
Abstract
Pore-forming toxins (PFTs) are key virulence determinants produced and secreted by a variety of human bacterial pathogens. They disrupt the plasma membrane (PM) by generating stable protein pores, which allow uncontrolled exchanges between the extracellular and intracellular milieus, dramatically disturbing cellular homeostasis. In recent years, many advances were made regarding the characterization of conserved repair mechanisms that allow eukaryotic cells to recover from mechanical disruption of the PM membrane. However, the specificities of the cell recovery pathways that protect host cells against PFT-induced damage remain remarkably elusive. During bacterial infections, the coordinated action of such cell recovery processes defines the outcome of infected cells and is, thus, critical for our understanding of bacterial pathogenesis. Here, we review the cellular pathways reported to be involved in the response to bacterial PFTs and discuss their impact in single-cell recovery and infection.
Similar content being viewed by others
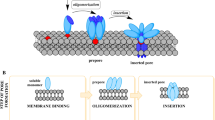
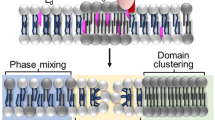
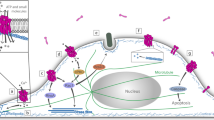
Avoid common mistakes on your manuscript.
Introduction
The plasma membrane (PM) constitutes a selective barrier between the intracellular and extracellular environment, defining the limits of every living eukaryotic cell [1, 2]. Its emergence was a critical event during evolution, and its integrity is crucial to maintain cellular homeostasis and support life [3]. Transient PM lesions still occur in healthy conditions, particularly, in tissues under high mechanical stress or biochemical stress such as muscle, skin, or gut epithelia [4, 5]. Conversely, persistent PM damage hallmarks several pathologies, such as heart failure, neurodegeneration, and infection [6,7,8,9].
Being the first protective cellular barrier, the PM is preferentially targeted by pathogens to exploit host intracellular nutrients, disrupt signaling, cross-tissue barriers, and/or kill immune cells [10,11,12,13,14]. In particular, bacteria secrete monomeric pore-forming toxins (PFTs), which oligomerize upon binding to the host PM and assemble into transmembrane stable pores that permeabilize cells to ions, metabolites, and proteins [11, 15,16,17,18,19,20,21], triggering a variety of coordinated host-cell responses. PFTs are both necessary and sufficient for the pathogenesis of several bacterial species [22,23,24]. Such proteins exist in virtually all the kingdoms of life, comprising different structural families, for which the mechanisms of oligomerization and pore formation have been extensively characterized (reviewed in [15]). In contrast, their effects at the cellular level and specific roles in disease development are by far less understood.
PM injury elicits multiple responses depending on the nature of the damage and the cell type involved. Despite certain specificities, these responses are based on conserved events that include sensing the damage, activating repair mechanisms, restoring homeostasis, and activating innate immunity, thereby alerting neighboring tissues. In recent years, considerable advances were made regarding the identification and characterization of PM repair mechanisms and single-cell recovery processes. Such studies have highlighted the sequential nature that underlies the spatio-temporal coordination of cell recovery, but have predominantly focused on localized mechanical- or laser-induced damage (reviewed recently in [5, 25]). Accordingly, although the mechanisms identified are still relevant in the context of PFT-mediated damage, the specific features of repairing stable protein pores and recovering cellular homeostasis following PM damage caused by bacterial PFTs remain poorly understood. Nevertheless, here, we review the large amount of work carried during the past years, concerning the multitude of cellular responses to various bacterial PFTs. We attempt to discuss such responses in light of our current understanding of general PM repair mechanisms and single-cell recovery mechanisms to provide a more complete view of the processes deployed by host cells to specifically face PFT-mediated intoxication. When adequate, we also discuss their relevance during infection.
Sensing the damage
PM disruption allows the influx of calcium and efflux of potassium, altering the intracellular-ion composition, which has long been recognized as the primary trigger for cell responses to PM damage (Figs. 1, 2) [5, 25, 26]. The influx of extracellular calcium activates PM repair pathways (Fig. 1) and protective cytoskeletal remodeling (Fig. 3) [27,28,29,30]. However, overwhelming and long-lasting calcium elevations (approximately > 20 µM) are toxic [31, 32] and compromise host-cell signaling, which can desensitize immune cells [33], destabilize tissue barriers [34], or induce cell death [35,36,37]. Thus, controlling the rise of cytosolic calcium levels appears fundamental to determine cell fate following PM disruption, either promoting survival or triggering cell death [32, 38, 39].

Proposed overview model depicting calcium-dependent PM repair mechanisms that protect host cells against PFTs. (1) Toxin oligomerization, pore formation, and calcium influx initiate the activation of calcium-dependent protective events. (2) Annexins are recruited to damaged areas according to their differential calcium sensitivity (gray scale) and assemble into 2D protein arrays to clog PM pores. (3) PM blebbing and shedding occur at damaged sites and involve recruitment of ESCRT subunits, which facilitate budding and ATP-dependent release of PM vesicles containing PFT pores, annexins, and ESCRT components. (4) In parallel, calcium influx triggers PM docking and exocytosis of cortical lysosomes. Upon assembly of SNARE complexes, the calcium sensor Synaptotagmin VII (Syt-VII) enables fusion of lysosomes with the PM and release of lysosomal enzymes, in particular ASM. (5) ASM hydrolyses PM sphingomyelin, producing ceramide domains, which facilitate membrane invagination and endocytosis of PFTs’ pores and incomplete pore structures. Ceramide domains may also contribute to annexin recruitment. (6) PFTs traffic to MVBs through ESCRT-dependent sorting and are degraded via MVB–lysosomal fusion. Toxins may also be recycled back to the PM and further secreted

Proposed model for potassium-dependent host-protective mechanisms against PFT intoxication. (1) Potassium efflux and release of ATP occur across PFT-assembled pores. Extracellular ATP activates the P2X7 receptor and cation channel, triggering further potassium efflux and calcium influx. These events activate several recovery processes that include: (2) PM translocation of lysosomal ASM, subsequent remodeling of PM lipid composition, and release of phosphatidylserine (PS)-enriched PM vesicles. This process may enable the secretion of cytokines and occurs downstream activation of p38. (3) Inflammasome activation, caspase-1 processing and activation of IL-1 beta secretion. Caspase-1 activation increases lipid metabolism and membrane biogenesis pathways. (4) Potassium efflux also activates MAPK signaling, in particular p38 and JNK, which further control the UPR and protective transcriptional responses required for survival against PFTs

Proposed model illustrating the protective mechanisms of actomyosin remodeling in response to PFT intoxication. (1, 2) Pore formation and the subsequent calcium influx induce cortical actomyosin remodeling by: disassembling actomyosin structures; activating the GTPases Rac1 and RhoA, and enhancing calpain activity which breaks cytoskeletal–PM contacts and disrupts interactions between actin and actin-binding proteins. Cortical lysosomal positioning is maintained by interactions between Rab3A and NMIIA. The rise in cytosolic calcium activates Rab3 and promotes actin remodeling contributing to binding, docking, and fusion of cortical lysosomes with the PM. (3) Actomyosin remodeling and lysosome secretion lower PM tension and modify the PM lipid composition causing PM blebbing, ruffling, and shedding or internalization of PFT pores. Actomyosin reorganization is regulated by RhoA and Rac1 and is stabilized by the formation of NMIIA cortical bundles. Concomitant ER expansion and ER–cytoskeletal interactions also contribute to stabilize the cortical actomyosin network. (4) Following shedding or internalization of PFT pores, cells re-establish cytoskeletal organization and recover normal cytosolic calcium levels
Potassium efflux promotes stress-activated and mitogen-activated protein kinase (MAPK) pathways that can protect against PFT activity in vitro and in vivo [40,41,42,43], but the drop in cytosolic potassium levels also alters the cellular metabolic state, triggers innate immune signaling, and may cause pro-inflammatory cell death (Fig. 2) [35, 41, 44].
The magnitude of ion imbalance depends on the dimension of the PM wound which, in the case of toxin-induced pores, varies greatly according to the stoichiometry and size of the pore [15]. Cholesterol-dependent cytolysins (CDCs), such as listeriolysin O (LLO) and streptolysin O (SLO), secreted by the Gram-positive bacteria Listeria monocytogenes and Streptococcus pyogenes, respectively, assemble into large (30–50 nm in diameter) heterogeneous pores, whereas smaller toxins, such as aerolysin from Aeromonas hydrophila or alpha-toxin from Staphylococcus aureus, originate pores of only ~ 2 nm in diameter [15].
The full recovery of PM integrity was shown in different cell types perforated by small or large PFTs [31, 41, 45], yet counter-intuitively, cells damaged by small toxin pores, appear to take longer to recover PM integrity (~ h), when compared to large CDC-induced damage (~ min). This has been largely attributed to the lower calcium permeability of small pores, and the consequent defect to efficiently trigger rapid calcium-dependent repair mechanisms. The recovery from damage caused by small toxin pores must rely mainly in potassium-dependent cellular responses, which, indeed, protect cells upon attack by a variety of small PFTs such as alpha-toxin [46], Vibrio cholerae cytolysin (VCC) [47, 48], and Cry5B [48]. In agreement with these observations, mutations that increase the channel width of the small PFT phobalysin from Photobacterium damselae, enhanced calcium-dependent repair, whereas bulky amino acid residues within VCC channels were shown to delay the recovery [49].
Not only the size of the pore, but also its structure, may determine the specificity of repair pathways. Damage caused by Sticholysin II, a small actinoporin produced by a sea anemone, is repaired with equivalent kinetics of CDCs [50]. CDCs originate both stable protein-lined and heterogeneous pores, which, at low stoichiometry, form less stable arc-like structures composed by proteins and lipids siding each other [51,52,53,54]. Comparably to such less stable CDC structures, actinoporins form intercalated protein–lipid pores [55, 56], also less stable than protein-lined pores formed by other small toxins [15, 51, 52]. Thus, heterogeneous pore-forming structures may be repaired more efficiently, as it occurs with electroporation-induced lipid wounds, indicating that the nature of the pore also determines its effective repair.
Alteration of the cytosolic ion composition upon PM damage relies on additional secondary events: certain PFTs can induce the opening of intracellular calcium stores [33] and allow the release of ATP to the extracellular environment. Host cells recognize extracellular ATP, which further enhances intracellular-ion imbalance [57], but may also trigger protective responses in neighboring cells (Fig. 2) [58,59,60]. These processes rely on the expression of P2X7 receptor (P2X7R), which is a cation channel. However, given the broad range of ATP sensitivities displayed by purinergic receptors [61, 62], it is possible that alternative P2X receptors also respond to ATP released from PFT-damaged cells.
Finally, the endogenous pore-forming activity of host proteins such as mixed lineage kinase-like (MLKL) or gasdermin D may also contribute to further promote ATP and potassium efflux or the influx of extracellular calcium [63]. Gasdermin D and MLKL form pores in the inner PM leaflet, during pro-inflammatory cell death mechanisms (pyroptosis and necroptosis, respectively), which can be activated by PFTs’ intoxication [35, 64]. Specific roles for gasdermin D or MLKL during cellular recovery from PFT intoxication have not been established. Nevertheless, activation of crucial pyroptotic and necroptotic effectors during PFT intoxication enhances alarmin release and promotes host inflammation during pneumonia caused by Serratia marcescens infection [64].
Overall, host responses to PFT intoxication will vary depending on the differential cytosolic ion gradients produced by structurally different PFTs, their concentration, and the release of additional cellular metabolites.
Plasma membrane repair
Clogging the pore
The influx of extracellular calcium following PM damage promotes the exocytosis of cortical vesicles (e.g., lysosomes) and the recruitment of protein arrays to PM wounds. These processes, through the formation of a patch of homotypically fused vesicles and a clog of fusogenic protein arrays, were proposed to limit the loss of cytosolic content and the rise of intracellular calcium to toxic levels during mechanical- or laser-induced PM damage [5]. Such calcium-mediated exocytosis also reduces membrane tension, which may contribute to the spontaneous resealing of lipid-based wounds [65]. However, stable protein pores, such those generated by PFTs, do not spontaneously reseal and must be actively removed.
Annexins, one of the major components of clogging protein complexes, are cytosolic calcium sensors with the capacity to aggregate, bind phospholipids, and promote membrane fusion in a calcium-regulated manner [66, 67]. They are promptly recruited to PM lesions in cells damaged by different CDCs (SLO and pneumolysin, PLY) [31]. Upon pore formation, annexins sequentially and reversibly translocate to the PM surface according to their different calcium sensitivities (Fig. 1) [68]. Annexins with high calcium sensitivity (A2 and A6) are early recruited to the sites of PM damage, and were detected in PM blebs and vesicular or tubular structures released by SLO- or PLY-intoxicated cells [68, 69]. In turn, annexins with low calcium sensitivity (A1 and A5) appear later around PM wounds and their translocation to the PM surface correlates with the inability of cells to recover from PM damage [68], presumably because the intracellular calcium concentration has reached a toxic threshold (~ 20 µM). Annexins (A2, A6, A1, and A5) exhibit protective roles upon mechanical- or laser-induced PM damage and in PM damage-related disorders [67]. Yet, how annexins clog a protein pore and protect cells during PFT intoxication remains unclear. Nevertheless, A1 localizes to PFT-damaged PM regions and is detected within large PM blebs that appear to compartmentalize cytoplasmic content. Moreover, similarly to what was observed upon mechanically induced damage of HeLa cells [70], A1 depletion or targeting with blocking antibodies increases susceptibility to CDCs, thus confirming a protective role against PFTs [71, 72]. Furthermore, cryo-electron tomography of vesicles released by PLY-damaged cells show high-density structures concentrated below toxin pores, resembling the A5 two-dimensional arrays that assemble at sites of laser-induced PM wounds [69, 73,74,75]. Mass spectrometry analysis confirmed that such vesicles are enriched in annexins [69]. Altogether these observations led to speculate that annexins assemble into two-dimensional arrays that clog PFT pores, avoiding the detrimental diffusion of calcium to the entire cell (Fig. 1) [32, 68, 69]. Such clog may also isolate damage within PM blebs [72].
Quarantining PM damage: blebbing
Blebbing is a universal cellular response to PM injury described in different processes such as cytokinesis, cell migration, and apoptosis [76, 77]. PM blebs require calcium-dependent actomyosin contraction and result from the disruption of PM–cytoskeleton interactions, which decreases PM tension and enables its expansion. Since vesicle exocytosis also reduces PM tension, it is possible that this process contributes to blebbing upon PM damage. Intriguingly, in the context of PFTs, blebbing may result from intrinsic properties of specific PM lipid domains that respond to toxin oligomerization and binding, before PM disruption [78, 79].
In PLY- or SLO-damaged cells, large PM blebs were proposed to create a confined space where calcium concentration is higher than in the cell body [31, 72]. Large blebs possibly protect the cell from deleterious calcium elevations and loss of cytosolic content. The majority of large PM blebs retract, supporting their role as clogging structures or, alternatively, as secondary events of the PFT-induced cortical cytoskeletal disruption. Nonetheless, different cell types were shown to release large (µm size) blebs, villi, or bleb-like structures containing cytoplasmic material in response to PM damage caused by protein pores (Fig. 4) [69, 80, 81]. Such release of large blebs may result from the inability of cells to repair overwhelming damaged areas and/or derive from the engagement of cell death pathways. Along with the shedding of smaller blebs, the release of large PM structures will likely allow the removal of PFT pores, the disposal of irreversibly damaged organelles, and also convey cytoplasmic inflammatory signals. Indeed, blebs produced by apoptotic cells carry and release cytoplasmic danger signals [82], and large cell particles shed by cancer cells, in vivo, are delivered to myeloid cells, thereby altering their behavior [83]. Drosophila melanogaster gut epithelial cells targeted by S. marcescens expressing the PFT ShlA also release bleb-like cytoplasmic extrusions containing damaged organelles [84]. This response maintains epithelial integrity and limits host injury, as flies infected with ShlA-deficient strains undergo amplified injury. Similarly, cytoplasm-containing blebs from Mycobacterium-infected cells also deliver intracellular bacteria to phagocytic cells (Fig. 5) [85]. On the other hand, bacteria can hijack PM blebs to promote dissemination. This is the case of Pseudomonas aeruginosa that utilizes type III secretion system (T3SS) pore-forming translocon proteins to induce PM blebbing and exploit blebs as niches for replication [86]; and of L. monocytogenes, which promotes LLO-dependent blebbing to support cell-to-cell spread within PM blebs (Fig. 5) [87].

Schematic representation of shedding of large cytoplasm-containing blebs or extrusions, and thinning of epithelia damaged by PFTs. (1) The ion imbalance generated by pore formation promotes apical actomyosin remodeling and vesicle secretion. (2) Both processes lower PM tension contributing to PM blebbing, remodeling, and shedding. The cytosolic ion imbalance alters organelle dynamics and causes organelle damage, including: lipid-droplet formation, mitochondria fission and enlargement, ER expansion and vacuolation, lysosomal secretion and rupture. (3) Damaged organelles are detected in the proximity of the PM and within cytoplasmic extrusions or large cell particles (e.g., blebs and villi) released by intoxicated cells. (4) Epithelial integrity and cellular homeostasis are maintained by transient (~ h) contraction and thinning of the epithelial actomyosin network and removal of damaged organelles via autophagy. Toxin pores are eliminated by PM shedding, endocytosis, and autophagic targeting

Model illustrating how the main host-protective responses to PFT intoxication influence the outcome of bacterial infections. (1) Lysosome exocytosis and actomyosin remodeling promote the secretion of lysosomal enzymes that alter PM lipid composition and enable activation of endocytic pathways which allow pathogen internalization. (2) The release of hydrolytic enzymes contributes for pathogen killing. (3) Alterations in PM tension caused by PFT-induced damage promote the formation and release of PM protrusions and/or blebs that allow the dissemination of L. monocytogenes in enclosed vesicles or allow the shedding of intracellular bacteria. Released bacteria-containing vesicles can also be subsequently engulfed and killed by recruited phagocytes (efferocytosis). (4) Large PM blebs may sustain the replication of intracellular P. aeruginosa. (5) Autophagy targets PFT-producing bacteria in the host cytosol, upon vacuolar escape, and either promotes pathogen killing or the formation of SLAPs, a niche for L. monocytogenes replication
Removing PM pores: shedding
PM blebbing enables the shedding of microvesicles, which is regarded as a major PM repair mechanism that protects from detergent-induced or mechanically induced wounds and against intoxication with small stable PFTs, such as alpha-toxin or Cry5B, and large CDCs [32, 69, 79, 88,89,90,91,92]. Elimination of PFTs within PM vesicles occurs in various cell types and may constitute an intrinsic protection mechanism, which is further enhanced by toxin oligomerization, pore formation, and calcium entry [79, 92]. This mechanism was recently proposed to benefit from the heterogeneous binding of certain PFTs to cholesterol-rich PM domains [93], and allows the rapid shedding of fully formed pores and incomplete pore-forming structures within small PM microvesicles (~ 100 to 200 nm) [69, 92]. PM shedding occurs at wound sites, and involves passive PM blebbing, active vesicle budding, and release of vesicular structures enriched in endosomal sorting complex required for transport (ESCRT) proteins, annexins, PFT pores, and other molecules (Fig. 1) [69, 78, 79, 90, 92]. ESCRT complexes have established roles in membrane deformation and scission processes [e.g., multivesicular body (MVB) biogenesis and viral budding], and ESCRT-III-mediated PM shedding was fully characterized in laser-wounded cells [88]. In this context, ESCRT-III adaptor proteins, the apoptosis linked gene (ALG)-2, and ALG-2-interacting protein X (ALIX) enable the rapid (~ min) calcium-dependent recruitment of ESCRT subunits to PM wounds [94]. The adenosine triphosphatase Vps4, responsible for ESCRT-III disassembly, is recruited subsequently and contributes to the budding and ATP-dependent pinching of PM microvesicles containing lesions, thereby protecting cells from small (< 100 nm) PM wounds [88, 94, 95]. In agreement, LLO-intoxicated cells show punctate distribution of the ESCRT-III component CHMP4B at the plasma membrane [88], and vesicles released by cells challenged with different CDCs are enriched in ESCRT assembly and disassembly subunits [69].
Interestingly, ALG-2 can interact with different annexins in vitro [96], and the recruitment of annexin A1 to the PM of mechanically or laser-damaged cancer cells is followed by PM shedding [97], suggesting an interplay between clogging and shedding PM injuries. The ESCRT-III complex also coordinates blebbing and shedding of PM vesicles promoted by MLKL activity during necroptosis [98], the major pro-inflammatory programmed cell death pathway promoted by PFTs in vivo [35]. Hence, ESCRT proteins have emerged as important mediators of cell-autonomous defenses and innate immunity against bacterial pathogens. However, in contrast to the well-established role of ESCRT proteins in viral budding, few reports assessed ESCRT-mediated repair during bacterial infections. Nonetheless, PM shedding occurs in vivo upon PFT intoxication, concomitantly with the other PM repair processes [48].
Extracellular ATP, which can be released by PFT-damaged cells, also triggers PM blebbing and shedding, via the activation of P2X7R channels, potentially protecting neighboring cells prior PFTs’ attack [99,100,101]. In line with these observations, artificial liposomes can reduce toxin binding to host cells in vitro and protect against PFT-dependent infections in vivo [102, 103]. Thus, PM shedding not only represents a major cell-autonomous defense but may also prevent recurrent toxin attacks by trapping free toxin molecules within secreted vesicles [72].
Removing PM pores: lysosome exocytosis and endocytic degradation
As mentioned above, PM ruptures trigger calcium-dependent exocytosis of peripheral vesicles, predominantly lysosomes [90, 104]. These vesicles were proposed to patch PM wounds [65] and reduce PM tension, contributing to the spontaneous resealing of lipid-based injuries, such as laser-induced or mechanically induced PM wounds [105]. Lysosome exocytosis upon PM damage, is reminiscent to the secretion of granules by cytotoxic lymphocytes or professional secretory cells and involves calcium-dependent interactions between the calcium sensor synaptotagmin VII, dysferlin, and lysosomal (e.g., VAMP-7) and PM (e.g., SNAP-23 and syntaxin-4) SNARES (N-ethylmaleimide-sensitive factor attachment protein receptors) (Fig. 1) [106,107,108,109]. In addition, small GTPases (Rab3, Rab10, and Arl8b), the Rab3 effector synaptotagmin-like protein 4a (Slp4-a) and the actin motor protein non-muscle myosin IIA (NMIIA), also control cortical lysosome positioning required for PM repair (Fig. 3) [110, 111]. Secretion of lysosomes allows the release of lysosomal enzymes, such as acid sphingomyelinase (ASM), and cathepsin B and L, which alter local PM composition promoting PFT removal via endocytosis (Fig. 1) [112,113,114]. In particular, ASM was shown to hydrolyze sphingomyelin into phosphorylcholine and ceramide domains, triggering the endocytosis of PFT pores within small (50–100 nm) lipid-rich PM invaginations termed caveolae [115,116,117]. Caveolin- or ASM-deficient cells have impaired ability to repair PFT-induced injuries [104, 118, 119] and extracellular ASM is sufficient to promote SLO internalization [104, 115]. However, to date, internalization of active pores by caveolar-dependent mechanisms has never been directly visualized and the role of endocytosis as a mechanism of PFT removal remains controversial. Indeed, blocking endocytosis does not compromise the removal of PFT pores from intoxicated cells [79]. In addition, endocytosed PFT pores would remain associated with the membrane of endosomes possibly leading to endosomal leakage and release of toxic enzymes to the cell cytosol. Nevertheless, it has been shown that toxin internalization occurs for various PFTs [112, 120, 121], which apparently are sorted to MVBs in a ubiquitination/ESCRT-dependent manner. Accordingly, poly-ubiquitinated proteins are observed close to laser-induced PM wounds in parallel to PM shedding [88]. PFTs are possibly degraded upon MVB–lysosomal fusion [112, 120] or can be re-routed to the extracellular milieu within exosome-like compartments (Fig. 1) [120]. Moreover, it has been increasingly clear that the endocytic machinery contributes to the survival of cells upon intoxication. Endocytic components such as caveolin-1 and GRAF1 were recently shown to play a role during PM repair of PFT pores by acting on the remodel and regeneration of the normal composition of the PM following repair [122]. Based on ultra-structural evidences, a recent report proposed a model where PFT-induced active pores are removed by the shedding of microvesicles, whereas endocytosis restores PM homeostasis by removing inactive PFT monomers and vesicles that failed to shed, once repair is complete [92].
Lysosome exocytosis may be linked to PM shedding. In glial cells, secretion of ASM promotes PM shedding of phosphatidylserine (PS)-rich vesicles downstream P2X7R activation (Fig. 2) [123]. This process requires Src kinase-dependent phosphorylation of the p38 MAPK [123] that is activated by all the tested PFTs and promotes the shedding of cell adhesion molecules in response to S. aureus PFTs [124]. Thus, it is possible that p38 activation per se explains the removal of small PFT pores, which do not trigger massive calcium-dependent repair. Yet, further work is required to address this hypothesis.
Off note, ASM-produced PM ceramide domains increase annexin A1 binding to the PM [125]. Thus, annexin clogging, PM shedding, lysosomal exocytosis, and further endocytosis of PFTs may likely benefit from the coordinated action of common molecular effectors which, altogether, protect upon intoxication by various PFTs. In agreement, increased endocytosis and PM shedding occur during Caenorhabditis elegans intoxication with different PFTs, through a process that depends on the small GTPases Rab5 and Rab11 [48], master endo- and exocytic regulators, respectively.
Fusion of cortical lysosomes with the PM and the consequent extracellular delivery of lysosomal enzymes is not only important for PM repair, but was also proposed as a conserved cell-autonomous defense against pathogens that damage the host-cell PM (Fig. 5) [106, 126]. In favor of such mechanism, Arl8 and NMIIA, which promote calcium-dependent lysosome exocytosis, contribute to bacterial clearance in vivo, or can reduce host-cell invasion by bacterial pathogens, respectively [110, 127]. Concurrently, Mycobacterium tuberculosis diminishes the expression of synaptotagmin VII to promote PM damage and necrotic-based dissemination [128].
Endocytosis following PM perforation can also be exploited by different pathogens to induce internalization [129, 130]. In fact, LLO is sufficient to promote calcium- and potassium-mediated bacterial internalization and endocytosis of inert beads [129, 131]. In the other hand, avoiding PFT-mediated cytotoxicity can also favor the pathogenesis of intracellular bacteria. In this context, it was recently shown that a specific C-terminal PEST-like sequence present in LLO mediates its removal from the inner face of the PM by endocytosis, preventing premature killing of infected cells, thereby favoring L. monocytogenes infection [132].
Altogether it is becoming evident that clogging of the pore, PM blebbing, shedding of PM vesicles, and lysosomal exocytosis and endocytosis are interconnected mechanisms that cooperate during PM repair upon PFTs attack, and determine the fate of intoxicated cells.
Intracellular responses
Cytoskeleton remodeling
Regardless the source of PM damage, cells undergo dramatic cytoskeletal alterations, which result from mechanical or biochemical cues that strongly affect the homeostatic tension properties of the cell [39, 133]. The remodeling of the actomyosin cytoskeleton reduces PM tension and facilitates vesicle recruitment and PM shedding, thus promoting PM repair [90, 134,135,136,137]. Accordingly, compounds that stabilize the actomyosin network, such as jasplakinolide and phalloidin, hinder the recovery of PM integrity upon mechanical-, laser-, and PFT-induced damage [90, 134, 138, 139], whereas actin-depolymerizing agents (e.g., cytochalasin D, latrunculin, and DNAse I) lead to faster repair [90, 105, 134, 135, 140, 141].
Although a direct interaction between PLY and actin has been reported [142], the targeting of the actomyosin cytoskeleton by PFTs occurs mainly through the sustained rise in cytosolic calcium levels caused by PFT-mediated damage. This process induces the activation of different cytoskeleton-modulating enzymes, which is sufficient to trigger actin cytoskeleton remodeling, facilitating cell adaptation, and recovery responses to different types of PM damage (Fig. 3) [143]. In particular, the calcium-dependent cysteine proteases [144], calpains, favor repair of mechanical and laser injuries [145,146,147], facilitate vesicle fusion with the PM [146], and can cleave actin-associated proteins ultimately leading to actin remodeling [144, 148]. In LLO-damaged cells, calpain-2 is recruited to cortical actomyosin bundles, which assemble around sites of PM damage and blebbing [89]. Moreover, during Streptococcus pneumoniae infection, PLY-dependent calpain activation promotes the release of the pro-inflammatory cytokine IL-1 beta by the infected macrophages [149], a process that can occur via PM shedding [59]. Interestingly, an evolutionary and biochemical link between ESCRT proteins and calpains has been proposed, which may have crucial roles during yeast adaption responses [150, 151].
The assembly and dynamics of actomyosin rings that provide purse-string forces to close laser-induced PM and epithelial wounds, in Xenopus oocytes or Drosophila embryos, involve the coordinated activity of Rho GTPases and NMII [138, 141, 152]. Rho GTPases are also activated during PLY intoxication of neuronal cells and promote calcium-dependent actin remodeling (Fig. 3) [153]. Pharmacological inhibition of actin polymerization delays the formation of an A6 PM clog in laser-wounded muscle cells [154], indicating a role for actin remodeling during PM clogging. Whether this occurs upon intoxication with PFTs is still unclear.
Nevertheless, NMIIA rearrangements and the assembly of stable cortical actomyosin bundles at sites of PM blebbing and PFT-induced damage correlate with increased cell survival following LLO intoxication [89]. This process involves the translocation of endoplasmic reticulum (ER) proteins to the cell surface and depends on the ER chaperone Gp96 [89]. Gp96 interacts with NMIIA and the actin adaptor Filamin A [89], and regulates NMIIA activity and cytoskeletal remodeling in response to PM damage [89]. Remarkably, both Gp96 and NMIIA protect cells against PFTs and Gp96 is critical for host survival during LLO-dependent L. monocytogenes infection [89].
Filamin A and Gp96 appear to coordinate actin organization through processes that may rely on ER–cytoskeleton interactions, ER dynamics, and polarized secretion [89, 155,156,157]. Of note, polarized lysosome secretion is controlled by NMIIA and is essential for PM repair of mechanically, laser-, or PFT-induced wounds [111, 136, 158]. The relevance of generalized actomyosin reorganization during PFT intoxication is unclear. However, a similar response was proposed to underlie acute morphological adaptations to PM damage, cell stress, or migration [143]. This mechanism is regulated by calcium and the ER-associated formin, IFN2, and involves IFN2-mediated turnover of cortical actin filaments and concurrent polymerization from the ER [143]. Altogether, these observations suggest a strong link between cytoskeleton remodeling, ER dynamics, and vesicle trafficking, supporting the complexity of PM repair pathways.
Actomyosin remodeling may also be important for epithelial integrity. During cytoplasmic extrusion, triggered by bacterial PFTs, gut epithelial cells contract, thinning the epithelial layer while maintaining barrier function. This process involves massive actomyosin rearrangements and recovery of normal cytoskeletal morphology, via CyclinJ-dependent signaling [84]. Similarly, following PFT intoxication of HeLa cells in vitro, the actomyosin network recovers the normal organization with similar kinetics (~ 4 to 8 h), which correlates with the occasional release of large cytoplasmic containing blebs [80, 89]. Whether both phenomena are functionally interrelated is unknown, yet it is tempting to speculate that the mechanisms underlying actomyosin remodeling observed in vitro following PFT intoxication, not only favor PM repair, but also impact general epithelial organization and integrity during tissue responses to bacterial-mediated damage.
Microtubule (MT) dynamics also influences PM repair. Mechanically induced PM damage promotes calcium-dependent disassembly of MTs around the wound sites [159, 160] which facilitates PM repair [105, 135, 147, 161]. MT rearrangements assist continuous vesicle and lipid trafficking to the wound sites from the Golgi, protecting against recurrent mechanically induced PM wounds [160]. In line with this, translocation of Golgi-derived vesicles to the PM is important to reduce M. tuberculosis-induced damage and diminish infection [128]. Contrarily, repair of SLO-intoxicated cells depends on intact MTs as the addition of nocodazole, an established MTs disruptor, was proposed to inhibit PM repair [45]. However, PLY was shown to induce MT stabilization in a process that depends on Src kinase and may be linked to cell damage during infection [162].
Cell survival pathways
Mitogen-activated protein kinases (MAPKs) are serine/threonine kinases that largely respond to stress stimuli and coordinate a variety of processes such as cell survival, metabolism, and proliferation [163]. Conventional MAPKs include the extracellular signal-regulated kinases 1 and 2 (ERK1/2), p38 isoforms (alpha, beta, gamma, and delta) and c-Jun amino (N)-terminal kinases 1, 2, and 3 (JNK1/2/3). Activation of MAPK pathways, in particular p38, is promoted by potassium efflux [40] and constitutes a widely conserved defense mechanism against PFT-mediated damage [4, 11, 164]. However, p38 activation is dispensable for the recovery of SLO-intoxicated keratinocytes [165]. p38 can be activated downstream the engagement of TLR4 receptor by different CDCs such as Anthrolysin (ALO) from Bacillus anthracis, which induces iNOS expression and promotes pro-inflammatory cytokines release [166, 167]. In parallel, LPS-mediated TLR4 stimulation activates p38 via the production of reactive oxygen species (ROS) and downstream the apoptosis signal-regulating kinase 1 (ASK1) activity [166, 167]. Whether such pathway is activated by PFTs is not known. However, during intoxication by alpha-toxin, ASK1 knockdown and ROS scavenging do not prevent p38 activation [165], suggesting that other factors may be involved.
During PFT intoxication, p38 activation is dependent on potassium efflux yet, how potassium depletion triggers MAPK signaling and which p38-regulated mechanisms promote cell recovery from PFT-induced damage remains unclear. Perturbations of potassium levels in different contexts such as low potassium diet [168] and skin wound healing [169] also trigger p38 activation. In addition, the decrease of cytosolic potassium levels activates the inflammasome NLRP3 [44] by a mechanism that involves the serine/threonine kinase Nek7, which regulates NLRP3 oligomerization and activation downstream of potassium efflux [170]. Interestingly, NLRP3 is activated during intoxication with the Clostridium tetani PFT, tetanolysin (TLO), and by Streptococcus pyogenes infection in an SLO-dependent manner, leading to caspase-1 activation and IL-1 beta release [171, 172]. However, the molecular details of a potential p38 and NLRP3 interconnection remain elusive.
To date, the activation of p38 was reported in response to all PFT tested—including Clostridium perfringens beta-toxin, aerolysin, alpha-toxin, VCC, HlyA, Cry toxins, and CDCs, such as PLY, SLO, LLO, ALO, Vaginolysin (VLY), and Inerolysin (INY) [40, 41, 50, 164, 165, 173,174,175,176]—in a variety of hosts including different mammalian cell lines, C. elegans, and various insect species [41,42,43, 177,178,179,180].
Activation of p38 and/or ERK1/2 induces the shedding of PM vesicles and receptors in microglia, platelets, and tumor cells [181,182,183]. Alpha-toxin- and ALO-induced p38 and ERK1/2 activation also promotes the shedding of cell adhesion and intracellular-contact molecules [124, 184]. Accordingly, MAPK-mediated PM shedding was thus proposed to contribute to tissue barrier disruption during PFT intoxication or PFT-mediated infection. Despite MAPK-mediated shedding of PFT pores was not yet identified, it is tempting to speculate that p38 activation could directly support PM repair by promoting the shedding of PFT pores. Indeed, p38 activity promotes recovery of potassium homeostasis and increases cell survival in response to PFT intoxication [41]. In addition, activation and transcriptional up-regulation of both p38 and JNK protect C. elegans from intoxication with Cry5B and large CDCs such as SLO [43, 179]. Both p38 and JNK regulate multiple downstream signals, which were shown to be protective upon PFT intoxication in vivo. Specifically, p38 up-regulates genes involved in the activation of the Unfolded Protein Response (UPR) and expression of putative cation efflux channels [42]. On the other hand, JNK was described as the master regulator of PFT-induced transcriptional responses, capable of up-regulating p38 specific pathways and innate immune signaling via the AP-1 transcription factor [179].
Reversely, MAPK activation during PFT intoxication may also have detrimental effects, as blocking p38 activity reduces PLY-mediated cytotoxicity in human SH-SY5Y neuroblastoma cells [185].
ER stress and organelle damage
Perturbations on ER homeostasis lead to the accumulation of unfolded proteins, which are recognized by ER stress sensors that signal for the re-establishment of normal ER conditions. This response involves the: (1) reduction of general protein translation; (2) up-regulation of chaperone expression to enhance protein folding; (3) activation of ER degradation pathways [186]. This process is called Unfolded Protein Response (UPR) and comprises three main branches defined by the specific stress sensor engaged: inositol requiring enzyme 1 (IRE1), protein kinase RNA (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6) [186]. Under irreversible ER stress, the UPR can ultimately induce apoptosis [187,188,189,190,191].
PFTs activate IRE1 downstream p38-MAPK signaling in vivo, and both IRE1 and ATF6 protect C. elegans against intoxication by Cry5B [42], yet, the mechanisms through which UPR activation protects against PFT intoxication remain unclear. During infection, UPR activation by PFTs, limits intracellular replication of L. monocytogenes [187], or diminishes the growth of extracellular Group A Streptococcus through the production of specific metabolites [192]. In the other hand, UPR activation may also lead to cell death [193], particularly upon prolonged dysregulation of calcium signaling [37, 194]. During S. pneumoniae infection in vivo, circulating PLY targets cardiomyocytes, leading to uncontrolled activation of PKCα-troponin and UPR pathways, loss of the contractile properties, and acute cardiac injury [37].
Upon PFTs’ challenge, the perturbations of calcium homeostasis can be due to calcium influx from the extracellular environment, subsequent release of calcium by intracellular calcium stores, and, possibly, from direct damage of ER compartments, all contributing to ER stress [38, 195, 196]. In this context, different PFTs such as LLO, Aerolysin, and ShlA were shown to induce ER expansion, fission, and vacuolation [84, 89, 197]. PFT-mediated ER vacuolation may result from ER damage or cell death [84, 89]. Nevertheless, ER vacuolation in LLO-damaged cells ranges from mild fission to disruption of the entire ER network with cells recovering normal ER morphology following toxin washout [89]. ER fission likely limits ion and protein diffusion, and thus, this process could help cells isolating intracellular calcium pools, and preventing overwhelming and deleterious elevations. On the other hand, limiting protein diffusion may compromise host-cell signaling and affect the responses to intoxication and ultimately to infection.
The morphology of post-ER compartments and the Golgi apparatus is not affected by aerolysin even in cells with dramatic ER fission and vacuolation [197]. However, VCC-induced vacuoles recruit proteins from the trans-Golgi network (TGN46), and co-localize with late-endocytic (Rab7 and LAMP1) [198] and autophagic (LC3) compartments [199, 200]. Accordingly, both vacuole turnover and recovery of ER morphology following PFT intoxication may rely in autophagic pathways. In addition, cells respond to PFT-induced potassium efflux by inducing the formation of lipid droplets [41] and promoting caspase-1-dependent activation of sterol-responsive element-binding proteins (SREBPs) [201], central regulators of lipid metabolism, and membrane biogenesis [202]. Both processes appear to promote cell survival against PFT intoxication. Intriguingly, however, the host pore-forming protein gasdermin D, which is activated downstream caspase-1, up-regulates liver lipogenesis and contributes to the excess inflammation in mouse models of fatty liver disease [203]. Accordingly, it is possible that, while caspase-1-promoted fatty acid metabolism may increase cell-autonomous survival during PFTs’ intoxication, such mechanism may also trigger unexpected inflammatory processes in vivo.
Different PFTs can also directly target mitochondria [204,205,206,207,208], either affecting mitochondrial permeability, morphology, or functioning [204, 205, 207,208,209,210,211]. The Helicobacter pylori PFT, VacA, promotes Drp1-mediated mitochondrial fission and causes release of cytochrome c, an event also promoted by HlyA and the staphylococcal toxin Panton–Valentine leukocidin (PVL) [204,205,206, 208]. Mitochondrial damage caused by PVL and PLY also induces the release of Smac [206] and the apoptosis-inducing factor (AIF) [207] and mtDNA [209], respectively. Ultimately, this may lead to the activation of pro-apoptotic caspase-3 and -9, and trigger cell death [204, 206, 208]. Accordingly, inhibition of Drp1 was shown to prevent mitochondrial permeabilization and cell death upon VacA intoxication [212]. During infection, LLO-mediated transient mitochondrial fragmentation does not lead to cell death but promotes L. monocytogenes replication [213]. Contrarily, the PLY-mediated release of mtDNA within secreted microvesicles contributes to pro-inflammatory responses against S. pneumoniae [209].
PFT-induced lysosomal damage was also observed in epithelial cells during L. monocytogenes infection [214]. LLO and other CDCs induce lysosomal permeabilization and release of the lysosomal aspartyl-protease cathepsin D, which remains transiently active in the cytosol, but it is not involved in PFTs’ degradation [214]. LLO-dependent degradation of the crucial SUMOylation conjugation enzyme, Ubc9, promotes L. monocytogenes infection and is partially blocked by an aspartyl-protease inhibitor [215]. Impaired SUMOylation and alteration of the cellular proteome are promoted by LLO and PFO by decreasing the abundance of a variety of ubiquitilome-related proteins [216]. This process involves only post-transcriptional mechanisms and may also be beneficial for L. monocytogenes infection.
Altogether, PFT-induced organelle damage is linked to alterations of ion imbalance, cellular metabolism, and cell death. Nevertheless, in different model systems, cells have been shown to recover from such stress by coordinating different processes that may involve: rescuing pathways such as the UPR, putative calcium sequestration mechanisms, increased membrane synthesis and lipid metabolism, and purging or recycling of damage compartments by autophagy (Fig. 4).
Autophagy
Autophagy constitutes a central defense mechanism against both extracellular and invading bacterial pathogens [12, 121, 217,218,219]. It is activated upon PFT-induced PM damage to sustain metabolic homeostasis as PM perforation causes nutrient and energy depletion. In particular, SLO, alpha-toxin and VCC, promote autophagy activation downstream AMPK or elF2alpha-kinase 4, known as energy- and nutrient-depletion sensors, respectively [220, 221]. Autophagy was also proposed to promote PM repair in C. elegans fed with Escherichia coli expressing Cry5B. The co-localization of Cry5B with autophagic markers suggests that autophagy participates in the degradation of PFTs [121]. In agreement, worms intoxicated with Cry5B, Cry21A, and SLO display decreased survival upon autophagy inhibition [121]. VacA stability is also promoted by the inhibition of autophagy [222]. VCC activates autophagy and induces the formation of large intracellular vacuoles, which are targeted by the autophagic machinery [199, 223]. In addition, inhibition of autophagy in VCC-intoxicated cells impairs cell survival [199]. Whether PFT-induced vacuoles are effectively removed by autophagy has never been confirmed. Nevertheless, autophagy plays a crucial role in ER remodeling [224], mitochondria degradation [225], and detection of damaged endosomes and lysosomes [226, 227]. Since PFTs promote ER vacuolation and can damage mitochondria and lysosomes, one can hypothesize that autophagy also protects intoxicated cells by removing damaged organelles (Fig. 4).
During infection, autophagy protects the host-cell cytosol against the activity of PFTs such as alpha-toxin and HlyA, which allow bacterial escape from intracellular vacuoles [12, 228], and limits VacA-induced cellular damage [222]. In contrast, the activation of autophagy during L. monocytogenes infection depends on LLO [229], but enables the formation of replication vacuoles (SLAPs—spacious Listeria-containing phagosomes), which may provide a niche for persistent infections [230, 231]. Thus, autophagy acts as an antimicrobial defense mechanism, but can also be exploited by PFT-expressing bacterial pathogens to promote their replication [232].
Although this review focuses on the mechanisms that protect host cells against PFTs, it is worth to mention that lytic concentrations or permanent exposure to PFTs cause irreversible PM permeability, culminating into uncontrolled necrotic cell death [11]. At sub-lytic doses, PFTs can induce different programmed cell death pathways, in particular pyroptosis and necroptosis, which significantly affect the outcome of infection in vivo [4, 11, 35, 64, 233, 234]. PFT-producing pathogens activate different cell death pathways [64]; however, the mechanisms underlying the activation of a specific cell death pathway during PFT intoxication or infection vary considerably according to the cell type or model organism and PFT tested. Whether the different cell death responses are either beneficial for the host—protecting it—or for the pathogen promoting infection needs further investigation.
Open questions and future directions
PFTs were for long considered potent unsophisticated virulence factors whose solely function was to form PM holes in host cells. However, in the recent years, several studies revealed PFTs as multifaceted factors inducing a plethora of cellular responses. At sub-lytic concentrations, PFTs allow bacteria to manipulate host-cellular functions, not necessarily to kill the host cells but to promote overall pathogenesis [4, 235]. Studies performed in cultured cell lines in vitro generated an extensive amount of data but, paradoxically, failed to establish mechanisms through which sub-lytic concentrations of PFTs may promote systemic infections. Efforts should now be directed to further understand the complexity of PFTs activity and the host response during disease.
The cellular responses to sub-lytic concentrations of PFTs were assessed individually for specific PFTs, using several cultured cell lines and a range of different concentrations of purified toxins. Given that PFTs are widespread among bacterial pathogens and share essential functions and effects, in the future, PFTs should be studied together with an emphasis on common features. In particular, a combination between ultra-structural, super resolution, and live imaging microscopy analysis of wound sites is now important to characterize the architecture of PFT-mediated injuries and localized repair mechanisms. This approach could reveal broad antimicrobial strategies to target PFTs and fight several bacterial infections.
As described in this review, a multitude of mechanisms were involved in PFTs’ defense at the cellular level. Whether these mechanisms are part of a coordinated response regulated in time and space needs to be addressed. In addition, the interdependence of the different defense events and the molecular basis for their activation require further investigation. Importantly, findings in the context of PFTs response will be of interest in the other pathological contexts in which plasma membrane damage is occurring.
PFTs’ defense at the organism level also deserves further exploration. Indeed, the exact mechanisms of action of cellular defense pathways, where they act and how they are protective in a complex organism, remain mainly unidentified. Host specificity needs to be considered and future efforts should focus on the development and use of valuable infection models, such as D. melanogaster, D. rerio, and C. elegans, which are extremely versatile and for which a large number of molecular tools are available. The understanding of PFT-induced host barrier dysfunction, inflammation, and immune response disruption is of critical importance in the perspective of systemic infection, and needs to be analyzed in vivo in the context of organized tissues composed by a variety of cell types. In addition, emerging three-dimensional (3D) cell culture models may also represent valuable tools to study host responses to PFT activity in the context of tissue properties or organ complexity.
Altogether, these studies should help to find novel treatments targeting PFTs from a broad range of slightly different toxins or enhancing host defense mechanisms.
Abbreviations
- A:
-
Annexin
- ALG-2:
-
Apoptosis-linked gene 2
- ALIX:
-
ALG-2-interacting protein X
- ALO:
-
Anthrolysin
- ASM:
-
Acid sphingomyelinase
- ATF6:
-
Activating transcription factor 6
- CDC:
-
Cholesterol-dependent cytolysin
- ER:
-
Endoplasmic reticulum
- ESCRT:
-
Endosomal sorting complex required for transport
- INY:
-
Inerolysin
- IRE1:
-
Inositol requiring enzyme 1
- LLO:
-
Listeriolysin O
- MAPK:
-
Mitogen-activated protein kinase
- MLKL:
-
Mixed lineage kinase-like
- MT:
-
Microtubule
- mtDNA:
-
Mitochondrial DNA
- MVB:
-
Multivesicular body
- NMIIA:
-
Non-muscle myosin IIA
- NMII:
-
Non-muscle myosin II
- PERK:
-
Protein kinase RNA (PKR)-like ER kinase
- PFO:
-
Perfringolysin O
- PFT:
-
Pore-forming toxin
- PLY:
-
Pneumolysin
- PM:
-
Plasma membrane
- PVL:
-
Panton–Valentine leukocidin
- SLAPs:
-
Spacious Listeria-containing phagosomes
- SLO:
-
Streptolysin O
- Slp4-a:
-
Synaptotagmin-like protein 4a
- SNARES:
-
N-ethylmaleimide sensitive factor attachment protein receptors
- SREBPs:
-
Sterol-responsive element-binding proteins
- T3SS:
-
Type-III secretion system
- TLO:
-
Tetanolysin
- UPR:
-
Unfolded protein response
- VCC:
-
Vibrio cholerae cytolysin
- VLY:
-
Vaginolysin
References
Bischofberger M, Gonzalez MR, van der Goot FG (2009) Membrane injury by pore-forming proteins. Curr Opin Cell Biol 21(4):589–595. https://doi.org/10.1016/j.ceb.2009.04.003
Cooper ST, McNeil PL (2015) Membrane repair: mechanisms and pathophysiology. Physiol Rev 95(4):1205–1240. https://doi.org/10.1152/physrev.00037.2014
Lane N, Martin WF (2012) The origin of membrane bioenergetics. Cell 151(7):1406–1416
Bischofberger M, Iacovache I, van der Goot FG (2012) Pathogenic pore-forming proteins: function and host response. Cell Host Microbe 12(3):266–275. https://doi.org/10.1016/j.chom.2012.08.005
Jimenez AJ, Perez F (2017) Plasma membrane repair: the adaptable cell life-insurance. Curr Opin Cell Biol 47:99–107. https://doi.org/10.1016/j.ceb.2017.03.011
Jimenez AJ, Perez F (2015) Physico-chemical and biological considerations for membrane wound evolution and repair in animal cells. Semin Cell Dev Biol 45:2–9. https://doi.org/10.1016/j.semcdb.2015.09.023
Han R (2011) Muscle membrane repair and inflammatory attack in dysferlinopathy. Skelet Muscle 1(1):10. https://doi.org/10.1186/2044-5040-1-10
Carmeille R, Croissant C, Bouvet F, Bouter A (2017) Membrane repair assay for human skeletal muscle cells. Methods Mol Biol 1668:195–207. https://doi.org/10.1007/978-1-4939-7283-8_14
Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL, Campbell KP (2003) Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 423(6936):168–172. https://doi.org/10.1038/nature01573
Lakey JH, van der Goot FG, Pattus F (1994) All in the family: the toxic activity of pore-forming colicins. Toxicology 87(1–3):85–108
Los FC, Randis TM, Aroian RV, Ratner AJ (2013) Role of pore-forming toxins in bacterial infectious diseases. Microbiol Mol Biol Rev 77(2):173–207. https://doi.org/10.1128/MMBR.00052-12
Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, Hamada S, Yoshimori T (2004) Autophagy defends cells against invading group A Streptococcus. Science 306(5698):1037–1040. https://doi.org/10.1126/science.1103966
Schnupf P, Portnoy DA (2007) Listeriolysin O: a phagosome-specific lysin. Microbes Infect 9(10):1176–1187. https://doi.org/10.1016/j.micinf.2007.05.005
Alli OA, Gao LY, Pedersen LL, Zink S, Radulic M, Doric M, Abu Kwaik Y (2000) Temporal pore formation-mediated egress from macrophages and alveolar epithelial cells by Legionella pneumophila. Infect Immun 68(11):6431–6440
Dal Peraro M, van der Goot FG (2016) Pore-forming toxins: ancient, but never really out of fashion. Nat Rev Microbiol 14(2):77–92. https://doi.org/10.1038/nrmicro.2015.3
Popoff MR (2014) Clostridial pore-forming toxins: powerful virulence factors. Anaerobe 30:220–238. https://doi.org/10.1016/j.anaerobe.2014.05.014
Reboul CF, Whisstock JC, Dunstone MA (2016) Giant MACPF/CDC pore forming toxins: a class of their own. Biochem Biophys Acta 1858 3:475–486. https://doi.org/10.1016/j.bbamem.2015.11.017
Johnson BB, Heuck AP (2014) Perfringolysin O structure and mechanism of pore formation as a paradigm for cholesterol-dependent cytolysins. Subcell Biochem 80:63–81. https://doi.org/10.1007/978-94-017-8881-6_5
Aroian R, van der Goot FG (2007) Pore-forming toxins and cellular non-immune defenses (CNIDs). Curr Opin Microbiol 10(1):57–61. https://doi.org/10.1016/j.mib.2006.12.008
Cassidy SK, O’Riordan MX (2013) More than a pore: the cellular response to cholesterol-dependent cytolysins. Toxins 5(4):618–636. https://doi.org/10.3390/toxins5040618
Draeger A, Schoenauer R, Atanassoff AP, Wolfmeier H, Babiychuk EB (2014) Dealing with damage: plasma membrane repair mechanisms. Biochimie 107(Pt A):66–72. https://doi.org/10.1016/j.biochi.2014.08.008
Cossart P (1988) The listeriolysin O gene: a chromosomal locus crucial for the virulence of Listeria monocytogenes. Infection 16(Suppl 2):S157–S159
Glomski IJ, Decatur AL, Portnoy DA (2003) Listeria monocytogenes mutants that fail to compartmentalize listerolysin O activity are cytotoxic, avirulent, and unable to evade host extracellular defenses. Infect Immun 71(12):6754–6765
Bielecki J, Youngman P, Connelly P, Portnoy DA (1990) Bacillus subtilis expressing a haemolysin gene from Listeria monocytogenes can grow in mammalian cells. Nature 345(6271):175–176. https://doi.org/10.1038/345175a0
Andrews NW, Corrotte M (2018) Plasma membrane repair. Curr Biol 28(8):R392–R397. https://doi.org/10.1016/j.cub.2017.12.034
McNeil PL, Steinhardt RA (1997) Loss, restoration, and maintenance of plasma membrane integrity. J Cell Biol 137(1):1–4
Heilbrunn LV (1958) The dynamics of living protoplasm. Academic Press, New York, p 634
Chambers RC, Champers EL (1961) Explorations into the nature of the living cell. Harvard University Press, Cambridge, p 352
Tam C, Flannery AR, Andrews N (2013) Live imaging assay for assessing the roles of Ca2+ and sphingomyelinase in the repair of pore-forming toxin wounds. J Vis Exp 78:e50531. https://doi.org/10.3791/50531
Wippel C, Fortsch C, Hupp S, Maier E, Benz R, Ma J, Mitchell TJ, Iliev AI (2011) Extracellular calcium reduction strongly increases the lytic capacity of pneumolysin from Streptococcus pneumoniae in brain tissue. J Infect Dis 204(6):930–936. https://doi.org/10.1093/infdis/jir434
Wolfmeier H, Schoenauer R, Atanassoff AP, Neill DR, Kadioglu A, Draeger A, Babiychuk EB (2015) Ca(2)(+)-dependent repair of pneumolysin pores: a new paradigm for host cellular defense against bacterial pore-forming toxins. Biochem Biophys Acta 1853 9:2045–2054. https://doi.org/10.1016/j.bbamcr.2014.09.005
Babiychuk EB, Monastyrskaya K, Potez S, Draeger A (2009) Intracellular Ca(2+) operates a switch between repair and lysis of streptolysin O-perforated cells. Cell Death Differ 16(8):1126–1134. https://doi.org/10.1038/cdd.2009.30
Gekara NO, Groebe L, Viegas N, Weiss S (2008) Listeria monocytogenes desensitizes immune cells to subsequent Ca2+ signaling via listeriolysin O-induced depletion of intracellular Ca2+ stores. Infect Immun 76(2):857–862
Chen F, Kumar S, Yu Y, Aggarwal S, Gross C, Wang Y, Chakraborty T, Verin AD, Catravas JD, Lucas R, Black SM, Fulton DJR (2014) PKC-dependent phosphorylation of eNOS at T495 regulates eNOS coupling and endothelial barrier function in response to G+-toxins. PLoS One 9(7):e99823
Gonzalez-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, Dube PH, Bergman MA, Orihuela CJ (2015) Pore-forming toxins induce macrophage necroptosis during acute bacterial pneumonia. PLoS Pathog 11(12):e1005337. https://doi.org/10.1371/journal.ppat.1005337
Kennedy CL, Smith DJ, Lyras D, Chakravorty A, Rood JI (2009) Programmed cellular necrosis mediated by the pore-forming alpha-toxin from Clostridium septicum. PLoS Pathog 5(7):e1000516. https://doi.org/10.1371/journal.ppat.1000516
Alhamdi Y, Neill DR, Abrams ST, Malak HA, Yahya R, Barrett-Jolley R, Wang G, Kadioglu A, Toh CH (2015) Circulating pneumolysin is a potent inducer of cardiac injury during pneumococcal infection. PLoS Pathog 11(5):e1004836. https://doi.org/10.1371/journal.ppat.1004836
Gekara NO, Westphal K, Ma B, Rohde M, Groebe L, Weiss S (2007) The multiple mechanisms of Ca2+ signalling by listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Cell Microbiol 9(8):2008–2021. https://doi.org/10.1111/j.1462-5822.2007.00932.x
Boucher E, Mandato CA (2015) Plasma membrane and cytoskeleton dynamics during single-cell wound healing. Biochim Biophys Acta 1853 10(Pt A):2649–2661. https://doi.org/10.1016/j.bbamcr.2015.07.012
Kloft N, Busch T, Neukirch C, Weis S, Boukhallouk F, Bobkiewicz W, Cibis I, Bhakdi S, Husmann M (2009) Pore-forming toxins activate MAPK p38 by causing loss of cellular potassium. Biochem Biophys Res Commun 385(4):503–506. https://doi.org/10.1016/j.bbrc.2009.05.121
Gonzalez MR, Bischofberger M, Freche B, Ho S, Parton RG, van der Goot FG (2011) Pore-forming toxins induce multiple cellular responses promoting survival. Cell Microbiol 13(7):1026–1043. https://doi.org/10.1111/j.1462-5822.2011.01600.x
Bischof LJ, Kao CY, Los FC, Gonzalez MR, Shen Z, Briggs SP, van der Goot FG, Aroian RV (2008) Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog 4(10):e1000176. https://doi.org/10.1371/journal.ppat.1000176
Huffman DL, Abrami L, Sasik R, Corbeil J, van der Goot FG, Aroian RV (2004) Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc Natl Acad Sci USA 101(30):10995–11000. https://doi.org/10.1073/pnas.0404073101
Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G (2013) K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38(6):1142–1153. https://doi.org/10.1016/j.immuni.2013.05.016
Walev I, Bhakdi SC, Hofmann F, Djonder N, Valeva A, Aktories K, Bhakdi S (2001) Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc Natl Acad Sci USA 98(6):3185–3190. https://doi.org/10.1073/pnas.051429498
Walev I, Martin E, Jonas D, Mohamadzadeh M, Muller-Klieser W, Kunz L, Bhakdi S (1993) Staphylococcal alpha-toxin kills human keratinocytes by permeabilizing the plasma membrane for monovalent ions. Infect Immun 61(12):4972–4979
Zitzer A, Wassenaar TM, Walev I, Bhakdi S (1997) Potent membrane-permeabilizing and cytocidal action of Vibrio cholerae cytolysin on human intestinal cells. Infect Immun 65(4):1293–1298
Los FC, Kao CY, Smitham J, McDonald KL, Ha C, Peixoto CA, Aroian RV (2011) RAB-5- and RAB-11-dependent vesicle-trafficking pathways are required for plasma membrane repair after attack by bacterial pore-forming toxin. Cell Host Microbe 9(2):147–157. https://doi.org/10.1016/j.chom.2011.01.005
von Hoven G, Rivas AJ, Neukirch C, Meyenburg M, Qin Q, Parekh S, Hellmann N, Husmann M (2017) Repair of a bacterial small beta-barrel toxin pore depends on channel width. mBio. https://doi.org/10.1128/mbio.02083-16
Cabezas S, Ho S, Ros U, Lanio ME, Alvarez C, van der Goot FG (2017) Damage of eukaryotic cells by the pore-forming toxin sticholysin II: consequences of the potassium efflux. Biochem Biophys Acta 1859 5:982–992. https://doi.org/10.1016/j.bbamem.2017.02.001
Ros U, Garcia-Saez AJ (2015) More than a pore: the interplay of pore-forming proteins and lipid membranes. J Membr Biol 248(3):545–561. https://doi.org/10.1007/s00232-015-9820-y
Gilbert RJ (2016) Protein–lipid interactions and non-lamellar lipidic structures in membrane pore formation and membrane fusion. Biochem Biophys Acta 1858 1858(3):487–499. https://doi.org/10.1016/j.bbamem.2015.11.026
Sonnen AF, Plitzko JM, Gilbert RJ (2014) Incomplete pneumolysin oligomers form membrane pores. Open Biol 4:140044. https://doi.org/10.1098/rsob.140044
Gilbert RJ, Dalla Serra M, Froelich CJ, Wallace MI, Anderluh G (2014) Membrane pore formation at protein–lipid interfaces. Trends Biochem Sci 39(11):510–516. https://doi.org/10.1016/j.tibs.2014.09.002
Mancheno JM, Martin-Benito J, Martinez-Ripoll M, Gavilanes JG, Hermoso JA (2003) Crystal and electron microscopy structures of sticholysin II actinoporin reveal insights into the mechanism of membrane pore formation. Structure 11(11):1319–1328
Tanaka K, Caaveiro JM, Morante K, Gonzalez-Manas JM, Tsumoto K (2015) Structural basis for self-assembly of a cytolytic pore lined by protein and lipid. Nat Commun 6:6337. https://doi.org/10.1038/ncomms7337
Skals M, Leipziger J, Praetorius HA (2011) Haemolysis induced by alpha-toxin from Staphylococcus aureus requires P2X receptor activation. Pflug Arch 462(5):669–679
Verhoef PA, Estacion M, Schilling W, Dubyak GR (2003) P2X7 receptor-dependent blebbing and the activation of Rho-effector kinases, caspases, and IL-1 beta release. J Immunol 170(11):5728–5738
MacKenzie A, Wilson HL, Kiss-Toth E, Dower SK, North RA, Surprenant A (2001) Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity 15(5):825–835
Solle M, Labasi J, Perregaux DG, Stam E, Petrushova N, Koller BH, Griffiths RJ, Gabel CA (2001) Altered cytokine production in mice lacking P2X(7) receptors. J Biol Chem 276(1):125–132. https://doi.org/10.1074/jbc.M006781200
Di Virgilio F, Adinolfi E (2017) Extracellular purines, purinergic receptors and tumor growth. Oncogene 36(3):293–303. https://doi.org/10.1038/onc.2016.206
Khakh BS, North RA (2012) Neuromodulation by extracellular ATP and P2X receptors in the CNS. Neuron 76(1):51–69. https://doi.org/10.1016/j.neuron.2012.09.024
Russo HM, Rathkey J, Boyd-Tressler A, Katsnelson MA, Abbott DW, Dubyak GR (2016) Active caspase-1 induces plasma membrane pores that precede pyroptotic lysis and are blocked by lanthanides. J Immunol 197(4):1353–1367. https://doi.org/10.4049/jimmunol.1600699
Gonzalez-Juarbe N, Bradley KM, Riegler AN, Reyes LF, Brissac T, Park SS, Restrepo MI, Orihuela CJ (2018) Bacterial pore-forming toxins promote the activation of caspases in parallel to necroptosis to enhance alarmin release and inflammation during pneumonia. Sci Rep 8(1):5846. https://doi.org/10.1038/s41598-018-24210-8
McNeil PL, Vogel SS, Miyake K, Terasaki M (2000) Patching plasma membrane disruptions with cytoplasmic membrane. J Cell Sci 113(Pt 11):1891–1902
Gerke V, Creutz CE, Moss SE (2005) Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol 6(6):449–461. https://doi.org/10.1038/nrm1661
Boye TL, Nylandsted J (2016) Annexins in plasma membrane repair. Biol Chem 397(10):961–969. https://doi.org/10.1515/hsz-2016-0171
Potez S, Luginbuhl M, Monastyrskaya K, Hostettler A, Draeger A, Babiychuk EB (2011) Tailored protection against plasmalemmal injury by annexins with different Ca2+ sensitivities. J Biol Chem 286(20):17982–17991. https://doi.org/10.1074/jbc.M110.187625
Wolfmeier H, Radecke J, Schoenauer R, Koeffel R, Babiychuk VS, Drucker P, Hathaway LJ, Mitchell TJ, Zuber B, Draeger A, Babiychuk EB (2016) Active release of pneumolysin prepores and pores by mammalian cells undergoing a Streptococcus pneumoniae attack. Biochim Biophys Acta 1860 11(Pt A):2498–2509. https://doi.org/10.1016/j.bbagen.2016.07.022
McNeil AK, Rescher U, Gerke V, McNeil PL (2006) Requirement for annexin A1 in plasma membrane repair. J Biol Chem 281(46):35202–35207. https://doi.org/10.1074/jbc.M606406200
Monastyrskaya K, Babiychuk EB, Draeger A, Burkhard FC (2013) Down-regulation of annexin A1 in the urothelium decreases cell survival after bacterial toxin exposure. J Urol 190(1):325–333. https://doi.org/10.1016/j.juro.2013.01.088
Babiychuk EB, Monastyrskaya K, Potez S, Draeger A (2011) Blebbing confers resistance against cell lysis. Cell Death Differ 18(1):80–89. https://doi.org/10.1038/cdd.2010.81
Bouter A, Gounou C, Berat R, Tan S, Gallois B, Granier T, d’Estaintot BL, Poschl E, Brachvogel B, Brisson AR (2011) Annexin-A5 assembled into two-dimensional arrays promotes cell membrane repair. Nat Commun 2:270. https://doi.org/10.1038/ncomms1270
Carmeille R, Degrelle SA, Plawinski L, Bouvet F, Gounou C, Evain-Brion D, Brisson AR, Bouter A (2015) Annexin-A5 promotes membrane resealing in human trophoblasts. Biochem Biophys Acta 1853 9:2033–2044. https://doi.org/10.1016/j.bbamcr.2014.12.038
Carmeille R, Bouvet F, Tan S, Croissant C, Gounou C, Mamchaoui K, Mouly V, Brisson AR, Bouter A (2016) Membrane repair of human skeletal muscle cells requires annexin-A5. Biochem Biophys Acta 1863 9:2267–2279. https://doi.org/10.1016/j.bbamcr.2016.06.003
Charras GT (2008) A short history of blebbing. J Microsc 231(3):466–478. https://doi.org/10.1111/j.1365-2818.2008.02059.x
Charras G, Paluch E (2008) Blebs lead the way: how to migrate without lamellipodia. Nat Rev Mol Cell Biol 9(9):730–736. https://doi.org/10.1038/nrm2453
Skocaj M, Yu Y, Grundner M, Resnik N, Bedina Zavec A, Leonardi A, Krizaj I, Guella G, Macek P, Kreft ME, Frangez R, Veranic P, Sepcic K (2016) Characterisation of plasmalemmal shedding of vesicles induced by the cholesterol/sphingomyelin binding protein, ostreolysin A-mCherry. Biochem Biophys Acta 1858 11:2882–2893
Keyel PA, Loultcheva L, Roth R, Salter RD, Watkins SC, Yokoyama WM, Heuser JE (2011) Streptolysin O clearance through sequestration into blebs that bud passively from the plasma membrane. J Cell Sci 124(Pt 14):2414–2423. https://doi.org/10.1242/jcs.076182
Mesquita FS, Brito C, Cabanes D, Sousa S (2017) Control of cytoskeletal dynamics during cellular responses to pore forming toxins. Commun Integr Biol 10(5–6):e1349582. https://doi.org/10.1080/19420889.2017.1349582
Moskovich O, Fishelson Z (2007) Live cell imaging of outward and inward vesiculation induced by the complement c5b–9 complex. J Biol Chem 282(41):29977–29986. https://doi.org/10.1074/jbc.M703742200
Wickman GR, Julian L, Mardilovich K, Schumacher S, Munro J, Rath N, Zander SA, Mleczak A, Sumpton D, Morrice N, Bienvenut WV, Olson MF (2013) Blebs produced by actin–myosin contraction during apoptosis release damage-associated molecular pattern proteins before secondary necrosis occurs. Cell Death Differ 20(10):1293–1305. https://doi.org/10.1038/cdd.2013.69
Headley MB, Bins A, Nip A, Roberts EW, Looney MR, Gerard A, Krummel MF (2016) Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature 531(7595):513–517. https://doi.org/10.1038/nature16985
Lee KZ, Lestradet M, Socha C, Schirmeier S, Schmitz A, Spenle C, Lefebvre O, Keime C, Yamba WM, Bou Aoun R, Liegeois S, Schwab Y, Simon-Assmann P, Dalle F, Ferrandon D (2016) Enterocyte purge and rapid recovery is a resilience reaction of the gut epithelium to pore-forming toxin attack. Cell Host Microbe 20(6):716–730. https://doi.org/10.1016/j.chom.2016.10.010
Martin CJ, Peters KN, Behar SM (2014) Macrophages clean up: efferocytosis and microbial control. Curr Opin Microbiol 17:17–23
Angus AA, Lee AA, Augustin DK, Lee EJ, Evans DJ, Fleiszig SM (2008) Pseudomonas aeruginosa induces membrane blebs in epithelial cells, which are utilized as a niche for intracellular replication and motility. Infect Immun 76(5):1992–2001. https://doi.org/10.1128/IAI.01221-07
Czuczman MA, Fattouh R, van Rijn JM, Canadien V, Osborne S, Muise AM, Kuchroo VK, Higgins DE, Brumell JH (2014) Listeria monocytogenes exploits efferocytosis to promote cell-to-cell spread. Nature 509(7499):230–234. https://doi.org/10.1038/nature13168
Jimenez AJ, Maiuri P, Lafaurie-Janvore J, Divoux S, Piel M, Perez F (2014) ESCRT machinery is required for plasma membrane repair. Science 343(6174):1247136. https://doi.org/10.1126/science.1247136
Mesquita FS, Brito C, Mazon Moya MJ, Pinheiro JC, Mostowy S, Cabanes D, Sousa S (2017) Endoplasmic reticulum chaperone Gp96 controls actomyosin dynamics and protects against pore-forming toxins. EMBO Rep 18(2):303–318. https://doi.org/10.15252/embr.201642833
Atanassoff AP, Wolfmeier H, Schoenauer R, Hostettler A, Ring A, Draeger A, Babiychuk EB (2014) Microvesicle shedding and lysosomal repair fulfill divergent cellular needs during the repair of streptolysin O-induced plasmalemmal damage. PLoS One 9(2):e89743. https://doi.org/10.1371/journal.pone.0089743
Walev I, Palmer M, Valeva A, Weller U, Bhakdi S (1995) Binding, oligomerization, and pore formation by streptolysin O in erythrocytes and fibroblast membranes: detection of nonlytic polymers. Infect Immun 63(4):1188–1194
Romero M, Keyel M, Shi G, Bhattacharjee P, Roth R, Heuser JE, Keyel PA (2017) Intrinsic repair protects cells from pore-forming toxins by microvesicle shedding. Cell Death Differ 24(5):798–808. https://doi.org/10.1038/cdd.2017.11
Drucker P, Bachler S, Wolfmeier H, Schoenauer R, Koffel R, Babiychuk VS, Dittrich PS, Draeger A, Babiychuk EB (2018) Pneumolysin-damaged cells benefit from non-homogeneous toxin binding to cholesterol-rich membrane domains. Biochem Biophys Acta 1863 8:795–805. https://doi.org/10.1016/j.bbalip.2018.04.010
Scheffer LL, Sreetama SC, Sharma N, Medikayala S, Brown KJ, Defour A, Jaiswal JK (2014) Mechanism of Ca(2)(+)-triggered ESCRT assembly and regulation of cell membrane repair. Nat Commun 5:5646. https://doi.org/10.1038/ncomms6646
Olmos Y, Carlton JG (2016) The ESCRT machinery: new roles at new holes. Curr Opin Cell Biol 38:1–11. https://doi.org/10.1016/j.ceb.2015.12.001
Satoh H, Nakano Y, Shibata H, Maki M (2002) The penta-EF-hand domain of ALG-2 interacts with amino-terminal domains of both annexin VII and annexin XI in a Ca2+-dependent manner. Biochem Biophys Acta 1600(1–2):61–67
Jaiswal JK, Lauritzen SP, Scheffer L, Sakaguchi M, Bunkenborg J, Simon SM, Kallunki T, Jaattela M, Nylandsted J (2014) S100A11 is required for efficient plasma membrane repair and survival of invasive cancer cells. Nat Commun 5:3795. https://doi.org/10.1038/ncomms4795
Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, Green DR (2017) ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169(2):286–300 e216. https://doi.org/10.1016/j.cell.2017.03.020
Schoenauer R, Atanassoff AP, Wolfmeier H, Pelegrin P, Babiychuk EB, Draeger A (2014) P2X7 receptors mediate resistance to toxin-induced cell lysis. Biochem Biophys Acta 1843 5:915–922. https://doi.org/10.1016/j.bbamcr.2014.01.024
North RA (2002) Molecular physiology of P2X receptors. Physiol Rev 82(4):1013–1067. https://doi.org/10.1152/physrev.00015.2002
Fujii Y, Nomura T, Yokoyama R, Shinoda S, Okamoto K (2003) Studies of the mechanism of action of the aerolysin-like hemolysin of Aeromonas sobria in stimulating T84 cells to produce cyclic AMP. Infect Immun 71(3):1557–1560
Henry BD, Neill DR, Becker KA, Gore S, Bricio-Moreno L, Ziobro R, Edwards MJ, Muhlemann K, Steinmann J, Kleuser B, Japtok L, Luginbuhl M, Wolfmeier H, Scherag A, Gulbins E, Kadioglu A, Draeger A, Babiychuk EB (2015) Engineered liposomes sequester bacterial exotoxins and protect from severe invasive infections in mice. Nat Biotechnol 33(1):81–88
Koffel R, Wolfmeier H, Larpin Y, Besancon H, Schoenauer R, Babiychuk VS, Drucker P, Pabst T, Mitchell TJ, Babiychuk EB, Draeger A (2018) Host-derived microvesicles carrying bacterial pore-forming toxins deliver signals to macrophages: a novel mechanism of shaping immune responses. Front Immunol 9:1688. https://doi.org/10.3389/fimmu.2018.01688
Tam C, Idone V, Devlin C, Fernandes MC, Flannery A, He X, Schuchman E, Tabas I, Andrews NW (2010) Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J Cell Biol 189(6):1027–1038. https://doi.org/10.1083/jcb.201003053
Togo T, Krasieva TB, Steinhardt RA (2000) A decrease in membrane tension precedes successful cell-membrane repair. Mol Biol Cell 11(12):4339–4346
Czibener C, Sherer NM, Becker SM, Pypaert M, Hui E, Chapman ER, Mothes W, Andrews NW (2006) Ca2+ and synaptotagmin VII-dependent delivery of lysosomal membrane to nascent phagosomes. J Cell Biol 174(7):997–1007. https://doi.org/10.1083/jcb.200605004
Reddy A, Caler EV, Andrews NW (2001) Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell 106(2):157–169
Chakrabarti S, Kobayashi KS, Flavell RA, Marks CB, Miyake K, Liston DR, Fowler KT, Gorelick FS, Andrews NW (2003) Impaired membrane resealing and autoimmune myositis in synaptotagmin VII-deficient mice. J Cell Biol 162(4):543–549. https://doi.org/10.1083/jcb.200305131
Defour A, Van der Meulen JH, Bhat R, Bigot A, Bashir R, Nagaraju K, Jaiswal JK (2014) Dysferlin regulates cell membrane repair by facilitating injury-triggered acid sphingomyelinase secretion. Cell Death Dis 5:e1306. https://doi.org/10.1038/cddis.2014.272
Michelet X, Tuli A, Gan H, Geadas C, Sharma M, Remold HG, Brenner MB (2018) Lysosome-mediated plasma membrane repair is dependent on the small GTPase Arl8b and determines cell death type in Mycobacterium tuberculosis infection. J Immunol 200(9):3160–3169. https://doi.org/10.4049/jimmunol.1700829
Encarnacao M, Espada L, Escrevente C, Mateus D, Ramalho J, Michelet X, Santarino I, Hsu VW, Brenner MB, Barral DC, Vieira OV (2016) A Rab3a-dependent complex essential for lysosome positioning and plasma membrane repair. J Cell Biol 213(6):631–640. https://doi.org/10.1083/jcb.201511093
Corrotte M, Fernandes MC, Tam C, Andrews NW (2012) Toxin pores endocytosed during plasma membrane repair traffic into the lumen of MVBs for degradation. Traffic 13(3):483–494. https://doi.org/10.1111/j.1600-0854.2011.01323.x
Draeger A, Babiychuk EB (2013) Ceramide in plasma membrane repair. Handb Exp Pharmacol 216:341–353. https://doi.org/10.1007/978-3-7091-1511-4_17
Castro-Gomes T, Corrotte M, Tam C, Andrews NW (2016) Plasma membrane repair is regulated extracellularly by proteases released from lysosomes. PLoS One 11(3):e0152583. https://doi.org/10.1371/journal.pone.0152583
Corrotte M, Almeida PE, Tam C, Castro-Gomes T, Fernandes MC, Millis BA, Cortez M, Miller H, Song W, Maugel TK, Andrews NW (2013) Caveolae internalization repairs wounded cells and muscle fibers. eLife 2:e00926. https://doi.org/10.7554/elife.00926
Parton RG, Simons K (2007) The multiple faces of caveolae. Nat Rev Mol Cell Biol 8(3):185–194. https://doi.org/10.1038/nrm2122
Idone V, Tam C, Goss JW, Toomre D, Pypaert M, Andrews NW (2008) Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J Cell Biol 180(5):905–914. https://doi.org/10.1083/jcb.200708010
Schuchman EH (2010) Acid sphingomyelinase, cell membranes and human disease: lessons from Niemann-Pick disease. FEBS Lett 584(9):1895–1900. https://doi.org/10.1016/j.febslet.2009.11.083
Schoenauer R, Larpin Y, Babiychuk EB, Drucker P, Babiychuk VS, Avota E, Schneider-Schaulies S, Schumacher F, Kleuser B, Koffel R, Draeger A (2018) Down-regulation of acid sphingomyelinase and neutral sphingomyelinase-2 inversely determines the cellular resistance to plasmalemmal injury by pore-forming toxins. FASEB J. https://doi.org/10.1096/fj.201800033r
Husmann M, Beckmann E, Boller K, Kloft N, Tenzer S, Bobkiewicz W, Neukirch C, Bayley H, Bhakdi S (2009) Elimination of a bacterial pore-forming toxin by sequential endocytosis and exocytosis. FEBS Lett 583(2):337–344. https://doi.org/10.1016/j.febslet.2008.12.028
Chen HD, Kao CY, Liu BY, Huang SW, Kuo CJ, Ruan JW, Lin YH, Huang CR, Chen YH, Wang HD, Aroian RV, Chen CS (2017) HLH-30/TFEB-mediated autophagy functions in a cell-autonomous manner for epithelium intrinsic cellular defense against bacterial pore-forming toxin in C. elegans. Autophagy 13(2):371–385. https://doi.org/10.1080/15548627.2016.1256933
Nygard Skalman L, Holst MR, Larsson E, Lundmark R (2018) Plasma membrane damage caused by listeriolysin O is not repaired through endocytosis of the membrane pore. Biol Open. https://doi.org/10.1242/bio.035287
Bianco F, Perrotta C, Novellino L, Francolini M, Riganti L, Menna E, Saglietti L, Schuchman EH, Furlan R, Clementi E, Matteoli M, Verderio C (2009) Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J 28(8):1043–1054. https://doi.org/10.1038/emboj.2009.45
Park PW, Foster TJ, Nishi E, Duncan SJ, Klagsbrun M, Chen Y (2004) Activation of syndecan-1 ectodomain shedding by Staphylococcus aureus alpha-toxin and beta-toxin. J Biol Chem 279(1):251–258. https://doi.org/10.1074/jbc.M308537200
Babiychuk EB, Monastyrskaya K, Draeger A (2008) Fluorescent annexin A1 reveals dynamics of ceramide platforms in living cells. Traffic 9(10):1757–1775. https://doi.org/10.1111/j.1600-0854.2008.00800.x
Roy D, Liston DR, Idone VJ, Di A, Nelson DJ, Pujol C, Bliska JB, Chakrabarti S, Andrews NW (2004) A process for controlling intracellular bacterial infections induced by membrane injury. Science 304(5676):1515–1518. https://doi.org/10.1126/science.1098371
Almeida MT, Mesquita FS, Cruz R, Osorio H, Custodio R, Brito C, Vingadassalom D, Martins M, Leong JM, Holden DW, Cabanes D, Sousa S (2015) Src-dependent tyrosine phosphorylation of non-muscle myosin heavy chain-IIA restricts Listeria monocytogenes cellular infection. J Biol Chem 290(13):8383–8395. https://doi.org/10.1074/jbc.M114.591313
Divangahi M, Chen M, Gan H, Desjardins D, Hickman TT, Lee DM, Fortune S, Behar SM, Remold HG (2009) Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat Immunol 10(8):899–906. https://doi.org/10.1038/ni.1758
Vadia S, Arnett E, Haghighat A-C, Wilson-Kubalek EM, Tweten RK, Seveau S (2011) The pore-forming toxin listeriolysin O mediates a novel entry pathway of L. monocytogenes into human hepatocytes. PLoS Pathog 7(11):e1002356
Fernandes MC, Cortez M, Flannery AR, Tam C, Mortara RA, Andrews NW (2011) Trypanosoma cruzi subverts the sphingomyelinase-mediated plasma membrane repair pathway for cell invasion. J Exp Med 208(5):909–921
Vadia S, Seveau S (2014) Fluxes of Ca2+ and K+ are required for the listeriolysin O-dependent internalization pathway of Listeria monocytogenes. Infect Immun 82(3):1084–1091. https://doi.org/10.1128/IAI.01067-13
Chen C, Nguyen BN, Mitchell G, Margolis SR, Ma D, Portnoy DA (2018) The listeriolysin O PEST-like sequence co-opts AP-2-mediated endocytosis to prevent plasma membrane damage during Listeria infection. Cell Host Microbe 23(6):786–795 e785. https://doi.org/10.1016/j.chom.2018.05.006
Boucher EK, Kato T, Mandato CA (2016) How plasma membrane and cytoskeletal dynamics influence single-cell wound healing: mechanotransduction, tension and tensegrity. In: Alexandrescu VA (ed) Wound healing—new insights into ancient challenges. Intechopen Publications, Rijieka, pp 197–225. https://doi.org/10.5772/63765
Miyake K, McNeil PL, Suzuki K, Tsunoda R, Sugai N (2001) An actin barrier to resealing. J Cell Sci 114(Pt 19):3487–3494
Togo T, Alderton JM, Bi GQ, Steinhardt RA (1999) The mechanism of facilitated cell membrane resealing. J Cell Sci 112(Pt 5):719–731
Togo T, Steinhardt RA (2004) Nonmuscle myosin IIA and IIB have distinct functions in the exocytosis-dependent process of cell membrane repair. Mol Biol Cell 15(2):688–695. https://doi.org/10.1091/mbc.E03-06-0430
McNeil PL, Kirchhausen T (2005) An emergency response team for membrane repair. Nat Rev Mol Cell Biol 6(6):499–505. https://doi.org/10.1038/nrm1665
Abreu-Blanco MT, Verboon JM, Parkhurst SM (2014) Coordination of Rho family GTPase activities to orchestrate cytoskeleton responses during cell wound repair. Curr Biol 24(2):144–155. https://doi.org/10.1016/j.cub.2013.11.048
Godin LM, Vergen J, Prakash YS, Pagano RE, Hubmayr RD (2011) Spatiotemporal dynamics of actin remodeling and endomembrane trafficking in alveolar epithelial type I cell wound healing. Am J Physiol Lung Cell Mol Physiol 300(4):L615–L623. https://doi.org/10.1152/ajplung.00265.2010
Walev I, Hombach M, Bobkiewicz W, Fenske D, Bhakdi S, Husmann M (2002) Resealing of large transmembrane pores produced by streptolysin O in nucleated cells is accompanied by NF-kappaB activation and downstream events. FASEB J 16(2):237–239. https://doi.org/10.1096/fj.01-0572fje
Benink HA, Bement WM (2005) Concentric zones of active RhoA and Cdc42 around single cell wounds. J Cell Biol 168(3):429–439. https://doi.org/10.1083/jcb.200411109
Hupp S, Fortsch C, Wippel C, Ma J, Mitchell TJ, Iliev AI (2013) Direct transmembrane interaction between actin and the pore-competent, cholesterol-dependent cytolysin pneumolysin. J Mol Biol 425(3):636–646. https://doi.org/10.1016/j.jmb.2012.11.034
Wales P, Schuberth CE, Aufschnaiter R, Fels J, Garcia-Aguilar I, Janning A, Dlugos CP, Schafer-Herte M, Klingner C, Walte M, Kuhlmann J, Menis E, Hockaday Kang L, Maier KC, Hou W, Russo A, Higgs HN, Pavenstadt H, Vogl T, Roth J, Qualmann B, Kessels MM, Martin DE, Mulder B, Wedlich-Soldner R (2016) Calcium-mediated actin reset (CaAR) mediates acute cell adaptations. eLife 5:1–31
Ono Y, Sorimachi H (2012) Calpains: an elaborate proteolytic system. Biochem Biophys Acta 1824 1:224–236. https://doi.org/10.1016/j.bbapap.2011.08.005
Mellgren RL, Zhang W, Miyake K, McNeil PL (2007) Calpain is required for the rapid, calcium-dependent repair of wounded plasma membrane. J Biol Chem 282(4):2567–2575. https://doi.org/10.1074/jbc.M604560200
Redpath GM, Woolger N, Piper AK, Lemckert FA, Lek A, Greer PA, North KN, Cooper ST (2014) Calpain cleavage within dysferlin exon 40a releases a synaptotagmin-like module for membrane repair. Mol Biol Cell 25(19):3037–3048. https://doi.org/10.1091/mbc.E14-04-0947
Xie XY, Barrett JN (1991) Membrane resealing in cultured rat septal neurons after neurite transection: evidence for enhancement by Ca(2+)-triggered protease activity and cytoskeletal disassembly. J Neurosci 11(10):3257–3267
Potter DA, Tirnauer JS, Janssen R, Croall DE, Hughes CN, Fiacco KA, Mier JW, Maki M, Herman IM (1998) Calpain regulates actin remodeling during cell spreading. J Cell Biol 141(3):647–662
Fang R, Wu R, Du H, Jin M, Liu Y, Lei G, Jiang B, Lei Z, Peng Y, Nie K, Tsuchiya K (2017) Pneumolysin-dependent calpain activation and interleukin-1alpha secretion in macrophages infected with Streptococcus pneumoniae. Infect Immun. https://doi.org/10.1128/iai.00201-17
Kullas AL, Li M, Davis DA (2004) Snf7p, a component of the ESCRT-III protein complex, is an upstream member of the RIM101 pathway in Candida albicans. Eukaryot Cell 3(6):1609–1618. https://doi.org/10.1128/EC.3.6.1609-1618.2004
Maki M (2012) ESCRT and calpain—old and new relationships. FEBS J 279(8):1398. https://doi.org/10.1111/j.1742-4658.2012.08561.x
Davenport NR, Sonnemann KJ, Eliceiri KW, Bement WM (2016) Membrane dynamics during cellular wound repair. Mol Biol Cell 27(14):2272–2285. https://doi.org/10.1091/mbc.E16-04-0223
Iliev AI, Djannatian JR, Nau R, Mitchell TJ, Wouters FS (2007) Cholesterol-dependent actin remodeling via RhoA and Rac1 activation by the Streptococcus pneumoniae toxin pneumolysin. Proc Natl Acad Sci USA 104(8):2897–2902. https://doi.org/10.1073/pnas.0608213104
Demonbreun AR, Quattrocelli M, Barefield DY, Allen MV, Swanson KE, McNally EM (2016) An actin-dependent annexin complex mediates plasma membrane repair in muscle. J Cell Biol 213(6):705–718. https://doi.org/10.1083/jcb.201512022
Ghosh S, Shinogle HE, Galeva NA, Dobrowsky RT, Blagg BS (2016) Endoplasmic reticulum-resident heat shock protein 90 (HSP90) isoform glucose-regulated protein 94 (GRP94) regulates cell polarity and cancer cell migration by affecting intracellular transport. J Biol Chem 291(16):8309–8323. https://doi.org/10.1074/jbc.M115.688374
van Vliet AR, Giordano F, Gerlo S, Segura I, Van Eygen S, Molenberghs G, Rocha S, Houcine A, Derua R, Verfaillie T, Vangindertael J, De Keersmaecker H, Waelkens E, Tavernier J, Hofkens J, Annaert W, Carmeliet P, Samali A, Mizuno H, Agostinis P (2017) The ER stress sensor PERK coordinates ER-plasma membrane contact site formation through interaction with filamin-A and F-actin remodeling. Mol Cell 65(5):885–899 e886. https://doi.org/10.1016/j.molcel.2017.01.020
Hong F, Mohammad Rachidi S, Lundgren D, Han D, Huang X, Zhao H, Kimura Y, Hirano H, Ohara O, Udono H, Meng S, Liu B, Li Z (2017) Mapping the interactome of a major mammalian endoplasmic reticulum heat shock protein 90. PLoS One 12(1):e0169260. https://doi.org/10.1371/journal.pone.0169260
Lin P, Zhu H, Cai C, Wang X, Cao C, Xiao R, Pan Z, Weisleder N, Takeshima H, Ma J (2012) Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated cell membrane repair. FASEB J 26(5):1875–1883. https://doi.org/10.1096/fj.11-188599
O’Brien ET, Salmon ED, Erickson HP (1997) How calcium causes microtubule depolymerization. Cell Motil Cytoskelet 36(2):125–135. https://doi.org/10.1002/(SICI)1097-0169(1997)36:2%3c125:AID-CM3%3e3.0.CO;2-8
Togo T (2006) Disruption of the plasma membrane stimulates rearrangement of microtubules and lipid traffic toward the wound site. J Cell Sci 119(Pt 13):2780–2786. https://doi.org/10.1242/jcs.03006
Shen SS, Tucker WC, Chapman ER, Steinhardt RA (2005) Molecular regulation of membrane resealing in 3T3 fibroblasts. J Biol Chem 280(2):1652–1660. https://doi.org/10.1074/jbc.M410136200
Iliev AI, Djannatian JR, Opazo F, Gerber J, Nau R, Mitchell TJ, Wouters FS (2009) Rapid microtubule bundling and stabilization by the Streptococcus pneumoniae neurotoxin pneumolysin in a cholesterol-dependent, non-lytic and Src-kinase dependent manner inhibits intracellular trafficking. Mol Microbiol 71(2):461–477
Cargnello M, Roux PP (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 75(1):50–83. https://doi.org/10.1128/MMBR.00031-10
Porta H, Cancino-Rodezno A, Soberon M, Bravo A (2011) Role of MAPK p38 in the cellular responses to pore-forming toxins. Peptides 32(3):601–606. https://doi.org/10.1016/j.peptides.2010.06.012
Husmann M, Dersch K, Bobkiewicz W, Beckmann E, Veerachato G, Bhakdi S (2006) Differential role of p38 mitogen activated protein kinase for cellular recovery from attack by pore-forming S. aureus alpha-toxin or streptolysin O. Biochem Biophys Res Commun 344(4):1128–1134. https://doi.org/10.1016/j.bbrc.2006.03.241
Park JM, Ng VH, Maeda S, Rest RF, Karin M (2004) Anthrolysin O and other gram-positive cytolysins are toll-like receptor 4 agonists. J Exp Med 200(12):1647–1655. https://doi.org/10.1084/jem.20041215
Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, Koyasu S, Matsumoto K, Takeda K, Ichijo H (2005) ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol 6(6):587–592. https://doi.org/10.1038/ni1200
Babilonia E, Li D, Wang Z, Sun P, Lin DH, Jin Y, Wang WH (2006) Mitogen-activated protein kinases inhibit the ROMK (Kir 1.1)-like small conductance K channels in the cortical collecting duct. J Am Soc Nephrol 17(10):2687–2696. https://doi.org/10.1681/asn.2006050426
Shim JH, Lim JW, Kim BK, Park SJ, Kim SW, Choi TH (2015) KCl mediates K(+) channel-activated mitogen-activated protein kinases signaling in wound healing. Arch Plast Surg 42(1):11–19. https://doi.org/10.5999/aps.2015.42.1.11
He Y, Zeng MY, Yang D, Motro B, Nunez G (2016) NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530(7590):354–357. https://doi.org/10.1038/nature16959
Chu J, Thomas LM, Watkins SC, Franchi L, Nunez G, Salter RD (2009) Cholesterol-dependent cytolysins induce rapid release of mature IL-1beta from murine macrophages in a NLRP3 inflammasome and cathepsin B-dependent manner. J Leukoc Biol 86(5):1227–1238. https://doi.org/10.1189/jlb.0309164
Harder J, Franchi L, Munoz-Planillo R, Park JH, Reimer T, Nunez G (2009) Activation of the Nlrp3 inflammasome by Streptococcus pyogenes requires streptolysin O and NF-kappa B activation but proceeds independently of TLR signaling and P2X7 receptor. J Immunol 183(9):5823–5829. https://doi.org/10.4049/jimmunol.0900444
Ratner AJ, Hippe KR, Aguilar JL, Bender MH, Nelson AL, Weiser JN (2006) Epithelial cells are sensitive detectors of bacterial pore-forming toxins. J Biol Chem 281(18):12994–12998. https://doi.org/10.1074/jbc.M511431200
Gelber SE, Aguilar JL, Lewis KL, Ratner AJ (2008) Functional and phylogenetic characterization of Vaginolysin, the human-specific cytolysin from Gardnerella vaginalis. J Bacteriol 190(11):3896–3903. https://doi.org/10.1128/JB.01965-07
Rampersaud R, Planet PJ, Randis TM, Kulkarni R, Aguilar JL, Lehrer RI, Ratner AJ (2011) Inerolysin, a cholesterol-dependent cytolysin produced by Lactobacillus iners. J Bacteriol 193(5):1034–1041. https://doi.org/10.1128/JB.00694-10
Nagahama M, Shibutani M, Seike S, Yonezaki M, Takagishi T, Oda M, Kobayashi K, Sakurai J (2013) The p38 MAPK and JNK pathways protect host cells against Clostridium perfringens beta-toxin. Infect Immun 81(10):3703–3708. https://doi.org/10.1128/IAI.00579-13
Aguilar JL, Kulkarni R, Randis TM, Soman S, Kikuchi A, Yin Y, Ratner AJ (2009) Phosphatase-dependent regulation of epithelial mitogen-activated protein kinase responses to toxin-induced membrane pores. PLoS One 4(11):e8076. https://doi.org/10.1371/journal.pone.0008076
Cancino-Rodezno A, Alexander C, Villasenor R, Pacheco S, Porta H, Pauchet Y, Soberon M, Gill SS, Bravo A (2010) The mitogen-activated protein kinase p38 is involved in insect defense against Cry toxins from Bacillus thuringiensis. Insect Biochem Mol Biol 40(1):58–63. https://doi.org/10.1016/j.ibmb.2009.12.010
Kao CY, Los FC, Huffman DL, Wachi S, Kloft N, Husmann M, Karabrahimi V, Schwartz JL, Bellier A, Ha C, Sagong Y, Fan H, Ghosh P, Hsieh M, Hsu CS, Chen L, Aroian RV (2011) Global functional analyses of cellular responses to pore-forming toxins. PLoS Pathog 7(3):e1001314. https://doi.org/10.1371/journal.ppat.1001314
Qiu L, Fan J, Liu L, Zhang B, Wang X, Lei C, Lin Y, Ma W (2017) Knockdown of the MAPK p38 pathway increases the susceptibility of Chilo suppressalis larvae to Bacillus thuringiensis Cry1Ca toxin. Sci Rep 7:43964. https://doi.org/10.1038/srep43964
Canault M, Duerschmied D, Brill A, Stefanini L, Schatzberg D, Cifuni SM, Bergmeier W, Wagner DD (2010) p38 mitogen-activated protein kinase activation during platelet storage: consequences for platelet recovery and hemostatic function in vivo. Blood 115(9):1835–1842
Muralidharan-Chari V, Clancy J, Plou C, Romao M, Chavrier P, Raposo G, D’Souza-Schorey C (2009) ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr Biol 19(22):1875–1885
Turola E, Furlan R, Bianco F, Matteoli M, Verderio C (2012) Microglial microvesicle secretion and intercellular signaling. Front Physiol 3:149
Popova TG, Millis B, Bradburne C, Nazarenko S, Bailey C, Chandhoke V, Popov SG (2006) Acceleration of epithelial cell syndecan-1 shedding by anthrax hemolytic virulence factors. BMC Microbiol 6:8. https://doi.org/10.1186/1471-2180-6-8
Stringaris AK, Geisenhainer J, Bergmann F, Balshusemann C, Lee U, Zysk G, Mitchell TJ, Keller BU, Kuhnt U, Gerber J, Spreer A, Bahr M, Michel U, Nau R (2002) Neurotoxicity of pneumolysin, a major pneumococcal virulence factor, involves calcium influx and depends on activation of p38 mitogen-activated protein kinase. Neurobiol Dis 11(3):355–368
Grootjans J, Kaser A, Kaufman RJ, Blumberg RS (2016) The unfolded protein response in immunity and inflammation. Nat Rev Immunol 16(8):469–484. https://doi.org/10.1038/nri.2016.62
Pillich H, Loose M, Zimmer KP, Chakraborty T (2012) Activation of the unfolded protein response by Listeria monocytogenes. Cell Microbiol 14(6):949–964. https://doi.org/10.1111/j.1462-5822.2012.01769.x
Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L, Truitt M, McManus MT, Ruggero D, Goga A, Papa FR, Oakes SA (2012) IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic caspase-2. Science 338(6108):818–822. https://doi.org/10.1126/science.1226191
Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, Heintz N, Greengard P, Hui S, Tang Q, Trusina A, Oakes SA, Papa FR (2012) IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab 16(2):250–264. https://doi.org/10.1016/j.cmet.2012.07.007
Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, Kilberg MS, Sartor MA, Kaufman RJ (2013) ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15(5):481–490. https://doi.org/10.1038/ncb2738
Ghosh R, Wang L, Wang ES, Perera BG, Igbaria A, Morita S, Prado K, Thamsen M, Caswell D, Macias H, Weiberth KF, Gliedt MJ, Alavi MV, Hari SB, Mitra AK, Bhhatarai B, Schurer SC, Snapp EL, Gould DB, German MS, Backes BJ, Maly DJ, Oakes SA, Papa FR (2014) Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 158(3):534–548. https://doi.org/10.1016/j.cell.2014.07.002
Baruch M, Belotserkovsky I, Hertzog BB, Ravins M, Dov E, McIver KS, Le Breton YS, Zhou Y, Cheng CY, Hanski E (2014) An extracellular bacterial pathogen modulates host metabolism to regulate its own sensing and proliferation. Cell 156(1–2):97–108. https://doi.org/10.1016/j.cell.2013.12.007
Akazawa Y, Isomoto H, Matsushima K, Kanda T, Minami H, Yamaghchi N, Taura N, Shiozawa K, Ohnita K, Takeshima F, Nakano M, Moss J, Hirayama T, Nakao K (2013) Endoplasmic reticulum stress contributes to Helicobacter pylori VacA-induced apoptosis. PLoS One 8(12):e82322. https://doi.org/10.1371/journal.pone.0082322
Cywes Bentley C, Hakansson A, Christianson J, Wessels MR (2005) Extracellular group A Streptococcus induces keratinocyte apoptosis by dysregulating calcium signalling. Cell Microbiol 7(7):945–955. https://doi.org/10.1111/j.1462-5822.2005.00525.x
Crowley KS, Liao S, Worrell VE, Reinhart GD, Johnson AE (1994) Secretory proteins move through the endoplasmic reticulum membrane via an aqueous, gated pore. Cell 78(3):461–471
Hamman BD, Hendershot LM, Johnson AE (1998) BiP maintains the permeability barrier of the ER membrane by sealing the lumenal end of the translocon pore before and early in translocation. Cell 92(6):747–758
Abrami L, Fivaz M, Glauser PE, Parton RG, van der Goot FG (1998) A pore-forming toxin interacts with a GPI-anchored protein and causes vacuolation of the endoplasmic reticulum. J Cell Biol 140(3):525–540
Moschioni M, Tombola F, de Bernard M, Coelho A, Zitzer A, Zoratti M, Montecucco C (2002) The Vibrio cholerae haemolysin anion channel is required for cell vacuolation and death. Cell Microbiol 4(7):397–409
Saka HA, Gutierrez MG, Bocco JL, Colombo MI (2007) The autophagic pathway: a cell survival strategy against the bacterial pore-forming toxin Vibrio cholerae cytolysin. Autophagy 3(4):363–365
Figueroa-Arredondo P, Heuser JE, Akopyants NS, Morisaki JH, Giono-Cerezo S, Enriquez-Rincon F, Berg DE (2001) Cell vacuolation caused by Vibrio cholerae hemolysin. Infect Immun 69(3):1613–1624
Gurcel L, Abrami L, Girardin S, Tschopp J, van der Goot FG (2006) Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 126(6):1135–1145. https://doi.org/10.1016/j.cell.2006.07.033
Sato R (2009) SREBPs: protein interaction and SREBPs. FEBS J 276(3):622–627. https://doi.org/10.1111/j.1742-4658.2008.06807.x
Xu B, Jiang M, Chu Y, Wang W, Chen D, Li X, Zhang Z, Zhang D, Fan D, Nie Y, Shao F, Wu K, Liang J (2017) Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J Hepatol. https://doi.org/10.1016/j.jhep.2017.11.040
Galmiche A, Rassow J, Doye A, Cagnol S, Chambard JC, Contamin S, de Thillot V, Just I, Ricci V, Solcia E, Van Obberghen E, Boquet P (2000) The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J 19(23):6361–6370. https://doi.org/10.1093/emboj/19.23.6361
Willhite DC, Blanke SR (2004) Helicobacter pylori vacuolating cytotoxin enters cells, localizes to the mitochondria, and induces mitochondrial membrane permeability changes correlated to toxin channel activity. Cell Microbiol 6(2):143–154
Genestier AL, Michallet MC, Prevost G, Bellot G, Chalabreysse L, Peyrol S, Thivolet F, Etienne J, Lina G, Vallette FM, Vandenesch F, Genestier L (2005) Staphylococcus aureus Panton–Valentine leukocidin directly targets mitochondria and induces Bax-independent apoptosis of human neutrophils. J Clin Investig 115(11):3117–3127. https://doi.org/10.1172/JCI22684
Braun JS, Hoffmann O, Schickhaus M, Freyer D, Dagand E, Bermpohl D, Mitchell TJ, Bechmann I, Weber JR (2007) Pneumolysin causes neuronal cell death through mitochondrial damage. Infect Immun 75(9):4245–4254. https://doi.org/10.1128/IAI.00031-07
Bielaszewska M, Ruter C, Kunsmann L, Greune L, Bauwens A, Zhang W, Kuczius T, Kim KS, Mellmann A, Schmidt MA, Karch H (2013) Enterohemorrhagic Escherichia coli hemolysin employs outer membrane vesicles to target mitochondria and cause endothelial and epithelial apoptosis. PLoS Pathog 9(12):e1003797. https://doi.org/10.1371/journal.ppat.1003797
Nerlich A, Mieth M, Letsiou E, Fatykhova D, Zscheppang K, Imai-Matsushima A, Meyer TF, Paasch L, Mitchell TJ, Tonnies M, Bauer TT, Schneider P, Neudecker J, Ruckert JC, Eggeling S, Schimek M, Witzenrath M, Suttorp N, Hippenstiel S, Hocke AC (2018) Pneumolysin induced mitochondrial dysfunction leads to release of mitochondrial DNA. Sci Rep 8(1):182. https://doi.org/10.1038/s41598-017-18468-7
Goldmann O, Sastalla I, Wos-Oxley M, Rohde M, Medina E (2009) Streptococcus pyogenes induces oncosis in macrophages through the activation of an inflammatory programmed cell death pathway. Cell Microbiol 11(1):138–155. https://doi.org/10.1111/j.1462-5822.2008.01245.x
Fennessey CM, Ivie SE, McClain MS (2012) Coenzyme depletion by members of the aerolysin family of pore-forming toxins leads to diminished ATP levels and cell death. Mol BioSyst 8(8):2097–2105. https://doi.org/10.1039/c2mb25142f
Jain P, Luo ZQ, Blanke SR (2011) Helicobacter pylori vacuolating cytotoxin A (VacA) engages the mitochondrial fission machinery to induce host cell death. Proc Natl Acad Sci USA 108(38):16032–16037. https://doi.org/10.1073/pnas.1105175108
Stavru F, Bouillaud F, Sartori A, Ricquier D, Cossart P (2011) Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc Natl Acad Sci USA 108(9):3612–3617. https://doi.org/10.1073/pnas.1100126108
Malet JK, Cossart P, Ribet D (2017) Alteration of epithelial cell lysosomal integrity induced by bacterial cholesterol-dependent cytolysins. Cell Microbiol. https://doi.org/10.1111/cmi.12682
Ribet D, Hamon M, Gouin E, Nahori MA, Impens F, Neyret-Kahn H, Gevaert K, Vandekerckhove J, Dejean A, Cossart P (2010) Listeria monocytogenes impairs SUMOylation for efficient infection. Nature 464(7292):1192–1195. https://doi.org/10.1038/nature08963
Malet JK, Impens F, Carvalho F, Hamon MA, Cossart P, Ribet D (2018) Rapid remodeling of the host epithelial cell proteome by the listeriolysin O pore-forming toxin. Mol Cell Proteom. https://doi.org/10.1074/mcp.ra118.000767
Kuo CJ, Hansen M, Troemel E (2018) Autophagy and innate immunity: insights from invertebrate model organisms. Autophagy 14(2):233–242. https://doi.org/10.1080/15548627.2017.1389824
Mathieu J (2015) Interactions between autophagy and bacterial toxins: targets for therapy? Toxins 7(8):2918–2958. https://doi.org/10.3390/toxins7082918
Yuan K, Huang C, Fox J, Laturnus D, Carlson E, Zhang B, Yin Q, Gao H, Wu M (2012) Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. J Cell Sci 125(Pt 2):507–515. https://doi.org/10.1242/jcs.094573
Kloft N, Neukirch C, Bobkiewicz W, Veerachato G, Busch T, von Hoven G, Boller K, Husmann M (2010) Pro-autophagic signal induction by bacterial pore-forming toxins. Med Microbiol Immunol 199(4):299–309. https://doi.org/10.1007/s00430-010-0163-0
He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93. https://doi.org/10.1146/annurev-genet-102808-114910
Terebiznik MR, Raju D, Vazquez CL, Torbricki K, Kulkarni R, Blanke SR, Yoshimori T, Colombo MI, Jones NL (2009) Effect of Helicobacter pylori’s vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy 5(3):370–379
Gutierrez MG, Saka HA, Chinen I, Zoppino FC, Yoshimori T, Bocco JL, Colombo MI (2007) Protective role of autophagy against Vibrio cholerae cytolysin, a pore-forming toxin from V. cholerae. Proc Natl Acad Sci USA 104(6):1829–1834. https://doi.org/10.1073/pnas.0601437104
Yorimitsu T, Klionsky DJ (2007) Eating the endoplasmic reticulum: quality control by autophagy. Trends Cell Biol 17(6):279–285. https://doi.org/10.1016/j.tcb.2007.04.005
Goldman SJ, Taylor R, Zhang Y, Jin S (2010) Autophagy and the degradation of mitochondria. Mitochondrion 10(4):309–315. https://doi.org/10.1016/j.mito.2010.01.005
Hung YH, Chen LM, Yang JY, Yang WY (2013) Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat Commun 4:2111. https://doi.org/10.1038/ncomms3111
Chen X, Khambu B, Zhang H, Gao W, Li M, Chen X, Yoshimori T, Yin XM (2014) Autophagy induced by calcium phosphate precipitates targets damaged endosomes. J Biol Chem 289(16):11162–11174. https://doi.org/10.1074/jbc.M113.531855
Mestre MB, Fader CM, Sola C, Colombo MI (2010) Alpha-hemolysin is required for the activation of the autophagic pathway in Staphylococcus aureus-infected cells. Autophagy 6(1):110–125
Birmingham CL, Canadien V, Gouin E, Troy EB, Yoshimori T, Cossart P, Higgins DE, Brumell JH (2007) Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy 3(5):442–451
Birmingham CL, Canadien V, Kaniuk NA, Steinberg BE, Higgins DE, Brumell JH (2008) Listeriolysin O allows Listeria monocytogenes replication in macrophage vacuoles. Nature 451(7176):350–354. https://doi.org/10.1038/nature06479
Birmingham CL, Higgins DE, Brumell JH (2008) Avoiding death by autophagy: interactions of Listeria monocytogenes with the macrophage autophagy system. Autophagy 4(3):368–371
Mostowy S, Cossart P (2012) Bacterial autophagy: restriction or promotion of bacterial replication? Trends Cell Biol 22(6):283–291. https://doi.org/10.1016/j.tcb.2012.03.006
Di Paolo NC, Doronin K, Baldwin LK, Papayannopoulou T, Shayakhmetov DM (2013) The transcription factor IRF3 triggers “defensive suicide” necrosis in response to viral and bacterial pathogens. Cell Rep 3(6):1840–1846. https://doi.org/10.1016/j.celrep.2013.05.025
McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, Smeaton S, El-Rachkidy R, McLoughlin RM, Mori A, Moran B, Fitzgerald KA, Tschopp J, Petrilli V, Andrew PW, Kadioglu A, Lavelle EC (2010) Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog 6(11):e1001191. https://doi.org/10.1371/journal.ppat.1001191
Iacovache I, Bischofberger M, van der Goot FG (2010) Structure and assembly of pore-forming proteins. Curr Opin Struct Biol 20(2):241–246. https://doi.org/10.1016/j.sbi.2010.01.013
Acknowledgements
We apologize to those researchers whose work we could not cite due to space limitations and gratefully acknowledge their contributions to the field. Work in the Group of Molecular Microbiology is supported by national funds through FCT—Fundação para a Ciência e a Tecnologia/MEC—Ministério da Educação e Ciência and co-funded by FEDER—Fundo Europeu de Desenvolvimento Regional through NORTE 2020 within the project NORTE-01-0145-FEDER-000012—Structured program on bioengineered therapies for infectious diseases, and Portugal 2020 and COMPETE 2020—Operacional Programme for Competitiveness and Internationalization (POCI) within the project POCI-01-0145-FEDER-030863. Publication Fees were supported by ICBAS, University of Porto. C.B. and F.S.M. were supported by FCT fellowships (SFRH/BD/112217/2015, SFRH/BPD/94458/2013, respectively). S.S. received support from FCT Investigator program (COMPETE, POPH, and FCT).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Brito, C., Cabanes, D., Sarmento Mesquita, F. et al. Mechanisms protecting host cells against bacterial pore-forming toxins. Cell. Mol. Life Sci. 76, 1319–1339 (2019). https://doi.org/10.1007/s00018-018-2992-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-018-2992-8