
Abstract
Cisplatin-induced renal tubular injury largely restricts the wide-spread usage of cisplatin in the treatment of malignancies. Identifying the key signaling pathways that regulate cisplatin-induced renal tubular injury is thus clinically important. PARVB, a focal adhesion protein, plays a crucial role in tumorigenesis. However, the function of PARVB in kidney disease is largely unknown. To investigate whether and how PARVB contributes to cisplatin-induced renal tubular injury, a mouse model (PARVB cKO) was generated in which PARVB gene was specifically deleted from proximal tubular epithelial cells using the Cre-LoxP system. In this study, we found depletion of PARVB in proximal tubular epithelial cells significantly attenuates cisplatin-induced renal tubular injury, including tubular cell death and inflammation. Mechanistically, PARVB associates with transforming growth factor-β-activated kinase 1 (TAK1), a central regulator of cell survival and inflammation that is critically involved in mediating cisplatin-induced renal tubular injury. Depletion of PARVB promotes cisplatin-induced TAK1 degradation, inhibits TAK1 downstream signaling, and ultimately alleviates cisplatin-induced tubular cell damage. Restoration of PARVB or TAK1 in PARVB-deficient cells aggravates cisplatin-induced tubular cell injury. Finally, we demonstrated that PARVB regulates TAK1 protein expression through an E3 ligase ITCH-dependent pathway. PARVB prevents ITCH association with TAK1 to block its ubiquitination. Our study reveals that PARVB deficiency protects against cisplatin-induced tubular injury through regulation of TAK1 signaling and indicates targeting this pathway may provide a novel therapeutic strategy to alleviate cisplatin-induced kidney damage.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cisplatin is one of the most efficient and widely used anti-tumor drug in the clinic [1]. However, the benefits of cisplatin are often hampered because of its side effects on various organs, especially the kidney [2, 3]. Approximately 30% of cancer patients that receive cisplatin treatment develops acute kidney injury (AKI) and is associated with high mortality [4]. The renal proximal tubules are the major targets for the nephrotoxicity induced by cisplatin [5]. Cisplatin accumulates in renal proximal tubular cells, resulting in inflammation, cell necrosis or apoptosis, followed by acute renal dysfunction and damage [6,7,8]. However, the causes of cisplatin-induced renal toxicity are not clear, which hinders the strategies for cisplatin-induced AKI prevention or therapy. Therefore, identification of the pathogenic mechanism of cisplatin-induced renal tubular injury has clinical significance.
PARVB (also known as β-parvin or affixin), a focal adhesion protein, links integrins with intracellular signaling pathways to regulate a number of cellular behaviors, including cell spreading, adhesion and survival [9,10,11]. PARVB is highly expressed in several tissues and maintains their normal functions. In cardiomyocytes, PARVB interacts with the transcriptional activator STAT3 and participates in blood vessel formation [12]. PARVB also regulates cardiomyocyte elongation and plays a crucial role in exercise-induced cardiac hypertrophy through PIX/Rac1-signaling [13]. Besides this, PARVB is involved in platelet aggregation by association with ILK and increasing ILK expression [14,15,16]. Apart from the maintenance of normal tissue functions, PARVB has also been reported to be involved in cancer progression and metastasis [12, 17, 18]. In breast cancer, PARVB promotes primary breast tumor formation and metastasis by activation of ILK/β-parvin/cofilin pathway [19]. However, the function of PARVB in kidney diseases, specifically in cisplatin-induced renal tubular injury is unknown. In this study, to elucidate whether and how PARVB plays roles on cisplatin-induced renal tubular injury, we generated proximal tubules-specific PARVB knockout mice (PARVB cKO mice) to assess the effect of PARVB deficiency on cisplatin-induced renal nephrotoxicity.
TAK1 (transforming growth factor-β-activated kinase 1, also called MAP3K7), is a member of the MAPKKK (mitogen-activated protein kinase kinase kinase) family, and functions as a central signalosome to mediate a series of biological processes [20]. Numerous knockout studies in mice have demonstrated that TAK1 regulates a wide range of physiological and pathological processes through activation of its downstream signaling pathways, including mitogen-activated protein kinases (MAPKs), NFκB and receptor-interacting protein kinase 1 (RIPK1) pathways [21,22,23]. Global depletion of TAK1 gene in mice causes embryonic lethality due to neural tube developmental abnormalities [24]. Tissue-specific deletion of TAK1 also results in a variety of tissue dysfunctions because of cell death and inflammation [25, 26]. For example, epithelial cell-specific deletion of TAK1 leads to epithelial cell apoptosis and severe inflammation. In kidney, depletion or inhibition of TAK1 prevents cisplatin-induced acute kidney injury in vivo and in vitro by inhibiting cisplatin-induced cell apoptosis and inflammation [23, 27]. These studies indicate the importance of an appropriate TAK1 expression level for maintaining tissue homeostasis. Therefore, the identification of factors that regulate TAK1 expression is necessary for the alleviation of TAK1-related injuries and disorders.
In this study, we demonstrate that cisplatin-induced nephrotoxicity depends on to a large extent on PARVB expression. Reducing PARVB expression alleviates cisplatin-induced acute kidney injury in vivo and in vitro. Moreover, our results reveal that PARVB associates with TAK1 to prevent ITCH-dependent TAK1 degradation. Therefore, depletion of PARVB in tubular cells decreases TAK1 expression, inhibits its downstream signaling and consequently alleviates cisplatin-induced tubular cell damage. Our findings provide evidence suggesting that targeting PARVB-TAK1 signaling could be a novel therapeutic strategy against cisplatin-induced acute kidney disease.
Materials and methods
Animal studies
PARVBfl/fl mice (C57BL/6J background) and γ-GT1-Cre transgenic mice (C57BL/6J background) were purchased from Cyagen Biosciences (Suzhou, China). PARVBfl/fl mice were crossed with γ-GT1-Cre mice to generate offspring with specific deletion of PARVB in proximal tubular epithelial cells (PARVB cKO). Genotyping was performed by PCR using DNA extracted from mouse tails. The primers used for genotyping were as follows: (1) γ-GT1-Cre transgene, sense: 5′-AGGTGTAGAGAAGGCACTTAGC-3′ and antisense: 5′-CTAATCGCCATCTTCCAGCAGG-3′, which generating a 202 bp fragment; (2) PARVB gene, sense: 5′-CCTAGATCCATCCCATGATACTTGC-3′ and anti-sense: 5′-TCTCAGATCCCATTTCGTAGCATTG-3′, which yielding 191 bp and 152 bp fragments for the floxed and wild-type alleles. All animals were born normally at the expected Mendelian frequency. A total of 131 offspring born from PARVBfl/+;γ-GT1-Cre+/+ intercross, 34 were WT mice (PARVB+/+;γ-GT1-Cre+/+), 65 were heterogenous mice (PARVBfl/+;γ-GT1-Cre+/+), 32 were PARVB cKO mice (PARVBfl/fl;γ-GT1-Cre+/+). The observed ratio is close to the expected Mendelian frequency 1:2:1. All experimental mice were fed in the standard animal cages and provided with regular water and food, without any special treatment except for the indicated experimental procedures. CO2 inhalation was used to euthanize all experimental mice.
To generate cisplatin-induced acute kidney injury mouse model [28,29,30,31,32,33], 10 male WT or 10 PARVB cKO mice at eight weeks of age weighing 25 ~ 30 g were administered with cisplatin (1 mg/mL solution in sterile saline; Sigma-Aldrich, P4394) at 15 mg/kg in a single intraperitoneal injection. 10 male WT or 10 PARVB cKO mice at eight weeks of age were administered with saline as control. Mice were sacrificed at 72 h after cisplatin or saline injection. The blood samples, urine samples and kidney tissues were collected for further analysis. In addition, 5 male WT and 5 PARVB cKO mice at eight weeks of age or 5 male WT and 5 PARVB cKO mice at twenty-four weeks of age without cisplatin treatment were analyzed by body weight and then were killed for further studies.
All animal experiments were performed following the guidelines and regulations and approved by the Institutional Animal Care and Use Committee at the Southern University of Science and Technology of China.
Histopathology and immunohistochemical staining
Kidney tissues of mice were fixed in 10% paraformaldehyde solution for 48 h immediately after the sacrifice, and the tissue samples were embedded in paraffin. Section (3 μm thick) were stained with Hematoxylin and Eosin (H&E) and Periodic Acid Schiff (PAS) according to the manufacturer’s instructions. A score was assigned according to the percentage of tissues in the corticomedullary region exhibiting renal acute injury per 10 high-power fields as follows [34]: 0 = 0%, 1 = < 10%, 2 = 10% ~ 25%, 3 = 25% ~ 50%, 4 = 50% ~ 75%, 5 = > 75%. Immunohistochemical staining (IHC) was performed with different antibodies against PARVB (Invitrogen, PA5-106425, 1:400), KIM-1(R&D, AF1750, 1:500), cleaved caspase-3 (CST, 9661 S, 1:500), p-MLKL (CST, D6E3G, 1:500), F4/80 (CST, 70076 S, 1:1000), CD11b (CST, 93169 S, 1:500) and TAK1(ABclonal, A12022, 1:200). After incubation with primary antibodies at 4 °C overnight, the slides were then stained with the MaxVisionTM HRP-Polymer anti-Mouse/Rabbit secondary antibody for 1 h at room temperature. Non-immune normal IgG was used to substitute primary antibodies as a negative control. Slides were scanned by Nanozoomer-S60. Sections of at least three individual kidney tissues in each group were taken for analysis. IHC images were analyzed by Image J software in at least ten randomly selected fields per section under 20 × objective.
Blood sample and tissue collection
Blood samples were collected from the hearts of mice. Briefly, the mice were deeply anesthetized with 2.5% Avertin (0.2 mL/10 g), and a cardiac puncture was performed to expose the heart. Using a 1 mL syringe (DOUBLE DOVE, 0102), 300 ~ 500 µL of fresh blood was drawn from the right ventricle and transferred into a 1.5 mL Eppendorf (EP) tube. The blood samples were then allowed to rest at 4 °C for 24 h to facilitate separation. Subsequently, the samples were centrifuged at 4 °C (5000 × g, 20 min) and the supernatant was collected and stored at -80 °C for further analysis. To harvest kidney tissue, the mice were put to death by inhalation of CO2. Both left and right kidneys were removed by a midline abdominal incision.
Urine sample collection
24 h urine samples were collected using metabolic cages, transferred to 1.5 mL EP tubes and centrifuged at 4 °C (5000 × g for 10 min). The supernatant was stored at -80 °C for further analysis. For the saline-treated mice, approximately 1 mL of clear and yellow urine was collected. In contrast, the urine from cisplatin-treated mice was less in volume, darker in color, and turbid.
Biochemical tests
Urine and blood samples were collected for biochemical analysis. Urine neutrophil gelatinase-associated lipocalin (NGAL) was measured in duplicate using a commercial mouse albumin ELISA quantitation kit, according to the manufacturer’s protocol (R&D, DY1857). Serum creatinine (SCr) was measured in duplicate using a commercial ELISA quantitation kit (Sigma-Aldrich, MAK080) following the manufacturer’s protocol. Serum TNF-α was measured in duplicate using a commercial ELISA quantitation kit, according to the manufacturer’s protocol (MULTI SCIENCES, EK182).
Cell culture
Human kidney tubular epithelial cell line (HK2) was purchased from Pricella (CL-0109). The cell lines were authenticated and performed mycoplasma contamination testing at the beginning of this study. HK2 cells were cultured in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12) (Gibco, 11320-033) supplemented with 10% FBS (CellorLab, CY103) and 100 U/mL penicillin and 100 µg/mL streptomycin (Invitrogen, 15140-122). HK2 cells were incubated at 37 °C with 5% CO2. HK2 cells were incubated with 15 µM cisplatin in culture medium for 24–48 h.
RNA interference assay
Small interfering RNA (siRNA) oligonucleotides against PARVB and ITCH were designed and synthesized by GenePharma (Suzhou, China). The siRNA sequences were as follows: (1) PARVB siRNA1, sense: 5′-GGUUCGAGCGGGAUGCCUUTT-3′ and antisense: 5′ -AAGGCAUCCCGCUCG AACCTT − 3′; (2) PARVB siRNA2, sense: 5′ - GGUGCUGGAAGCAGUACAUTT-3′ and antisense: 5′-AUGUACUGCUUCC AGCACTT-3′; (3) ITCH-siRNA, sense: 5′-GGAUCACAACUUGGUUCAATT-3′ and antisense: 5′-UUGAACCAAGUUGUGAUCCTT-3′; (4) Negative control siRNA, sense: 5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense: 5′-ACGUGACA CGUUCGGAGAATT-3′. HK2 cells were transfected with 20 µM siRNAs using Lipofectamine RNAiMax transfection reagent (Invitrogen, 13778150) according to the manufacturer’s instructions. After 24–48 h, proteins were extracted and transfection efficiency was evaluated by western blotting.
Flow cytometry
The extent of programmed cell death was examined using an Annexin-V-FITC apoptosis detection kit (Goonie, 100–101) according to the manufacturer’s instructions. HK2 cells were trypsinized and collected from the suspension by centrifugation. The cells were resuspended in 500 µL of Annexin V binding buffer and stained with 5 µL Annexin V-FITC and 5 µL propidium iodide (PI) in the dark. Apoptotic cells were detected using a flow cytometer (Beckman, USA), and the data were analyzed using FlowJo 7.6 software (TreeStar, USA).
RT-qPCR
Total RNA was extracted from kidney tissues or HK2 cells using Trizol reagent (Life Technology, Rockville, MD, USA). 1 µg of RNA was reverse transcribed into cDNA with a reverse transcription system kit according to the manufacturer’s instructions (TOYOBO, 037400) according to standard protocols. cDNA samples were subjected to quantitative RT-PCR using LightCycler 480 SYBR® Green I Master (Roche, 04887352001) with an Applied Biosystems 7500 Real-Time PCR system. The primer sequences used for RT-PCR were as follows: (1) GAPDH: sense: 5′-GTCTCCT CTGACTTCAACAGCG-3′ and antisense: 5′-ACCACCCTGTTGCTGTAGCCAA-3′; (2) PARVB (mouse): sense: 5′-GACATCCCACCCTATTCTTAACTCTGC-3′ and antisense: 5′- TCTCAGATCCCATTTCGTAGCATTG′-3′; (3) PARVB (human): sense: 5′-GGUCAGUGUAGUUGAUGUA-3′ and antisense: 5′-UACAUCAACUACAC UGACC-3′; (4) TAK1: sense: 5′-CAGAGCAACTCTGCCACCAGTA-3′ and antisense: 5′-CATTTGTGGCAGGAACTTGCTCC-3′; (5) TNF-α: sense: 5′-CTCTTCTGCCT GCTGCACTTTG-3′ and antisense: 5′-ATGGGCTACAGGCTTGTCACTC-3′; (6) IL-1β: sense: 5′-CCACAGACCTTCCAGGAGAATG-3′ and antisense: 5′-GTGCAGTTCAGTGATCGTACAGG-3′.
Western blotting
Proteins from HK2 cells were extracted with lysis buffer (Beyotime, P0013). 10 ~ 30 µg protein samples were separated by 10% ~ 12% SDS- polyacrylamide gel and transferred onto PVDF (polyvinylidene difluoride) membranes (Millipore Corp, ISEQ00010). PVDF membranes were then incubated with primary antibodies against GAPDH (ABclonal, AC035, 1:10000), PARVB (Bioswamp, 31388, 1:1,000), TAK1 (CST, 61-7300, 1:1000), P38 (CST, 8690 S, 1:1000), p-P38 (CST, 4511 S, 1:1000), ERK (CST, 4695 S, 1:1000), p-ERK (CST, 4370 S, 1:1000), RIPK3 (Abcam, 305054, 1:1000), p-RIPK3 (Abcam, 209384, 1:1000) and cleaved-caspase-3 (CST, 9664 S, 1:1000) overnight at 4 °C followed by incubation with secondary antibodies (Invitrogen, 1:10000) for 1 h at room temperature. The protein bands were visualized with an ECL kit (YAME, SQ101). The band intensities were quantified using Image J gel analysis software. All experiments were repeated at least three times.
Nano LC-MS/MS analysis
HK2 cell lysates (~ 6 mg) were immunoprecipitated with a mouse anti-PARVB antibody (Merck Millipore, MAB2620) or mouse control IgG (Santa Cruz biotechnology, SC2025). Each immunoprecipitated sample was then prepared for LC-MS/MS analysis as previously described [35, 36]. Briefly, samples were washed three times with 1 mL 50 mM ammonium bicarbonate (ABC). Following reduction with 10 mM Tris (2-carboxyethyl) phosphine hydrochloride (TCEP) and alkylation with 15 mM iodoacetamide (IAA) at 37 °C for 30 min, the beads were washed with 1 mL 50 mM ABC twice and digested with 10 µl 0.25 mg/mL trypsin (Promega) overnight at 37 °C. The digested peptides were washed with 60 µl 1% (v/v) formic acid (FA), dried in a speed vacuum and redissolved in 1% FA and desalted by C18-StageTips. The Tips were washed sequentially with 60 µL methanol, 60 µL solution A (80% (v/v) acetonitrile (ACN), 0.5% (v/v) acetic acid (AcOH)) and 60 µL 1% FA twice. Peptides were washed with 60 µL 1% FA twice and eluted by 80 µL solution A. Finally, eluted peptide mixtures were lyophilized to dryness and redissolved in 16 µL 0.1% FA for nano LC-MS/MS analysis. The raw data were searched using MaxQuant software (version 1.5.5.1). All statistical and bioinformatics analyses were performed using Perseus software (version 1.5.5.3).
Co-immunoprecipitation
Proteins from HK2 cells were extracted with lysis buffer (Beyotime, P0013) supplemented with protease inhibitors. Samples were incubated for 30 min at 4℃ and centrifuged (14,000 × g for 15 min at 4 °C) to collect supernatant. Equal amounts of total lysates (4 ~ 6 mg) were incubated with primary antibody and 40 µL protein A/G Sepharose beads overnight at 4 °C. Then beads were rinsed three times in 1× PBS with protease inhibitors. Samples were then processed for western blotting.
Statistical analyses
All data represent as mean ± SEM. Two-tailed Student’s t test was used to compare two groups of samples. One-way ANOVA was used for multiple comparisons, followed by with Sidak Post Hoc Test. P values less than 0.05 were considered significant. Prism 8 (GraphPad) was used for statistical analysis.
Results
PARVB expression was down-regulated in cisplatin-induced tubular injury
To explore the potential role of PARVB in renal tubular injury, we assessed PARVB protein expression in mouse renal tubulointerstitial sections by using a cisplatin-induced acute kidney injury (AKI) mouse model. Immunohistochemical staining showed that the protein level of PARVB was reduced in mouse tubulointerstitial sections after cisplatin treatment (Fig. 1A). Particularly, PARVB expression was largely decreased in the proximal tubules of cisplatin-treated mice (Fig. 1A). To verify these findings, we mimicked tubular injury in vitro by treating a widely used human-derived proximal tubular epithelial cells (HK2) with cisplatin. The results showed that both PARVB mRNA and protein expression levels were markedly down-regulated at multiple time points after cisplatin induction in HK2 cells (Fig. 1, B and C). Collectively, these observations imply that PARVB expression could be potentially involved in renal tubular injury.

PARVB expression is down-regulated in mouse tissues or human tubular cells treated with cisplatin. (A) Eight-week-old wild-type mice were treated with vehicle or cisplatin (15 mg/kg) for three days. Kidney sections from vehicle or cisplatin-treated mice were stained with antibodies against PARVB. Representative images and quantification analysis were shown. Scale bar, 50 μm. **P < 0.01 vs. Vehicle, n = 3 mice for each group. (B) Immunoblotting analysis of PARVB protein expression in HK2 cells treated with cisplatin at different time points (left panel). Quantification analysis was shown in the right panel. *P < 0.05, ***P < 0.001 vs. Vehicle. n = 4 independent experiments. (C) qPCR analysis of PARVB mRNA expression in HK2 cells treated with cisplatin at different time points. **P < 0.01 vs. Vehicle. n = 3 independent experiments
PARVB deficiency alleviates cisplatin-induced tubular injury
To better understand the function of PARVB in renal tubular injury, we generated proximal tubules-specific PARVB knockout mice (referred to as PARVB cKO hereafter) by using the Cre-LoxP system (γ-glutamyltransferase 1, γ-GT1-Cre) that targets exon 4 of the PARVB allele (Fig. 2A). Mice of all genotypes were viable and born at the expected Mendelian frequency. PARVB cKO mice, and control littermates, PARVB+/+; γ-GT1-Cre (referred to as WT hereafter), were identified by mouse tail genotyping (Fig. 2B). qPCR analysis indicated PARVB mRNA expression was successfully diminished in primary proximal tubules isolated from PARVB cKO mice compared to that from WT mice (Fig. 2C). Immunohistochemical staining of mouse renal sections further confirmed that PARVB was knocked out in proximal tubules of PARVB cKO mice (Fig. 2D). We then evaluated the global phenotypes of PARVB cKO mice. PARVB cKO and WT mice gave birth with normal body sizes (~ 1 g at day 1) and displayed normal behaviors. Moreover, PARVB cKO mice had the same body weights as WT mice at eight and twenty-four weeks of age (Fig. 2E). In addition, analyzing the whole kidneys isolated from 8-week-old and twenty-four-week-old PARVB cKO mice and WT littermates showed that knockout of PARVB in proximal tubules did not impair normal mouse kidney development (Supplementary Fig. 1A). Both renal histologic analysis and functional tests further confirmed these conclusions (Fig. 2, F-I and Supplementary Fig. 1B). These data indicated that proximal tubules-specific depletion of PARVB does not affect normal kidney function. Next, we investigated the consequence of PARVB depletion in cisplatin-induced renal tubular injury. As reported previously [7, 37], cisplatin significantly induced acute tubular injury in WT mice, characterized by tubular dilatation, tubular cell loss and proteinaceous casts formation (Fig. 2F). However, depletion of PARVB alleviated cisplatin-induced tubular damages analyzed by H&E and Periodic Acid-Schiff (PAS) staining (Fig. 2F). The tubular injury score in PARVB cKO mice was also decreased compared to that in WT littermates (Fig. 2G). Additionally, urine neutrophil gelatinase-associated lipocalin (NGAL), and serum creatinine (SCr), predictive biomarkers for acute kidney injury [38,39,40,41], were assessed. As expected, the levels of NGAL and SCr were significantly elevated in WT mice after cisplatin induction, whereas knockout of PARVB in renal proximal tubules reduced these levels (Fig. 2, H and I), suggesting that PARVB plays an important role in regulating cisplatin-induced nephropathy.

PARVB deficiency alleviates cisplatin-induced renal tubular injury in mice. (A) The diagram depicts the strategy for the generation of PARVB cKO mice. Mice expressing γ-GT1-Cre were crossed with mice carrying floxed PARVB locus (exon 4). (B) Representative PCR analysis of extracted genomic DNA from tail clippings. PCR product of floxed (191 bp) and wild-type (152 bp) were shown. Cre PCR product (202 bp) was also indicated. (C) qPCR analysis of PARVB mRNA expression in isolated primary tubular cells from wild-type mice (WT) and PARVB cKO mice (cKO). ***P < 0.001 vs. WT. n = 3 mice for each group. (D) Representative images of kidney sections stained with antibody against PARVB in WT and PARVB cKO mice Scale bar: 50 μm. (E) PARVB cKO mice showed no significant difference on body weight compared to WT mice at 8 and 24 weeks of age. n = 5 mice for each group. (F) Kidney sections from WT and PARVB cKO mice treated with vehicle or cisplatin (15 mg/kg) for three days were subjected to Haematoxylin and Eosin (H&E) or Periodic acid-Schiff (PAS) staining. Arrows indicated renal tubular dilatation and proteinaceous casts. Scale bar, 20 μm. (G) Quantification of renal histopathologic injury score in WT and PARVB cKO mice treated with or without cisplatin (15 mg/kg) for three days. ***P < 0.001 vs. WT + Cisplatin. n = 6 mice for each group. (H) Quantification of urinary Neutrophil gelatinase-associated lipocalin (NGAL) level in WT and PARVB cKO mice treated with or without cisplatin (15 mg/kg) for three days. ***P < 0.001 vs. WT + Cisplatin. n = 6 mice for each group. (I) Quantification of serum creatinine (SCr) in WT and PARVB cKO mice treated with or without cisplatin (15 mg/kg). **P < 0.01 vs. WT + Cisplatin. n = 6 mice for each group
PARVB deficiency prevents cisplatin-induced tubular cell death
Cisplatin has been reported to induce renal epithelial cell apoptosis and necroptosis, and eventually leads to renal dysfunction [42]. To examine whether PARVB ablation alleviates cisplatin-induced renal tubular injury through hindering tubular cell death, we performed immunohistochemical (IHC) staining of mouse kidney sections using antibodies against kidney injury molecule-1 (KIM-1) and cleaved-caspase-3, two well-known markers for renal cell injury and apoptosis. As shown in Fig. 3A and B, injured cells or apoptotic cells were barely detectable in the renal tissues of both WT and PARVB cKO mice without cisplatin treatment. However, cisplatin induction triggered renal cell injury and cell death in WT mice, and PARVB depletion significantly attenuated kidney injury and renal cell apoptosis (Fig. 3, A and B). To further confirm a role for PARVB in cisplatin-induced renal tubular cell death, we tested the effect of PARVB ablation on cell death in human-derived proximal tubular cells (HK2 cells). The results showed that the knockdown of PARVB in HK2 cells indeed reduced the percentage of cisplatin-induced dead cells, as measured by flow cytometry analysis (Fig. 3, C and D). Moreover, immunoblotting analysis revealed that knockdown of PARVB reduced cisplatin-induced cleaved-caspase 3, phosphor-RIPK3 and phosphor-RIPK1 levels, the central regulators of apoptosis and necroptosis [43,44,45,46] (Fig. 3, E-G). To further verify these findings, we isolated kidney sections from WT and PARVB cKO mice with or without cisplatin treatment and performed immunohistochemical staining with anti-p-MLKL (p-MLKL as a marker of necrosis [45]). As expected, the results showed that p-MLKL expression was hardly detected in the renal tissues from both WT and PARVB cKO mice without cisplatin treatment. However, cisplatin treatment increased p-MLKL expression in WT mice, while depletion of PARVB markedly reduced cisplatin-induced p-MLKL densities (revised Fig. 3H). Collectively, these results indicate that PARVB depletion significantly prevents cisplatin-induced tubular cell death.

PARVB deficiency attenuates cisplatin-induced renal cell death. (A) Representative images of kidney sections stained with antibody against KIM-1, a biomarker of renal tubular injury, in WT and PARVB cKO mice after cisplatin (15 mg/kg) or vehicle treatment. Scale bar: 20 μm. Quantification of the percentage of KIM-1 positive area was shown in the lower panel. ***P < 0.001 vs. WT + Cisplatin. n = 4 mice for each group. (B) Representative images of kidney sections stained with antibody against cleaved-caspase-3 in WT and PARVB cKO mice after cisplatin (15 mg/kg) or vehicle treatment. Scale bar: 10 μm. Quantification of the ratio of cleaved caspase-3-positive cell number to total cell number was shown in the lower panel. ***P < 0.001 vs. WT + Cisplatin. n = 6 mice for each group. (C) Flow cytometry analysis of the apoptotic cells in control (Nc siRNA) and PARVB knockdown (PB siRNA1 and PB siRNA2) HK2 cells with or without cisplatin (15 µM) treatment. CP, cisplatin. (D) Quantification analysis of (C) was shown. ***P < 0.001 vs. HK2 + Cisplatin, n = 4 independent experiments. (E) Immunoblotting analysis of the expression level of RIPK3, phosphorylated (p)-RIPK3 and cleaved-caspase-3 in control (Nc siRNA) and PARVB knockdown (PB siRNA1 and PB siRNA2) HK2 cells with or without cisplatin (15 µM) treatment. (F) Quantification analysis in (E) was shown. ***P < 0.001 vs. HK2 + Cisplatin. n = 3 independent experiments. (G) Immunoblotting analysis of the expression level of RIPK1, phosphorylated (p)-RIPK1 in control (Nc siRNA) and PARVB knockdown (PB siRNA1 and PB siRNA2) HK2 cells with or without cisplatin (15 µM) treatment. Quantification analysis was shown in the right panel. **P < 0.01 vs. HK2 + Cisplatin. n = 3 independent experiments. (H) Representative images of kidney sections stained with antibody against phosphorylated (p)-MLKL in WT and PARVB cKO mice after cisplatin (15 mg/kg) or vehicle treatment. Scale bar: 50 μm. Quantification of the percentage of p-MLKL positive area was shown in the right panel. ***P < 0.001 vs. WT + Cisplatin. n = 3 mice for each group
PARVB deficiency ameliorates cisplatin-induced renal inflammation
Cisplatin causes tubular injury not only because it induces tubular cell death but also because it aggravates interstitial inflammation [47]. Therefore, to investigate the role of PARVB in regulating cisplatin-induced renal inflammation, we analyzed the number of CD11b+-labelled myeloid cells and F4/80+-labelled macrophages in renal tissues. The results showed that the infiltration of CD11b+ myeloid cells and F4/80+ macrophages was largely reduced in renal tissues from PARVB cKO mice compared to those from WT mice after cisplatin induction (Fig. 4, A and B). These findings were further validated by measurement of pro-inflammatory cytokine expression, such as TNF-ɑ, in mouse serum isolated from WT and PARVB cKO mice (Fig. 4C). An obvious reduction of serum TNF-ɑ was found in cisplatin-treated PARVB cKO mice (Fig. 4C). To verify these in vivo data, we analyzed the effect of PARVB on cisplatin-induced inflammatory response in HK2 cells. Consistently, the knockdown of PARVB decreased the production of pro-inflammatory cytokines, TNF-ɑ and IL-1β, in HK2 cells in response to cisplatin (Fig. 4, D and E). Taken together, these data support a protective role of PARVB deficiency in cisplatin-induced inflammation.

PARVB deficiency protects against cisplatin-induced renal inflammation. (A) Representative images of kidney sections stained with antibody against CD11b (a myeloid cell marker) in WT and PARVB cKO mice after cisplatin (15 mg/kg) or vehicle treatment. Scale bar: 50 μm. Quantification analysis was shown in the right panel. ***P < 0.001 vs. WT + Cisplatin. n = 3 mice for each group. (B) Representative images of kidney sections stained with antibody against F4/80 (a macrophage marker) in WT and PARVB cKO mice after cisplatin (15 mg/kg) or vehicle treatment. Scale bar: 20 μm. Quantification analysis was shown in the right panel. ***P < 0.001 vs. WT + Cisplatin. n = 6 mice for each group. (C) ELISA analysis of TNF-ɑ level in mouse serum isolated from WT or PARVB cKO mice with or without cisplatin (15 mg/kg) treatment. **P < 0.01 vs. WT + Cisplatin. n = 5 mice for each group. (D-E) qPCR analysis of TNF-ɑ(D) and IL-1β(E) mRNA expression in control (Nc siRNA) and PARVB knockdown (PB siRNA1 and PB siRNA2) HK2 cells with or without cisplatin (15 µM) treatment. *P < 0.05, **P < 0.01 vs. HK2 + Cisplatin, n = 3 independent experiments
PARVB deficiency attenuates cisplatin-induced tubular injury through regulation of TAK1 expression
To comprehensively understand the molecular mechanism by which PARVB regulates cisplatin-induced tubular injury, we performed RNA sequencing (RNA-seq) analysis to elucidate the pathways affected by PARVB. Results showed that 1343 genes were down-regulated and 1898 genes were up-regulated in the PARVB knockdown cells compared to WT cells after cisplatin treatment. The result of Kyoto Encyclopedia of Genes and Genomes (KEGG) analyzed by OmicShare tools (http://www.omicshare.com/tools) revealed that these differentially expressed genes were associated with apoptotic pathways and inflammation, such as MAPK, TGF-β and TNF signaling pathways (Fig. 5, A and B). To better exploit the mechanism underlying the regulation of tubular cell death and inflammation by PARVB, we sought to identify proteins that are associated with PARVB in HK2 cells treated with cisplatin by Nanoscale liquid chromatography coupled to tandem mass spectrometry (nano LC-MS/MS) approach (Fig. 5C) [48, 49]. One of the identified PARVB-associated proteins is TAK1. Because TAK1 is a central regulator of the MAPK pathway that ultimately controls cell death and inflammation and is involved in cisplatin-induced tubular injury [20, 23, 25, 50], we chose it for further study. PARVB-TAK1 interaction was first verified by sequential co-immunoprecipitation in HK2 cells (Fig. 5, D and E). We then explored the functional influence of PARVB on TAK1. Given the critical role of TAK1 protein level in the pathogenesis of cisplatin-induced tubular injury [23], we sought to investigate whether PARVB could regulate TAK1 expression. Interestingly, the knockdown of PARVB in HK2 cells resulted in a remarkable reduction of TAK1 protein expression upon cisplatin treatment (Fig. 5F). This observation was further confirmed by TAK1 IHC staining in renal tissues isolated from PARVB cKO mice and WT mice that received cisplatin (Fig. 5G). However, the mRNA level of TAK1 was not altered in PARVB knockdown cells compared to wild-type cells with or without cisplatin (Supplementary Fig. 2), suggesting that the regulation of TAK1 by PARVB is likely at the post-transcriptional level. Previous studies indicate that TAK1 undergoes proteasome-mediated degradation upon extracellular stimuli [51, 52]. Therefore, we sought to test whether PARVB stabilized TAK1 protein expression by regulating its degradation. To do this, we treated wild-type and PARVB knockdown HK2 cells with proteasomal inhibitor MG132. As predicted, the proteasomal inhibitor effectively reversed PARVB deficiency-induced TAK1 degradation after cisplatin treatment (Fig. 5H). Further analysis confirmed that knockdown of PARVB increased the ubiquitination level of TAK1 upon MG132 treatment (Fig. 5I). Collectively, these data strongly imply that PARVB associates with TAK1 and thereby prevents proteasome-mediated degradation of TAK1 in renal tubular cells. TAK1 is well-known to control cell death and inflammation by regulating its downstream effectors such as MAPKs [20, 53]. Given the aforementioned RNA-seq data showed that depletion of PARVB downregulated MAPK signaling (Fig. 5B), we then confirmed the effect of PARVB on MAPK signaling by immunoblotting analysis. As expected, PARVB depletion efficiently reduced the cisplatin-induced phosphorylation level of ERK and p38 MAPKs (Fig. 5J). Together, these results suggest that depletion of PARVB accelerates TAK1 protein degradation and restrains its downstream MAPK signaling activation, thereby attenuating cisplatin-induced tubular cell death and inflammation.

PARVB promotes cisplatin-induced renal cell death and inflammation through regulation of TAK1 protein expression. (A) Volcano plot of differentially expressed genes in cells as indicated in the figure. (B) KEGG analysis of the enriched differentially expressed genes in control (Nc siRNA) and PARVB knockdown (PB siRNA1) HK2 cells treated with cisplatin (15 µM). KEGG, Kyoto Encyclopedia of Genes and Genomes. (C) Volcano plot showing PARVB association proteins identified using PARVB immunoprecipitation followed by Mass Spectrometry (MS) in HK2 cells treated with cisplatin. The positions of PARVB and TAK1 were indicated. (D) HK2 cell lysates were immunoprecipitated with anti-PARVB antibody or mouse control IgG (mIgG) followed by immunoblotting with antibodies as indicated. (E) HK2 cell lysates were immunoprecipitated with anti-TAK1 antibody or rabbit control IgG (rIgG) followed by immunoblotting with antibodies as indicated. (F) Immunoblotting analysis of the expression level of TAK1 in control (Nc siRNA) and PARVB knockdown (PB siRNA1 and PB siRNA2) HK2 cells with or without cisplatin. Quantification analysis was shown in the right panel. **P < 0.01, ***P < 0.001 vs. HK2 + Cisplatin. n = 4 independent experiments. (G) Representative images of kidney sections stained with antibody against TAK1 in WT and PARVB cKO mice after cisplatin (15 mg/kg) or vehicle treatment. Scale bar: 50 μm. Quantification analysis was shown in the right panel. **P < 0.01 vs. WT + Cisplatin. n = 3 mice for each group. (H) Immunoblotting analysis of TAK1 protein level in control (Nc siRNA) or PARVB knockdown (PB siRNA1) HK2 cells treated with proteasomal inhibitor MG132 for 6 h and cisplatin for 48 h. MG132, 10 µM. Quantification analysis was shown in the lower panel. ***P < 0.001 vs. Nc siRNA + Vehicle. n = 3 independent experiments. (I) Control or PARVB knockdown HK2 cells were treated with MG132 for 6 h and cisplatin for 48 h, and then subjected to immunoprecipitated with anti-TAK1 antibody, followed by immunoblotting with antibodies as indicated. Ub: ubiquitin. (J) Immunoblotting analysis of the protein expression level of ERK, phosphorylated (p)-ERK, P38, phosphorylated (p)-P38 in control (Nc siRNA) and PARVB knockdown (PB siRNA1 and PB siRNA2) HK2 cells with or without cisplatin treatment. Quantification analysis of the ratio of p-ERK/ERK and p-P38/P38 was shown in the right panel. ***P < 0.001 vs. HK2 + Cisplatin. n = 3 independent experiments. CP, cisplatin
PARVB deficiency promotes ITCH E3 ligase-mediated TAK1 degradation
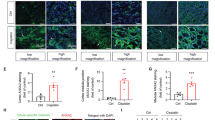
It has been previously reported that a set of E3 ubiquitin ligases, such as ITCH (Itchy E3 ubiquitin protein ligase), USP4 (Ubiquitin specific peptidase 4) and USP8 (Ubiquitin specific peptidase 8) can catalyze TAK1 polyubiquitination and promote its degradation after stimuli [50, 54,55,56,57,58,59]. Therefore, we speculate that PARVB deficiency promotes TAK1 degradation by regulating the function of E3 ligases. To determine this, we silenced a series of E3 ligases of TAK1 in PARVB-deficient HK2 cells. The results showed that ITCH silencing, but not silencing of other E3 ligases (USP4 and USP8), reversed the PARVB-deficiency-induced TAK1 degradation, suggesting ITCH is involved in the regulation of PARVB-mediated TAK1 degradation (Fig. 6A and Supplementary Fig. 3). More importantly, the depletion of PARVB significantly enhanced the interaction between TAK1 and ITCH, implying PARVB prevents TAK1 degradation by inhibiting the ITCH-TAK1 association (Fig. 6B). Proximity ligation analysis further confirmed this observation (Fig. 6C). Higher levels of TAK1-ITCH formation (red puncta) were detected in PARVB knockdown cells after treatment with cisplatin (Fig. 6C), suggesting that PARVB deficiency facilitates ITCH association with TAK1 and promotes TAK1 degradation.

PARVB blocks ITCH-dependent TAK1 degradation. ITCH protein level was silenced by siRNA in Control (Nc siRNA) or PARVB knockdown (PB siRNA1) HK2 cells. (A) Immunoblotting analysis of the protein expression level of ITCH or TAK1 in cells as specified in the figure with cisplatin treatment (15 µM). Quantification analysis was shown in the right panel. ***P < 0.001 vs. Nc siRNA + Cisplatin, n = 4 independent experiments. (B) Cell lysates were immunoprecipitated with anti-TAK1 antibody followed by immunoblotting with antibodies as indicated. (C) Representative images of in situ PLA analyses of the TAK1-ITCH interaction (red dots) in cells as specified in the figure were shown in the upper panel. Cell nuclei were visualized with DAPI (blue). Scale bar: 10 μm. Quantification of PLA puncta per cell/field was shown in the lower panel. ***P < 0.001 vs. Nc siRNA + Cisplatin, n = 6 independent experiments. (D) Immunoblotting analysis of the protein expression level of ERK, phosphorylated (p)-ERK and P38, phosphorylated (p)-P38 in cells as specified in the figure with cisplatin treatment (15 µM). Quantification analysis was shown in the right panel. **P < 0.01, ***P < 0.001 vs. Nc siRNA + Cisplatin, n = 3 independent experiments. (E) Flow cytometry analysis of the apoptotic cells in cells as specified in the figure with cisplatin treatment (15 µM). Quantification analysis was shown in the right panel. ***P < 0.001 vs. Nc siRNA + Cisplatin, n = 3 independent experiments. (F) Immunoblotting analysis of the expression level of RIPK3, phosphorylated (p)-RIPK3 and cleaved-caspase-3 in cells as specified in the figure with cisplatin treatment. Quantification analysis was shown in the right panel. ***P < 0.001 vs. Nc siRNA + Cisplatin. n = 3 independent experiments. (G) qPCR analysis of TNF-ɑ and IL-1β mRNA expression in cells as specified in the figure with or without cisplatin (15 µM) treatment. ***P < 0.001 vs. Nc siRNA + Cisplatin, n = 3 independent experiments. CP, Cisplatin
The next question we attempt to answer is whether ITCH-mediated TAK1 degradation is crucial for PARVB-regulated tubular cell death and inflammation. Immunoblotting analysis showed that ITCH silencing significantly reverses the restraint of MAPK signaling in PARVB knockdown cells upon cisplatin stimuli (Fig. 6D). Consequently, PARVB-deficiency-induced inhibition of tubular cell death and inflammation was notably restored by ITCH silencing (Fig. 6, E-G). Collectively, our data demonstrate that ITCH-dependent TAK1 degradation is critical for PARVB-mediated regulation of cisplatin nephrotoxicity.
Overexpression of TAK1 in PARVB-deficient cells aggravates cisplatin-induced renal injury
To further verify TAK1 protein level is important for PARVB-mediated cisplatin nephropathy, we re-expressed TAK1 or PARVB in PARVB knockdown HK2 cells. As expected, the re-expression of PARVB increased TAK1 protein in PARVB knockdown cells, confirming the above findings that PARVB regulates TAK1 protein levels (Fig. 7A). More importantly, overexpression of TAK1, like that of PARVB, effectively rescued PARVB-deficiency-inhibited MAPK signaling upon cisplatin treatment (Fig. 7A). In addition, overexpression of TAK1 or PARVB worsened cisplatin-induced tubular cell apoptosis and necroptosis in PARVB knockdown cells (Fig. 7, B and C). Pro-inflammatory response was exacerbated by overexpression of TAK1 or PARVB in PARVB knockdown cells as well (Fig. 7D), confirming PARVB-mediated cisplatin nephropathy, at least in part, through regulating TAK1 protein expression.

Overexpression of TAK1 in PARVB-deficient cells aggravates cisplatin-induced tubular injury. Control (Nc siRNA) or PARVB knockdown (PB siRNA1) HK2 cells were infected with lentiviral vectors encoding full-length PARVB, TAK1 or empty vector. (A) Immunoblotting analysis of the protein expression level of ERK, phosphorylated (p)-ERK and p38, phosphorylated (p)-p38, TAK1 or PARVB in cells as specified in the figure with cisplatin (15 µM) treatment. Quantification analysis was shown in the right panel. ***P < 0.001 vs. Nc siRNA + Cisplatin, n = 3 independent experiments. CP, Cisplatin. (B) Flow cytometry analysis of the apoptotic cells in cells as specified in the figure with cisplatin treatment (15 µM). Quantification analysis was shown in the right panel. **P < 0.01 vs. Nc siRNA + Cisplatin, n = 3 independent experiments. (C) Immunoblotting analysis of the expression level of RIPK3, phosphorylated (p)-RIPK3 and cleaved-caspase-3 in cells as specified in the figure with cisplatin treatment. Quantification analysis was shown in the right panel. ***P < 0.001 vs. Nc siRNA + Cisplatin. n = 3 independent experiments. (D) qPCR analysis of TNF-ɑ and IL-1β mRNA expression in cells as specified in the figure with cisplatin treatment. **P < 0.01, ***P < 0.001 vs. Nc siRNA + Cisplatin, n = 3 independent experiments. (E) Schematic illustration of the mechanism of PARVB regulation of cisplatin-induced acute renal injury
Discussion
Cisplatin-induced acute kidney injury is a significant clinical issue that limits cisplatin usage in the treatment of cancer patients [60]. Cisplatin tends to accumulate in the renal tubular cells and triggers tubular cell apoptosis and necrosis, impairing kidney function [61]. In addition, cisplatin can induce a tubular cell inflammatory response, which recruits immune cells to renal tissues, further exacerbating kidney damage [62]. Therefore, investigation of key molecular targets for preventing cisplatin-induced acute kidney injury has clinical significance. In this study, we identified a novel role for PARVB in cisplatin-induced acute kidney injury and provided evidence that targeting PARVB-TAK1 signaling is a promising therapeutic intervention for cisplatin-induced kidney damage.
TAK1 is a master regulator of various cellular processes, including inflammation and cell death [63, 64]. Aberrant expression of TAK1 has been implicated in multiple diseases, including cisplatin-induced kidney disease [23, 65,66,67]. Therefore, TAK1 expression is tightly controlled at multiple levels in normal cells, including transcriptional, post-transcriptional, and post-translational regulation [65]. Ubiquitination modification is one of the most effective and efficient regulations of TAK1 expression upon receiving extracellular signals. So far, several E3 ubiquitin enzymes, such as ITCH, USP4 and USP8 have been reported to regulate TAK1 expression and thus to modulate TAK1-mediated cell fate [50, 54,55,56,57,58,59]. However, how to maintain the homeostasis of TAK1 protein expression upon cellular stimuli was unclear. In the current study, we identified PARVB as a novel TAK1-interacting protein that blocks ITCH-dependent proteasomal degradation of TAK1. Depletion of PARVB reduced cisplatin-induced upregulation of TAK1 expression and effectively protected against cisplatin-induced tubular cell injury. Mechanistically, PARVB deficiency enhanced ITCH interaction with TAK1, further promoting ITCH-mediated TAK1 degradation and downstream events. However, how PARVB suppresses ITCH-dependent TAK1 degradation remains to be determined. One possibility is that PARVB might competitively inhibit ITCH association with TAK1. Alternatively, PARVB might regulate the function of other factors that recruit ITCH and target TAK1 for degradation. It will be important to investigate these possibilities in future studies.
How could PARVB alleviate cisplatin-induced kidney damage? In this study, we propose a model (Fig. 7E). In wild-type renal tubular cells with relatively high levels of PARVB, TAK1 is expressed at a relatively high level, as PARVB associates with TAK1 and prevents ITCH-mediated TAK1 degradation upon cisplatin stimuli. Thus, renal tubular cells suffer cell death and inflammation in response to cisplatin. However, in PARVB-depleted cells, reduced levels of PARVB allow increased ITCH association with TAK1, resulting in increased degradation of TAK1, downstream signaling inactivation and thereby preventing tubular cell injury.
In summary, we have demonstrated a novel role of PARVB in the regulation of cisplatin-induced acute kidney injury. We provide strong evidence suggesting that PARVB functions in this process through association with TAK1 and regulation of ITCH-dependent TAK1 degradation, thereby attenuating cisplatin-induced renal injury. Given the important role of PARVB-TAK1 axis in the cisplatin-induced AKI, targeting this signaling may provide a promising therapeutic strategy for alleviating cisplatin-caused nephrotoxicity. Although our current study focuses on cisplatin-induced kidney damage, it will be interesting to investigate in future studies whether the protective role of PARVB deficiency is applicable to other acute kidney injuries such as ischemia-reperfusion injury.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary files. Requests for materials should be addressed to YS.
References
Ghosh S, Cisplatin (2019) The first metal based anticancer drug. Bioorg Chem 88:102925
Yao X, Panichpisal K, Kurtzman N, Nugent K (2007) Cisplatin nephrotoxicity: a review. Am J Med Sci 334:115–124
Dasari S, Tchounwou PB (2014) Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol 740:364–378
Khairoun M, Uffen JW, Ocak G, Koopsen R, Haitjema S, Oosterheert JJ et al (2021) The incidence, mortality and renal outcomes of acute kidney injury in patients with suspected infection at the emergency department. PLoS ONE 16:e0260942
Dolman ME, Harmsen S, Storm G, Hennink WE, Kok RJ (2010) Drug targeting to the kidney: advances in the active targeting of therapeutics to proximal tubular cells. Adv Drug Deliv Rev 62:1344–1357
Ozkok A, Edelstein CL (2014) Pathophysiology of cisplatin-induced acute kidney injury. Biomed Res Int ; 2014: 967826
Holditch SJ, Brown CN, Lombardi AM, Nguyen KN, Edelstein CL (2019) Recent advances in models, mechanisms, biomarkers, and interventions in Cisplatin-Induced Acute kidney Injury. Int J Mol Sci ; 20
Manohar S, Leung N (2018) Cisplatin nephrotoxicity: a review of the literature. J Nephrol 31:15–25
Sepulveda JL, Wu C (2006) The parvins. Cell Mol Life Sci 63:25–35
Rosenberger G, Jantke I, Gal A, Kutsche K (2003) Interaction of alphaPIX (ARHGEF6) with beta-parvin (PARVB) suggests an involvement of alphaPIX in integrin-mediated signaling. Hum Mol Genet 12:155–167
Yamaji S, Suzuki A, Sugiyama Y, Koide Y, Yoshida M, Kanamori H et al (2001) A novel integrin-linked kinase-binding protein, affixin, is involved in the early stage of cell-substrate interaction. J Cell Biol 153:1251–1264
Papachristou DJ, Gkretsi V, Rao UN, Papachristou GI, Papaefthymiou OA, Basdra EK et al (2008) Expression of integrin-linked kinase and its binding partners in chondrosarcoma: association with prognostic significance. Eur J Cancer 44:2518–2525
Thievessen I, Suhr F, Vergarajauregui S, Böttcher RT, Brixius K, Rosenberger G et al (2022) The focal adhesion protein β-parvin controls cardiomyocyte shape and sarcomere assembly in response to mechanical load. Curr Biol 32:3033–
Stiegler AL, Draheim KM, Li X, Chayen NE, Calderwood DA, Boggon TJ (2012) Structural basis for Paxillin binding and focal adhesion targeting of β-Parvin. J Biol Chem 287:32566–32577
Yamaji S, Suzuki A, Kanamori H, Mishima W, Yoshimi R, Takasaki H et al (2004) Affixin interacts with alpha-actinin and mediates integrin signaling for reorganization of F-actin induced by initial cell-substrate interaction. J Cell Biol 165:539–551
Tu Y, Zhang YHY, Hua Y, Wu C (2001) A New Focal adhesion protein that interacts with integrin-linked kinase and regulates cell adhesion and spreading. J Cell Biol 153(3):585–598
Wu CF, Ng KF, Chen CS, Chang PL, Chuang CK, Weng WH et al (2010) Expression of parvin-beta is a prognostic factor for patients with urothelial cell carcinoma of the upper urinary tract. Br J Cancer 103:852–860
Geramoutsou C, Nikou S, Karavias D, Arbi M, Tavlas P, Tzelepi V et al (2022) Focal adhesion proteins in hepatocellular carcinoma: RSU1 a novel tumour suppressor with prognostic significance. Pathol Res Pract 235:153950
Shibue T, Brooks MW, Weinberg RA (2013) An integrin-linked machinery of cytoskeletal regulation that enables experimental tumor initiation and metastatic colonization. Cancer Cell 24:481–498
Mihaly SR, Ninomiya-Tsuji J, Morioka S (2014) TAK1 control of cell death. Cell Death Differ 21:1667–1676
Gao J, Liu Y, Chen J, Tong C, Wang Q, Piao Y (2022) Curcumin treatment attenuates cisplatin-induced gastric mucosal inflammation and apoptosis through the NF- kappa B and MAPKs signaling pathway. Hum Exp Toxicol 41:9603271221128738
Zhou J, Fan Y, Zhong J, Huang Z, Huang T, Lin S et al (2018) TAK1 mediates excessive autophagy via p38 and ERK in cisplatin-induced acute kidney injury. J Cell Mol Med 22:2908–2921
Zhou J, An CL, Jin XG, Hu ZY, Safirstein RL, Wang YL (2020) TAK1 deficiency attenuates cisplatin-induced acute kidney injury. Am J Physiol-Renal 318:F209–F215
Jeanes A, Coulthard LG, Mantovani S, Markham K, Woodruff TM (2015) Co-ordinated expression of innate immune molecules during mouse neurulation. Mol Immunol 68:253–260
Ma FY, Tesch GH, Ozols E, Xie M, Schneider MD, Nikolic-Paterson DJ (2011) TGF-beta1-activated kinase-1 regulates inflammation and fibrosis in the obstructed kidney. Am J Physiol Ren Physiol 300:F1410–1421
Simmons AN, Kajino-Sakamoto R, Ninomiya-Tsuji J (2016) TAK1 regulates Paneth cell integrity partly through blocking necroptosis. Cell Death Dis 7:e2196
Zhou J, Fan YL, Zhong JY, Huang ZX, Huang T, Lin S et al (2018) TAK1 mediates excessive autophagy via p38 and ERK in cisplatin-induced acute kidney injury. J Cell Mol Med 22:2908–2921
Li JZ, Xu Z, Jiang L, Mao JH, Zeng ZF, Fang L et al (2014) Rictor/mTORC2 protects against cisplatin-induced tubular cell death and acute kidney injury. Kidney Int 86:86–102
Li M, Li CM, Ye ZC, Huang JY, Li Y, Lai WY et al (2020) Sirt3 modulates fatty acid oxidation and attenuates cisplatin-induced AKI in mice. J Cell Mol Med 24:5109–5121
Chen CL, Wang WL, Raymond M, Ahmadinejad F, Poklis JL, Em B et al (2023) Genetic knockout of fatty acid Amide Hydrolase (FAAH) ameliorates cisplatin-induced Nephropathy in mice br. Mol Pharmacol ; 103
Maekawa H, Inoue T, Ouchi H, Jao TM, Inoue R, Nishi H et al (2019) Mitochondrial damage causes inflammation via cGAS-STING signaling in Acute kidney Injury. Cell Rep 29:1261–
Mitazaki S, Honma S, Suto M, Kato N, Hiraiwa K, Yoshida M et al (2011) Interleukin-6 plays a protective role in development of cisplatin-induced acute renal failure through upregulation of anti-oxidative stress factors. Life Sci 88:1142–1148
Liu J, Livingston MJ, Dong GE, Tang CY, Su YC, Wu GY et al (2018) Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis ; 9
Li Y, Shi L, Zhao F, Luo Y, Zhang M, Wu X et al (2024) PIM1 attenuates cisplatin-induced AKI by inhibiting Drp1 activation. Cell Signal 113:110969
Guo C, Ding Y, Yang A, Geng Y, Liu C, Zhou L et al (2022) CHILKBP protects against podocyte injury by preserving ZO-1 expression. Cell Mol Life Sci ; 80
Ma L, Tian Y, Qian T, Li W, Liu C, Chu B et al (2022) Kindlin-2 promotes src-mediated tyrosine phosphorylation of androgen receptor and contributes to breast cancer progression. Cell Death Dis ; 13
Perse M, Veceric-Haler Z (2018) Cisplatin-Induced Rodent Model of Kidney Injury: Characteristics and Challenges. Biomed Res Int ; 2018: 1462802
Khawaja S, Jafri L, Siddiqui I, Hashmi M, Ghani F (2019) The utility of neutrophil gelatinase-associated Lipocalin (NGAL) as a marker of acute kidney injury (AKI) in critically ill patients. Biomark Res 7:4
Marakala V (2022) Neutrophil gelatinase-associated lipocalin (NGAL) in kidney injury - A systematic review. Clin Chim Acta 536:135–141
Waikar SS, Bonventre JV (2008) Biomarkers for the diagnosis of acute kidney injury. Nephron Clin Pract 109:c192–197
Vaidya VS, Ferguson MA, Bonventre JV (2008) Biomarkers of acute kidney injury. Annu Rev Pharmacol Toxicol 48:463–493
Wang X, Zhou Y, Wang D, Wang Y, Zhou Z, Ma X et al (2023) Cisplatin-induced ototoxicity: from signaling network to therapeutic targets. Biomed Pharmacother 157:114045
Mifflin L, Ofengeim D, Yuan JY (2020) Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat Rev Drug Discovery 19:553–571
Degterev A, Ofengeim D, Yuan JY (2019) Targeting RIPK1 for the treatment of human diseases. Proc Natl Acad Sci USA 116:9714–9722
Ai YW, Meng YT, Yan B, Zhou QY, Wang XD (2024) The biochemical pathways of apoptotic, necroptotic, pyroptotic, and ferroptotic cell death. Mol Cell 84:170–179
Morgan MJ, Kim YS (2022) Roles of RIPK3 in necroptosis, cell signaling, and disease. Experimental Mol Med 54:1695–1704
Pabla N, Dong Z (2008) Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 73:994–1007
El Magdoub HM, Schaalan MF, Rahmo RM, Farag DB, Khedr LH (2020) Implications of miRNAs on TGF-beta/TAK1/mTOR pathway in mediating the renoprotective effects of pentoxifylline against cisplatin-induced nephrotoxicity in rats. Toxicol Appl Pharmacol 404:115184
Li G, Cheng Z (2021) miR-339-5p Inhibits Autophagy to Reduce the Resistance of Laryngeal Carcinoma on Cisplatin via Targeting TAK1. Biomed Res Int ; 2021: 9938515
Landstrom M (2010) The TAK1-TRAF6 signalling pathway. Int J Biochem Cell Biol 42:585–589
Hirata Y, Takahashi M, Morishita T, Noguchi T, Matsuzawa A (2017) Post-translational modifications of the TAK1-TAB complex. Int J Mol Sci ; 18
Liang L, Fan Y, Cheng J, Cheng D, Zhao Y, Cao B et al (2013) TAK1 ubiquitination regulates doxorubicin-induced NF-kappaB activation. Cell Signal 25:247–254
Mukhopadhyay H, Lee NY (2020) Multifaceted roles of TAK1 signaling in cancer. Oncogene 39:1402–1413
Ge QY, Chen J, Li GX, Tan XL, Song J, Ning D et al (2021) GRAMD4 inhibits tumour metastasis by recruiting the E3 ligase ITCH to target TAK1 for degradation in hepatocellular carcinoma. Clin Transl Med 11:e635
Yang Z, Xian H, Hu J, Tian S, Qin Y, Wang RF et al (2015) USP18 negatively regulates NF-kappaB signaling by targeting TAK1 and NEMO for deubiquitination through distinct mechanisms. Sci Rep 5:12738
Li Q, Yan J, Mao AP, Li C, Ran Y, Shu HB et al (2011) Tripartite motif 8 (TRIM8) modulates TNFalpha- and IL-1beta-triggered NF-kappaB activation by targeting TAK1 for K63-linked polyubiquitination. Proc Natl Acad Sci U S A 108:19341–19346
Zhang H, Han Y, Xiao W, Gao Y, Sui Z, Ren P et al (2023) USP4 promotes the proliferation, migration, and invasion of esophageal squamous cell carcinoma by targeting TAK1. Cell Death Dis 14:730
Zhang Y, Luo Y, Wang Y, Liu H, Yang Y, Wang Q (2018) Effect of deubiquitinase USP8 on hypoxia/reoxygenation–induced inflammation by deubiquitination of TAK1 in renal tubular epithelial cells. Int J Mol Med 42:3467–3476
Lei CQ, Wu X, Zhong X, Jiang L, Zhong B, Shu HB (2019) USP19 inhibits TNF-alpha- and IL-1beta-Triggered NF-kappaB activation by deubiquitinating TAK1. J Immunol 203:259–268
Volovat S, Apetrii M, Stefan A, Vlad C, Voroneanu L, Hogas M et al (2023) Cisplatin and AKI: an ongoing battle with new perspectives-a narrative review. Int Urol Nephrol 55:1205–1209
Peres LA, da Cunha AD Jr (2013) Acute nephrotoxicity of cisplatin: molecular mechanisms. J Bras Nefrol 35:332–340
Curry JN, McCormick JA (2022) Cisplatin-Induced kidney Injury: delivering the Goods. J Am Soc Nephrol 33:255–256
Delaney JR, Mlodzik M (2006) TGF-beta activated kinase-1: new insights into the diverse roles of TAK1 in development and immunity. Cell Cycle 5:2852–2855
Shim JH, Xiao C, Paschal AE, Bailey ST, Rao P, Hayden MS et al (2005) TAK1, but not table 1 or table 2, plays an essential role in multiple signaling pathways in vivo. Genes Dev 19:2668–2681
Lamothe B, Lai Y, Hur L, Orozco NM, Wang J, Campos AD et al (2012) Deletion of TAK1 in the myeloid lineage results in the spontaneous development of myelomonocytic leukemia in mice. PLoS ONE 7:e51228
Chauhan A, Hudobenko J, Al Mamun A, Koellhoffer EC, Patrizz A, Ritzel RM et al (2018) Myeloid-specific TAK1 deletion results in reduced brain monocyte infiltration and improved outcomes after stroke. J Neuroinflammation 15:148
Su W, Gao W, Zhang R, Wang Q, Li L, Bu Q et al (2023) TAK1 deficiency promotes liver injury and tumorigenesis via ferroptosis and macrophage cGAS-STING signalling. JHEP Rep 5:100695
Acknowledgements
We thank the SUSTech Animal Facility for the maintenance of mice and the SUSTech Core Facility for technical assistance for the use of flow cytometer and histology/pathology equipment. We thank Dr. Andrew Hutchins (SUSTech) for comments on the manuscript.
Funding
This work was supported, in part, by grants from the National Natural Science Foundation of China (8237100185, 82070728); the Natural Science Foundation of Guangdong Province (2024A1515013048, 2023B0303040004, 2021B1515120063 and 2017B030301018); the Shenzhen Innovation Committee of Science and Technology, China (JCYJ20200109141212325).
Author information
Authors and Affiliations
Contributions
Y.S. designed the study, supervised the project and wrote the manuscript; C.W. and Y.D. provided advice on some experiments; A.Y. performed the experiments and data analysis and wrote the manuscript; Y.D., C.G., C.L., Z.X., M.Q., P.B., R.C., B.L., G.L. performed some experiments; A.Y. and Y.S. take the responsibility for the integrity of the data analysis.
Corresponding author
Ethics declarations
Ethics approval
All animal experiments were performed following the guidelines and regulations as approved by the Institutional Animal Care and Use Committee at the Southern University of Science and Technology of China (Approval Number: SUSTech-JY202310108).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, A., Ding, Y., Guo, C. et al. PARVB deficiency alleviates cisplatin-induced tubular injury by inhibiting TAK1 signaling. Cell. Mol. Life Sci. 81, 385 (2024). https://doi.org/10.1007/s00018-024-05422-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-024-05422-w