO, Akt1 KO, IRS1 and IRS2 KO, IGF-1 KO | ↑ in GRGR KO and alpha-IRKO/GR KO, GRHep KO ↓ with ageing in alpha-IRKO, pancreas-specific IRS2 KO |
Nutrients and gene expression modulation | ↑ by high-fat diet, high-protein diet, infusion of glucose in rats ↓ after chronic hyperglycaemia and glucotoxicity | ↑ by high-fat diet and high-protein diet ↓ in IL-6 KO mice |
Placental hormones and gut hormones | ↑ in PRLR KO mice ↑ in GIPR KO (islet size) = GLP1R KO mice, but reduced number of large islets and enhanced susceptibility to beta cell apoptosis | = GLP1R KO mice, but altered more islets with centrally located alpha cells |
microRNAs | ↑ miR-132, miR-184 and miR-338-3p in insulin resistance and obesity ↓ miR- 21, miR- 34a, miR- 199a-5p, miR-199a-3p, miR-203, miR-210 and miR-383 during compensation failure to insulin resistance | – |
Insulin/IGF-1 and glucagon signalling
Genetically engineered knockout and transgenic mouse models and knockdown approaches targeting one or more proteins in the insulin/IGF-1 signalling pathway, including the insulin and IGF-1 receptors [66, 67], IRS-1 and IRS-2 [68, 69], Akt [70], phosphoinositide 3-kinase (PI3-kinase) [71] or FOXO1 [72], point to their significant roles in the regulation of islet biology. Several groups, including ours, have reported on the significance of insulin/IGF-1 signalling in the regulation of both beta and alpha cells [66, 28]. A role for insulin signalling in the regulation of alpha cell function and mass is evident from analyses of alpha-IRKO mice [28]. Interestingly, the mutant mice exhibited hyperglucagonaemia, glucose intolerance and an age-dependent progressive increase in beta cell area, while alpha cell area was unchanged leading to a decrease in relative alpha cell area [70]. The reduced relative alpha cell area in older alpha-IRKO mice may be secondary to attenuation of insulin signalling-mediated anti-apoptosis leading to increased alpha cell death. In humans, an increase in alpha cell relative area has been reported in insulin-resistant patients [73] and in patients with type 2 diabetes [74] or type 1 diabetes [75]; however, the mechanism(s) underlying this apparent increase in alpha cell mass remains unclear.
Disruptions in glucagon signalling induced in animal models by decreasing glucagon receptor (GR) expression [29, 76] or modulating proteins in the receptor signalling pathway [77] determine alpha cell hyperplasia. Studies on these disruptions suggest that, similar to the observations on beta cell hyperplasia in insulin resistance, an increase in alpha cell proliferation and hormone production occurs in states of glucagon resistance. In addition, the contribution of hyperinsulinaemia to alpha cell hyperplasia is supported by observations that IRS2 KO mice exhibit reduced alpha cell mass and glucagon secretion [69], while in vitro insulin treatment of alpha TC1 cells leads to increased alpha cell proliferation by triggering mTOR [78].
Among alpha cell regulators, recent findings suggest that a circulating factor, which is likely to be generated following disruption of hepatic GR signalling, can increase alpha cell proliferation independent of direct pancreatic input [79]. Thus, despite preservation of islet GR signalling, GRHep−/− mice develop hyperglucagonaemia and alpha cell hyperplasia. Identification of novel factors regulating alpha cell proliferation and mass may facilitate the generation and expansion of alpha cells for transdifferentiation into beta cells and the treatment of diabetes.
Studies in adult baboons indicated an obesity-induced increase in alpha cell mass that was significantly greater than the increase in beta cell mass, leading to an imbalance in the beta:alpha ratio. The authors suggest that an abnormal activation of IL-6 and TNF-α signalling systems is associated with the increased alpha cell mass [80] (see text box: ‘Contributors to maintenance of islet cell mass in adult rodents’).
Nutrients and microRNAs
In addition to being an essential nutrient as a primary energy source in the body, glucose has an effect on islet cell population dynamics. For example, in rats, glucose infusion increases beta cell numbers by 50% [81], with neogenesis of precursor cells being a dominant contributor to the increased mass [82]. Furthermore, glucose promotes beta cell survival by suppressing a constitutive apoptotic programme in vivo [83], and there is evidence that chronic changes in beta cell glucose metabolism induced by genetic or pharmacological manipulation of glucokinase can regulate beta cell mass regeneration in vivo [84]. Paradoxically, chronic hyperglycaemia promotes glucotoxicity which, in turn, exacerbates diabetes by increasing apoptosis [85]. Although it is now several decades since the concept of glucose toxicity [86] was first introduced, the precise cut-off point at which glucose levels transition from being useful and supportive of proliferation to assisting in the apoptosis and destruction of beta cells has still not been defined.
Excessive nutrient intake, such as high-protein [87] or high-fat diets [88], has also been described as modulating beta and alpha cell morphology. Increased levels of IL-6 in type 2 diabetes coupled with high expression of IL-6 receptors on alpha cells suggest that this cytokine could contribute to the increased alpha cell mass observed in diabetes [88]. This notion is supported by fasting hypoglucagonaemia and an inability of mice lacking IL-6 to increase their alpha cell mass.
miRNAs are powerful regulators of growth, differentiation and organ function and act by modulating gene expression at the post-transcriptional level [89]. The expression of several miRNAs is affected by prolonged exposure to elevated concentrations of glucose, NEFA and proinflammatory cytokines [90]. Moreover, alterations in the expression of several islet miRNAs have been reported in different models of diabetes [91, 92]. As recently proposed by Nesca et al [93], obesity and insulin resistance trigger changes in the levels of miR-132, miR-184 and miR-338-3p, which promote beta cell mass compensation. However, the authors also suggest that compensation failure further modifies the expression of additional miRNAs (e.g. miR-21, miR-34a, miR-199a-5p, miR-199a-3p, miR-203, miR-210, miR-383), with consequent negative effects on beta cell biology. Recently, miR-184 has been reported to be reduced in islets from type 2 diabetes patients. Poy and colleagues report that the reduced miR-184 promotes expression of Ago2 to form the microRNA-induced silencing complex in the regulation of beta cell proliferation [94]. Additional research is necessary to carefully dissect the interactions and hierarchy of miRNAs in the regulation of compensatory beta cell proliferation.
Among other miRNAs, miRNA-7a [95] has been reported to limit adult beta cell proliferation by inhibiting mammalian target of rapamycin (mTOR) signalling. Mice lacking miR-375 exhibit reduced beta cell mass as a result of impaired proliferation, hyperglucagonaemia and increased alpha cell numbers suggesting regulation of both beta and alpha cells [96]. Genetic deletion of miR-375 in ob/ob mice profoundly diminished the proliferative capacity of the endocrine pancreas and resulted in a severely diabetic state, indicating a role for the miRNA in beta cell compensation [97].
Placental and gut hormones
During pregnancy, placental hormones, particularly placental lactogen, are responsible for alterations in beta cell mass. These hormones stimulate beta cell proliferation in isolated islets, and beta cell mass is reduced by up to 42% in receptor-deficient mice [98]. It has recently been demonstrated that the pro-survival effects of lactogens are mediated by protection against apoptotic pathways controlled by members of the BCL2 gene family in human beta cells [99] and against glucolipotoxicity-induced cell death via Janus kinase 2 (JAK2)/signal transducer and activator of transcription 5 (STAT5) signalling [100]. While in vitro studies have demonstrated that human somatomammotropin stimulates glucagon release [101], in vivo studies in both rats and humans suggest that pregnancy does not affect alpha cell function [102] and that alpha cells might not be involved in the increased insulin demand in response to insulin resistance during normal human pregnancy [103].
Gastrointestinal hormones have long been recognised as regulating islet mass. GLP-1, GIP, cholecystokinin and gastrin are all secreted in response to nutrient intake and act to regulate digestion, insulin secretion, satiety and beta cell mass. GLP-1 promotes beta cell proliferation, islet neogenesis and beta cell survival in vivo [104], while GIP promotes beta cell survival in vivo but only promotes beta cell proliferation in vitro [105]. Cholecystokinin regulates beta cell apoptosis and mitogenesis in vivo in rats [106], and gastrin stimulates islet neogenesis [107]. The mechanism(s) adopted by GLP-1 and GIP to modulate cell proliferation and function implicates multiple signalling pathways. The pathways characterised include those acting via the G-protein-coupled receptors that lead to activation of cAMP, protein kinase A (PKA) and the cAMP response element-binding protein (CREB) cascade to directly regulate genes involved in proliferation and apoptosis, and other pathways determining phosphorylation of Akt and mitogen-activated protein kinases [108]. The ghrelin gene-derived peptides and exendin-4 exert cyto-protective effects in human pancreatic islet endothelial cells. These anti-apoptotic effects involve the phosphoinositide 3-kinase [PI3K]/Akt, extracellular signal-regulated kinase1/2 (ERK1/2) and cAMP/PKA pathways [109]. Although ageing is characterised by a low level of beta cell proliferation, mice treated with exendin-4 at different ages show low glucose levels, suggesting a potential beneficial effect in ageing [110].
The C-terminal VGF peptide, TLQP-21, potentiates glucose-stimulated insulin secretion in isolated rat islets and reduces glycaemic excursion in Wistar rats following a glucose challenge. Chronic administration of TLQP-21 to Zucker diabetic fatty rats delays the onset of overt diabetes by preserving islet cell mass [111] and acts in a manner similar to exendin-4 by blocking beta cell apoptosis.
GLP-1 and GIP exert opposing actions on glucagon secretion. Thus, GLP-1 receptor agonists and dipeptidyl peptidase 4 (DPP4) inhibitors inhibit glucagon secretion [112], while GIP has been shown to increase glucagon secretion in humans under hyperglycaemic conditions [113].
As extensively reviewed by Drucker [114], the precise effects of GLP-1 on alpha cell mass are still unclear. The recent report of alpha cell hyperplasia in pancreases from diabetic patients treated with GLP-1 receptor agonists and DPP4 inhibitors without evidence for alpha cell proliferation are difficult to interpret [115]. The latter study [115] has been a matter of considerable debate since the data indicate the opposite of several preclinical studies in rodents and non-human primates, wherein the use of different drugs and experimental models did not lead to detectable alpha cell hyperplasia, and in fact showed reduced numbers of alpha cells [116, 117]. Thus it is unclear how GLP-1 ‘directly’ regulates alpha cell hyperplasia independent of evidence of proliferation. In this context, it is notable that GR disruption increases alpha cell mass [79] by increased proliferation independent of GLP-1 receptor signalling [118].
Old age
The endocrine compartment of the pancreas is relatively well maintained with advancing age [119], and studies indicate that beta cell mass remains relatively constant from age 20 to 100 years in healthy humans, with no major alterations in cell size and apoptosis, suggesting that the cells are likely to be long lived [120].
The Harlan and Tisdale groups used two intriguing techniques to study human beta cell turnover in vivo: (1) iododeoxyuridine (IdU) or bromodeoxyuridine (BrdU) staining in pancreases from autopsies performed on patients who, under clinical protocols, were administered the thymidine analogue; and (2) the technique of carbon-14 dating of DNA extracted from beta cells isolated from recently deceased donors. The two groups suggested that human beta cell turnover is limited to the first three decades of life, since no replication (incorporation of IdU) of islet beta cells was detectable in the participants over 30 years of age [121].
Age-dependent alterations in cell cycle protein expression in islet cells include an increase in p16cip1 levels [122] and a reduction in centromere protein A (CENPA) [120], a protein required for chromosomal segregation in mitosis.
Among other factors, alterations in the rates of apoptosis and amyloid deposition and an imbalance between activation and inactivation of cell cycle proteins have all been topics of debate with regard to their contribution to the pathogenesis of type 2 diabetes. When evaluating islet plasticity it is important to consider the rate of cell death. A systematic analysis of autopsied pancreases from humans at different ages suggested that beta cell apoptosis is low and remains constant throughout life [21]. However, the apoptotic rate has been reported to be increased in obese and diabetic individuals compared with the rate in lean and non-diabetic groups [123]. In contrast, a study by Reers et al showed that a decline in beta cell replication with age was not associated with a change in the frequency of apoptosis [124]. The increase in the apoptotic rate has been explained, in part, by the accumulation of amyloid plaques [125], aggregates of islet amyloid polypeptide (IAPP, a hormone co-secreted with insulin), which are increased in islets of diabetic and obese non-diabetic individuals. Since rodent IAPP does not aggregate because it has a different structure to the human hormone, investigators have examined the effects of IAPP accumulation in islets in transgenic mice expressing human IAPP. Overexpression of human IAPP has been reported to cause early hyperglycaemia followed by accumulation of amylin plaques in the islets in ageing mice [126]. Janson et al [127] reported that intermediate-sized amyloid particles in human islets promote membrane damage and subsequent cell death (Fig. 2).

Schematic illustration of age-dependent changes of islet cell morphology from adulthood to old age in humans. BAD, Bcl-2-associated death promoter; BAX, BCL2-associated X protein; BMI1, B-lymphoma Moloney murine leukaemia virus insertion region-1; EZH2, enhancer of zeste homologue 2 (EZH2)
Similar to humans, islet amyloidosis was observed in a large number of baboons with normal fasting plasma glucose levels and in those with impaired fasting glucose or type 2 diabetes. Ageing was positively correlated with the severity of islet amyloidosis, and increased amyloidosis was associated with an increased rate of alpha cell replication and beta cell apoptosis [128].
Studies in murine models suggest that mature beta cells are sensitive to cell cycle perturbations. Specifically, the products of the Ink4a/Arf (Cdkn2a) locus, p16cip1 and p19, the expression of which increases with age [129], have been associated with reduced beta cell proliferation [130]. In this context it is notable that genome-wide association studies in humans link the CDKN2A locus to type 2 diabetes susceptibility [131–134], providing physiological relevance.
Epigenetic regulation of the CDKN2A locus determines a series of histone modifications to control beta cell proliferation during ageing. In particular, ageing de-represses this locus, leading to increased levels of cell cycle inhibitors (p16cip1 and p19), decreased B-lymphoma Moloney murine leukaemia virus insertion region-1 (BMI1) banding [135] and decreased levels of enhancer of zeste homologue 2 (EZH2) [136], a histone methyltransferase and member of the polycomb group of proteins.
Other stressors that influence the biology of the ageing beta cell, a major secretory cell include endoplasmic reticulum (ER) and oxidative stress. As an adaptive response to ER stress, pancreatic beta cells express high levels of the transducers inositol-requiring enzyme 1 (IRE1), PKR-like ER kinase (PERK) and activating transcription factor-6 (ATF6), which balance the protein synthesis and ER folding capacity of the cells. Despite an increase in unfolded protein response (UPR) signalling, which allows proinsulin synthesis by increasing the ER folding capacity after acute exposure to hyperglycaemia [137], chronic ER stress leads to a decrease in insulin synthesis and activation of the apoptotic cell death programme [138]. NEFA have been recognised as key contributors that can trigger the apoptotic programme, in part mediated by the ER stress response, and this highlights the significance of obesity and nutrients in the context of lipotoxic effects on beta cells [139].
Among other ER stress-related proteins, the spliced form of X box binding protein 1 (XBP1s), a protein involved in IRE1 activation, has been reported to regulate both alpha and beta cell function. Mice with alpha cell-specific disruption of XBP1 expression (alpha-XBP1KO) [140] exhibit altered glucagon secretion as a result of increased ER stress that is not directly associated with alpha cell mass or growth. Surprisingly, prolonged XBP1 overproduction in rat islet cells impaired glucose-stimulated insulin secretion, BAX (BCL2-associated X protein) translocation, cytochrome c release and activation of caspase 3 and beta cell apoptosis [141]. However, sustained production of XBP1 inhibits beta cell function and eventually triggers the mitochondrial pathway of apoptosis, probably secondary to inhibition of PDX1 and musculoaponeurotic fibrosarcoma oncogene homologue A (MAFA) expression [141]. In summary, ER stress and its related proteins seem to affect beta cell mass by inducing apoptotic death, while alpha cell mass remains relatively normal.
Mitochondrial dysfunction and, consequently, increasing oxidative stress with age are responsible for reduced beta cell function and survival [142]. Several growth factor pathways including insulin have been reported to modulate mitochondrial function and regulation of oxidative stress leading to beta cell apoptosis. The presence of a complex between the pro-apoptotic protein Bcl-2-associated death promoter (BAD) and glucokinase in mouse and human beta cell mitochondria may play a role in this process and contribute to defects in mitochondrial function [143].
It is important to note that a majority of studies exploring the capacity of endogenous renewal have been performed in young rodents and cannot be directly translated to an equivalent age in humans. For example, partial pancreatectomy in rodents promotes beta cell regeneration and increased insulin content [144]. In contrast, in one report, adult humans who underwent partial pancreatectomy did not exhibit evidence for increased regeneration [145] but exhibited long-term recovery of beta cell function [146].
In addition, the dramatic age-dependent decline of pancreatic beta cell replication in response to diverse insults (high-fat diet, streptozotocin administration or short-term treatment with the GLP-1 analogue exendin-4) has been correlated by Tschen et al [110] with an increase in Bmi1 levels, a polycomb group protein that regulates the Ink4a locus and modulates the ability of the beta cell mass to expand. Interestingly, a systemic factor has been considered responsible for increasing the proliferation rate of old pancreatic beta cells when parabiosed to young mice [147]. Whether this factor is similar to the circulating factor in insulin-resistant states [4] has not been fully explored.
Future directions
New therapies are urgently required to restrain beta cell death and promote beta cell function and regenerate healthy functional beta cells in diabetic patients. To date, several growth factors and transcription factors have been highlighted for their ability to increase beta cell mass and proliferation based on elegant genetically engineered transgenic and knockout approaches in rodents. However, the poor translation of the beta cell proliferative effects of potential factors and small molecules observed in young rodent to humans underscores the poor state of knowledge of signalling pathways in human beta cells [8, 148, 149] and urgently calls for a major scientific effort in this area. Investigating the modifications that occur during the ageing process provides a unique opportunity to identify possible targets to prevent and slow the progression of diabetes, and to maintain higher rates of proliferation and regeneration of insulin-producing cells in pathophysiological conditions. Current studies suggest that potential induction of replication of adult beta cells, neogenesis from progenitor cells and duct cells, transdifferentiation from alpha to beta cells and de-differentiation of beta cells are plausible mechanisms that could be harnessed to engineer a therapeutic increase in insulin-producing cells in humans. It will be exciting to see whether one or more of the aforementioned mechanisms, or perhaps a new discovery, holds the key to safely and specifically enhancing functional beta cell mass to treat diabetes.
Abbreviations
- Alpha-IRKO:
-
Alpha cell-specific knockout of insulin receptors
- CDK:
-
Cyclin-dependent kinase
- ER:
-
Endoplasmic reticulum
- FOX:
-
Forkhead box protein
- GIP:
-
Gastric inhibitory polypeptide
- GLP-1:
-
Glucagon-like peptide 1
- GR:
-
Glucagon receptor
- IAPP:
-
Islet amyloid polypeptide
- IdU:
-
Iododeoxyuridine
- IGF-1:
-
Insulin-like growth factor 1
- IRE1:
-
Inositol-requiring enzyme 1
- PDX1:
-
Pancreatic and duodenal homeobox 1
- XBP1:
-
X box binding protein 1
References
Weir GC, Bonner-Weir S (2013) Islet β cell mass in diabetes and how it relates to function, birth, and death. Ann N Y Acad Sci 1281:92–105
Teo AK, Wagers AJ, Kulkarni RN (2013) New opportunities: harnessing induced pluripotency for discovery in diabetes and metabolism. Cell Metab 18:775–791
Talchai C, Xuan S, Lin HV, Sussel L, Accili D (2012) Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 150:1223–1234
El Ouaamari A, Kawamori D, Dirice E et al (2013) Liver-derived systemic factors drive β cell hyperplasia in insulin-resistant states. Cell Rep 3:401–410
Yi P, Park JS, Melton DA (2013) Betatrophin: a hormone that controls pancreatic β cell proliferation. Cell 153:747–758
Dor Y, Brown J, Martinez OI, Melton DA (2004) Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429:41–46
Xu X, D’Hoker J, Stangé G et al (2008) Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 132:197–207
Kulkarni RN, Mizrachi EB, Ocana AG, Stewart AF (2012) Human β-cell proliferation and intracellular signaling: driving in the dark without a road map. Diabetes 61:2205–2213
Müller WA, Faloona GR, Aguilar-Parada E, Unger RH (1970) Abnormal alpha-cell function in diabetes. Response to carbohydrate and protein ingestion. N Engl J Med 283:109–115
Dunning BE, Gerich JE (2007) The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr Rev 28:253–283
Braun M, Rorsman P (2010) The glucagon-producing alpha cell: an electrophysiologically exceptional cell. Diabetologia 53:1827–1830
Collombat P, Xu X, Ravassard P et al (2009) The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell 138:449–462
Thorel F, Népote V, Avril I et al (2010) Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 464:1149–1154
Juhl K, Bonner-Weir S, Sharma A (2010) Regenerating pancreatic beta-cells: plasticity of adult pancreatic cells and the feasibility of in-vivo neogenesis. Curr Opin Organ Transplant 1581:79–85
Bonner-Weir S (2001) Beta-cell turnover: its assessment and implications. Diabetes 50(Suppl 1):S20–S24
Scaglia L, Cahill CJ, Finegood DT, Bonner-Weir S (1997) Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology 138:1736–1741
Quinn AR, Blanco CL, Perego C et al (2012) The ontogeny of the endocrine pancreas in the fetal/newborn baboon. J Endocrinol 214:289–299
Heremans Y, van de Casteele M, in’t Veld P et al (2002) Recapitulation of embryonic neuroendocrine differentiation in adult human pancreatic duct cells expressing neurogenin 3. J Cell Biol 159:303–312
Apelqvist A, Li H, Sommer L et al (1999) Notch signalling controls pancreatic cell differentiation. Nature 400:877–881
Bernard-Kargar C, Ktorza A (2001) Endocrine pancreas plasticity under physiological and pathological conditions. Diabetes 50(Suppl 1):S30–S35
Gregg BE, Moore PC, Demozay D et al (2012) Formation of a human β-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab 97:3197–3206
Meier JJ, Köhler CU, Alkhatib B et al (2010) Beta-cell development and turnover during prenatal life in humans. Eur J Endocrinol 162:559–568
Bouwens L, Lu WG, de Krijger R (1997) Proliferation and differentiation in the human fetal endocrine pancreas. Diabetologia 40:398–404
Ackermann AM, Gannon M (2007) Molecular regulation of pancreatic beta-cell mass development, maintenance, and expansion. J Mol Endocrinol 38:193–206
Zhang H, Ackermann AM, Gusarova GA et al (2006) The FoxM1 transcription factor is required to maintain pancreatic beta-cell mass. Mol Endocrinol 20:1853–1866
Prasadan K, Daume E, Preuett B et al (2002) Glucagon is required for early insulin-positive differentiation in the developing mouse pancreas. Diabetes 51:3229–3236
Gromada J, Franklin I, Wollheim CB (2007) Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr Rev 28:84–116
Kawamori D, Kurpad AJ, Hu J et al (2009) Insulin signaling in alpha cells modulates glucagon secretion in vivo. Cell Metab 9:350–361
Gelling RW, Du XQ, Dichmann DS et al (2003) Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci U S A 100:1438–1443
Vincent M, Guz Y, Rozenberg M et al (2003) Abrogation of protein convertase2 activity results in delayed islet cell differentiation and maturation, increased alpha-cell proliferation, and islet neogenesis. Endocrinology 144:4061–4069
Lu J, Herrera PL, Carreira C, Bonnavion R et al (2010) Alpha cell-specific Men1 ablation triggers the transdifferentiation of glucagon-expressing cells and insulinoma development. Gastroenterology 138:1954–1965
Wilcox CL, Terry NA, Walp ER, Lee RA, May CL (2013) Pancreatic α-cell specific deletion of mouse Arx leads to α-cell identity loss. PLoS One 8:e66214. doi:10.1371/journal.pone.0066214
Mastracci TL, Wilcox CL, Arnes L et al (2011) Nkx2.2 and Arx genetically interact to regulate pancreatic endocrine cell development and endocrine hormone expression. Dev Biol 359:1–11
Hill DJ (2011) Nutritional programming of pancreatic β-cell plasticity. World J Diabetes 2:119–126
Gesina E, Blondeau B, Milet A et al (2006) Glucocorticoid signalling affects pancreatic development through both direct and indirect effects. Diabetologia 49:2939–2947
Reusens B, Remacle C (2006) Programming of the endocrine pancreas by the early nutritional environment. Int J Biochem Cell Biol 38:913–922
Elmes MJ, Gardner DS, Langley-Evans SC (2007) Fetal exposure to a maternal low-protein diet is associated with altered left ventricular pressure response to ischaemia-reperfusion injury. Br J Nutr 98:93–100
Sathishkumar K, Elkins R, Yallampalli U, Yallampalli C (2009) Protein restriction during pregnancy induces hypertension and impairs endothelium-dependent vascular function in adult female offspring. J Vasc Res 46:229–239
Brissova M, Aamodt K, Brahmachary P et al (2014) Islet microenvironment, modulated by vascular endothelial growth factor-a signaling, promotes β cell regeneration. Cell Metab. doi:10.1016/j.cmet.2014.02.001
Nolan CJ, Damm P, Prentki M (2011) Type 2 diabetes across generations: from pathophysiology to prevention and management. Lancet 378:169–181
Banerji MA (2002) Impaired beta-cell and alpha-cell function in African-American children with type 2 diabetes mellitus—“Flatbush diabetes”. J Pediatr Endocrinol Metab 15(Suppl 1):493–501
SEARCH for Diabetes in Youth Study Group, Liese AD, D’Agostino RB Jr, Hamman RF et al (2006) The burden of diabetes mellitus among US youth: prevalence estimates from the SEARCH for Diabetes in Youth Study. Pediatrics 118:1510–1518
Baranowski T, Cooper DM, Harrell J et al (2006) Presence of diabetes risk factors in a large U.S. eighth-grade cohort. Diabetes Care 29:212–217
Goran MI, Gower BA (2001) Longitudinal study on pubertal insulin resistance. Diabetes 50:2444–2450
Caprio S, Plewe G, Diamond MP et al (1989) Increased insulin secretion in puberty: a compensatory response to reductions in insulin sensitivity. J Pediatr 114:963–967
Arslanian SA, Kalhan SC (1995) Correlations between fatty acid and glucose metabolism: potential explanation of insulin resistance of puberty. Diabetes 43:908–914
Amiel SA, Sherwin RS, Simonson DC, Lauritano AA, Tamborlane WV (1986) Impaired insulin action in puberty: a contributing factor to poor glycemic control in adolescents with diabetes. N Engl J Med 315:215–219
Merimee TJ, Burgess JA, Rabinowitz D (1967) Influence of growth hormone on insulin secretion. Diabetes 16:478–482
Nielsen JH (1982) Effects of growth hormone, prolactin, and placental lactogen on insulin content and release, and deoxyribonucleic acid synthesis in cultured pancreatic islets. Endocrinology 110:600–606
Meier JJ, Butler AE, Saisho Y et al (2008) Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 57:1584–1594
Saisho Y, Butler AE, Meier JJ et al (2007) Pancreas volumes in humans from birth to age one hundred taking into account sex, obesity, and presence of type-2 diabetes. Clin Anat 20:933–942
Kassem SA, Ariel I, Thornton PS, Scheimberg I, Glaser B (2000) Beta-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes 49:1325–1333
Gepts W, de Mey J (1978) Islet cell survival determined by morphology. An immunocytochemical study of the islets of Langerhans in juvenile diabetes mellitus. Diabetes 27(Suppl 1):251–261
Hill DJ, Hogg J (1991) Growth factor control of pancreatic B cell hyperplasia. Baillieres Clin Endocrinol Metab 5:689–698
Kawamori D, Akiyama M, Hu J, Hambro B, Kulkarni RN (2011) Growth factor signalling in the regulation of α-cell fate. Diabetes Obes Metab 13(Suppl 1):21–30
Martín J, Hunt SL, Dubus P et al (2003) Genetic rescue of Cdk4 null mice restores pancreatic beta-cell proliferation but not homeostatic cell number. Oncogene 22:5261–5269
Kushner JA, Ciemerych MA, Sicinska E et al (2005) Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol 25:3752–3762
Georgia S, Bhushan A (2004) Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest 114:963–968
Uchida T, Nakamura T, Hashimoto N et al (2005) Deletion of Cdkn1b ameliorates hyperglycemia by maintaining compensatory hyperinsulinemia in diabetic mice. Nat Med 11:175–182
Kassem SA, Ariel I, Thornton PS (2001) p57KIP2 expression in normal islet cells and in hyperinsulinism of infancy. Diabetes 50:2763–2769
Ackermann Misfeldt A, Costa RH, Gannon M (2008) Beta-cell proliferation, but not neogenesis, following 60% partial pancreatectomy is impaired in the absence of FoxM1. Diabetes 57:3069–3077
Zarrouki B, Benterki I, Fontés G et al (2014) Epidermal growth factor receptor signaling promotes pancreatic β-cell proliferation in response to nutrient excess in rats through mTOR and FOXM1. Diabetes 63:982–993
Crabtree JS, Scacheri PC, Ward JM et al (2003) Of mice and MEN1: insulinomas in a conditional mouse knockout. Mol Cell Biol 23:6075–6085
Karnik SK, Hughes CM, Gu X et al (2005) Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27kip1p27kip1 and p18INK4c. Proc Natl Acad Sci U S A 102:14659–14664
Al-Hasani K, Pfeifer A, Courtney M et al (2013) Adult duct-lining cells can reprogram into β-like cells able to counter repeated cycles of toxin-induced diabetes. Dev Cell 26:86–100
Kulkarni RN, Brüning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR (1999) Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 96:329–339
Kulkarni RN, Holzenberger M, Shih DQ et al (2002) Beta-cell-specific deletion of the Igf1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter beta-cell mass. Nat Genet 31:111–115
Withers DJ, Gutierrez JS, Towery H et al (1998) Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391:900–904
Cantley J, Choudhury AI, Asare-Anane H et al (2007) Pancreatic deletion of insulin receptor substrate 2 reduces beta and alpha cell mass and impairs glucose homeostasis in mice. Diabetologia 50:1248–1256
Cho H, Mu J, Kim JK et al (2001) Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ). Science 292:1728–1731
Hagiwara S, Sakurai T, Tashiro F et al (1995) An inhibitory role for phosphatidylinositol 3-kinase in insulin secretion from pancreatic B cell line MIN6. Biochem Biophys Res Commun 214:51–59
Martinez SC, Tanabe K, Cras-Méneur C, Abumrad NA, Bernal-Mizrachi E, Permutt MA (2008) Inhibition of Foxo1 protects pancreatic islet beta-cells against fatty acid and endoplasmic reticulum stress-induced apoptosis. Diabetes 57:846–859
Mezza T, Muscogiuri G, Sorice GP et al (2014) Insulin resistance alters islet morphology in non-diabetic humans. Diabetes 63:994–1007
Yoon KH, Ko SH, Cho JH et al (2003) Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab 88:2300–2308
Orci L, Baetens D, Rufener C et al (1976) Hypertrophy and hyperplasia of somatostatin-containing D-cells in diabetes. Proc Natl Acad Sci U S A 73:1338–1342
Liang Y, Osborne MC, Monia BP et al (2004) Reduction in glucagon receptor expression by an antisense oligonucleotide ameliorates diabetic syndrome in db/db mice. Diabetes 53:410–417
Chen M, Gavrilova O, Zhao WQ et al (2005) Increased glucose tolerance and reduced adiposity in the absence of fasting hypoglycemia in mice with liver-specific Gs alpha deficiency. J Clin Invest 115:3217–3227
Liu Z, Kim W, Chen Z et al (2011) Insulin and glucagon regulate pancreatic alpha cell proliferation. PLoS One 6:e16096
Longuet C, Robledo AM, Dean ED et al (2013) Liver-specific disruption of the murine glucagon receptor produces α-cell hyperplasia: evidence for a circulating α-cell growth factor. Diabetes 62:1196–1205
Guardado-Mendoza R, Jimenez-Ceja L, Majluf-Cruz A et al (2013) Impact of obesity severity and duration on pancreatic β- and α-cell dynamics in normoglycemic non-human primates. Int J Obes (Lond) 37:1071–1078
Bernard C, Berthault MF, Saulnier C, Ktorza A (1999) Neogenesis vs. apoptosis as main components of pancreatic beta cell mass changes in glucose-infused normal and mildly diabetic adult rats. FASEB J 13:1195–1205
Paris M, Bernard-Kargar C, Berthault MF, Bouwens L, Ktorza A (2003) Specific and combined effects of insulin and glucose on functional pancreatic beta-cell mass in vivo in adult rats. Endocrinology 144:2717–2727
Hoorens A, van de Casteele M, Klöppel G, Pipeleers D (1996) Glucose promotes survival of rat pancreatic beta cells by activating synthesis of proteins which suppress a constitutive apoptotic program. J Clin Invest 98:1568–1574
Porat S, Weinberg-Corem N, Tornovsky-Babaey S (2011) Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab 13:440–449
Maedler K, Sergeev P, Ris F et al (2002) Glucose-induced β cell production of IL-1β contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110:851–860
Rossetti L, Giaccari A, DeFronzo RA (1990) Glucose toxicity. Diabetes Care 13:610–630
Morley MG, Leiter EH, Eisenstein AB, Strack I (1982) Dietary modulation of alpha cell volume and function in strain 129/J mice. Am J Physiol 242:G354–G359
Ellingsgaard H, Ehses JA, Hammar EB et al (2008) Interleukin-6 regulates pancreatic alpha-cell mass expansion. Proc Natl Acad Sci U S A 105:13163–13168
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 16:281–297
Guay C, Jacovetti C, Nesca V, Motterle A, Tugay K, Regazzi R (2012) Emerging roles of non-coding RNAs in pancreatic β-cell function and dysfunction. Diabetes Obes Metab 14(Suppl 3):12–21
Esguerra JL, Bolmeson C, Cilio CM, Eliasson L (2011) Differential glucose-regulation of microRNAs in pancreatic islets of non-obese type 2 diabetes model Goto-Kakizaki rat. PLoS One 6:e18613
Jacovetti C, Abderrahmani A, Parnaud G et al (2012) MicroRNAs contribute to compensatory β cell expansion during pregnancy and obesity. J Clin Invest 122:3541–3551
Nesca V, Guay C, Jacovetti C et al (2013) Identification of particular groups of microRNAs that positively or negatively impact on beta cell function in obese models of type 2 diabetes. Diabetologia 56:2203–2212
Tattikota SG, Rathjen T, McAnulty SJ et al (2014) Argonaute2 mediates compensatory expansion of the pancreatic β cell. Cell Metab 19:122–134
Wang Y, Liu J, Liu C, Naji A, Stoffers DA (2013) MicroRNA-7 regulates the mTOR pathway and proliferation in adult pancreatic β-cells. Diabetes 62:887–895
Poy MN, Eliasson L, Krutzfeldt J et al (2004) A pancreatic islet-specific microRNA regulates insulin secretion. Nature 432:226–230
Poy MN, Hausser J, Trajkovski M et al (2009) miR-375 maintains normal pancreatic alpha- and beta-cell mass. Proc Natl Acad Sci U S A 106:5813–5818
Freemark M, Avril I, Fleenor D et al (2002) Targeted deletion of the PRL receptor: effects on islet development, insulin production, and glucose tolerance. Endocrinology 143:1378–1385
Terra LF, Garay-Malpartida MH, Wailemann RA, Sogayar MC, Labriola L (2011) Recombinant human prolactin promotes human beta cell survival via inhibition of extrinsic and intrinsic apoptosis pathways. Diabetologia 54:1388–1397
Kondegowda NG, Mozar A, Chin C, Otero A, Garcia-Ocaña A, Vasavada RC (2012) Lactogens protect rodent and human beta cells against glucolipotoxicity-induced cell death through Janus kinase-2 (JAK2)/signal transducer and activator of transcription-5 (STAT5) signalling. Diabetologia 55:1721–1732
Laube H, Fussgänger RD, Schröder KE, Pfeiffer EF (1972) Acute effects of human chorionic somatomammotropin on insulin and glucagon release in the isolated perfused pancreas. Diabetes 21:1072–1076
Koiter TR, Faas MM, Visscher A, Kievit C, Steffens AB, Schuiling GA (1992) Regulation of peripheral glucagon concentrations in cyclic, pregnant, and lactating rats. Physiol Behav 51:1173–1178
Luyckx AS, Gerard J, Gaspard U, Lefebvre PJ (1975) Plasma glucagon levels in normal women during pregnancy. Diabetologia 11:549–554
Farilla L, Hui H, Bertolotto C et al (2002) Glucagon-like peptide-1 promotes islet cell growth and inhibits apoptosis in Zucker diabetic rats. Endocrinology 143:4397–4408
Widenmaier SB, Kim SJ, Yang GK et al (2010) A GIP receptor agonist exhibits beta-cell anti-apoptotic actions in rat models of diabetes resulting in improved beta-cell function and glycemic control. PLoS One 5:e9590
Kuntz E, Pinget M, Damgé P (2004) Cholecystokinin octapeptide: a potential growth factor for pancreatic beta cells in diabetic rats. JOP 5:464–475
Rooman I, Lardon J, Bouwens L (2002) Gastrin stimulates beta-cell neogenesis and increases islet mass from transdifferentiated but not from normal exocrine pancreas tissue. Diabetes 51:686–690
Lavine JA, Attie AD (2010) Gastrointestinal hormones and the regulation of β-cell mass. Ann N Y Acad Sci 1212:41–58
Favaro E, Granata R, Miceli I et al (2012) The ghrelin gene products and exendin-4 promote survival of human pancreatic islet endothelial cells in hyperglycaemic conditions, through phosphoinositide 3-kinase/Akt, extracellular signal-related kinase (ERK)1/2 and cAMP/protein kinase A (PKA) signalling pathways. Diabetologia 55:1058–1070
Tschen SI, Dhawan S, Gurlo T, Bhushan A (2009) Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes 58:1312–1320
Stephens SB, Schisler JC, Hohmeier HE et al (2012) A VGF-derived peptide attenuates development of type 2 diabetes via enhancement of islet β-cell survival and function. Cell Metab 16:33–43
Akarte AS, Srinivasan BP, Gandhi S, Sole S (2012) Chronic DPP-IV inhibition with PKF-275-055 attenuates inflammation and improves gene expressions responsible for insulin secretion in streptozotocin induced diabetic rats. Eur J Pharm Sci 47:456–463
Chia CW, Carlson OD, Kim W et al (2009) Exogenous glucose-dependent insulinotropic polypeptide worsens post prandial hyperglycemia in type 2 diabetes. Diabetes 58:1342–1349
Drucker DJ (2013) Incretin action in the pancreas: potential promise, possible perils, and pathological pitfalls. Diabetes 62:3316–3323
Butler AE, Campbell-Thompson M et al (2013) Marked expansion of exocrine and endocrine pancreas with incretin therapy in humans with increased exocrine pancreas dysplasia and the potential for glucagon-producing neuroendocrine tumors. Diabetes 62:2595–2604
Nyborg NC, Molck AM, Madsen LW, Bjerre Knudsen L (2012) The human GLP-1 analog liraglutide and the pancreas: evidence for the absence of structural pancreatic changes in three species. Diabetes 61:1243–1249
Busch SJ, Hoffmann P, Sahota P et al (2013) Studies in rodents with the dipeptidyl peptidase-4 inhibitor vildagliptin to evaluate possible drug-induced pancreatic histological changes that are predictive of pancreatitis and cancer development in man. Diabetes Obes Metab 15:72–76
Ali S, Lamont BJ, Charron MJ, Drucker DJ (2011) Dual elimination of the glucagon and GLP-1 receptors in mice reveals plasticity in the incretin axis. J Clin Invest 121:1917–1929
Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC (2012) β-Cell mass and turnover in humans: effects of obesity and aging. Diabetes Care 36:111–117
Cnop M, Hughes SJ, Igoillo-Esteve M et al (2010) The long lifespan and low turnover of human islet beta cells estimated by mathematical modelling of lipofuscin accumulation. Diabetologia 53:321–330
Perl S, Kushner JA, Buchholz BA et al (2010) Significant human beta-cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J Clin Endocrinol Metab 95:E234–E239
Kohler CU, Olewinski M, Tannapfel A, Schmidt WE, Fritsch H, Meier JJ (2011) Cell cycle control of beta-cell replication in the prenatal and postnatal human pancreas. Am J Physiol Endocrinol Metab 300:E221–E230
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52:102–110
Reers C, Erbel S, Esposito I et al (2009) Impaired islet turnover in human donor pancreata with aging. Eur J Endocrinol 160:185–191
Law E, Lu S, Kieffer TJ et al (2010) Differences between amyloid toxicity in alpha and beta cells in human and mouse islets and the role of caspase-3. Diabetologia 53:1415–1427
Verchere CB, D’Alessio DA, Palmiter RD et al (1996) Islet amyloid formation associated with hyperglycemia in transgenic mice with pancreatic beta cell expression of human islet amyloid polypeptide. Proc Natl Acad Sci U S A 93:3492–3496
Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC (1999) The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes 48:491–498
Guardado-Mendoza R, Davalli AM, Chavez AO et al (2009) Pancreatic islet amyloidosis, beta-cell apoptosis, and alpha-cell proliferation are determinants of islet remodeling in type-2 diabetic baboons. Proc Natl Acad Sci U S A 106:13992–13997
Krishnamurthy J, Torrice C, Ramsey MR et al (2004) Ink4a/Arf expression is a biomarker of aging. J Clin Invest 114:1299–1307
Krishnamurthy J, Ramsey MR, Ligon KL et al (2006) p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 443:453–457
Omori S, Tanaka Y, Takahashi A et al (2008) Association of CDKAL1, IGF2BP2, CDKN2A/B, HHEX, SLC30A8, and KCNJ11 with susceptibility to type 2 diabetes in a Japanese population. Diabetes 57:791–795
Zeggini E, Weedon MN, Lindgren CM et al (2007) Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 316:1336–1341
Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research, Saxena R, Voight BF et al (2007) Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316:1331–1336
Grarup N, Rose CS, Andersson EA et al (2007) Studies of association of variants near the HHEX, CDKN2A/B, and IGF2BP2 genes with type 2 diabetes and impaired insulin release in 10,705 Danish subjects: validation and extension of genome-wide association studies. Diabetes 56:3105–3111
Dhawan S, Tschen SI, Bhushan A (2009) Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev 23:906–911
Chen H, Gu X, Su IH et al (2009) Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev 23:975–985
Harding HP, Novoa I, Zhang Y et al (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6:1099–1108
Hartman MG, Lu D, Kim ML et al (2004) Role for activating transcription factor 3 in stress-induced beta-cell apoptosis. Mol Cell Biol 24:5721–5732
Cnop M, Ladrière L, Igoillo-Esteve M, Moura RF, Cunha DA (2010) Causes and cures for endoplasmic reticulum stress in lipotoxic β-cell dysfunction. Diabetes Obes Metab 12(Suppl 2):76–82
Akiyama M, Liew CW, Lu S et al (2013) X-box binding protein 1 is essential for insulin regulation of pancreatic α-cell function. Diabetes 62:2439–2449
Allagnat F, Christulia F, Ortis F et al (2010) Sustained production of spliced X-box binding protein 1 (XBP1) induces pancreatic beta cell dysfunction and apoptosis. Diabetologia 53:1120–1130
Anello M, Lupi R, Spampinato D et al (2005) Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 48:282–289
Liu S, Okada T, Assmann A et al (2009) Insulin signaling regulates mitochondrial function in pancreatic beta-cells. PLoS One 4:e7983
Peshavaria M, Larmie BL, Lausier J et al (2006) Regulation of pancreatic beta-cell regeneration in the normoglycemic 60% partial-pancreatectomy mouse. Diabetes 55:3289–3298
Menge BA, Tannapfel A, Belyaev O et al (2008) Partial pancreatectomy in adult humans does not provoke beta-cell regeneration. Diabetes 57:142–149
Menge BA, Breuer TG, Ritter PR, Uhl W, Schmidt WE, Meier JJ (2012) Long-term recovery of β-cell function after partial pancreatectomy in humans. Metabolism 61:620–624
Salpeter SJ, Khalaileh A, Weinberg-Corem N, Ziv O, Glaser B, Dor Y (2013) Systemic regulation of the age-related decline of pancreatic β-cell replication. Diabetes 62:2843–2848
Bernal-Mizrachi E, Kulkarni RN, Scott DK, Mauvais-Jarvis F, Stewart AF, Garcia-Ocaña A (2014) Human β-cell proliferation and intracellular signaling part 2: still driving in the dark without a road map. Diabetes 63:819–831
Vetere A, Choudhary A, Burns SM, Wagner BK (2014) Targeting the pancreatic β-cell to treat diabetes. Nat Rev Drug Discov 13:278–289
Acknowledgements
We thank E. Dirice (Joslin Diabetes Center, Boston) and S. Kahraman (Joslin Diabetes Center, Boston) for providing islet pictures.
Funding
RNK acknowledges support from National Institutes of Health Grants RO1 DK67536, RO1 DK055523 and a grant from the Juvenile Diabetes Research Foundation.
TM acknowledges support from Reynolds Travel Fellowship by European Association for the Study of Diabetes and Fellowship Prize by the Società Italiana di Diabetologia and the European Society for Clinical Nutrition and Metabolism.
Duality of interest
The authors declare that there is no duality of interest associated with the manuscript.
Contribution statement
The authors were involved in the conception and design of the manuscript, drafting the article and revising it critically for important intellectual content. TM designed all illustrations. RNK and TM edited the manuscript and approved the version to be published.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mezza, T., Kulkarni, R.N. The regulation of pre- and post-maturational plasticity of mammalian islet cell mass. Diabetologia 57, 1291–1303 (2014). https://doi.org/10.1007/s00125-014-3251-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-014-3251-7